Back to Journals » OncoTargets and Therapy » Volume 13
Clinicopathological, Immunohistochemical and Molecular Genetic Study on Epithelioid Glioblastoma: A Series of Fifteen Cases with Literature Review
Authors Zeng Y, Zhu X, Wang Y, Liu B, Yang X, Wang Q, Du J, Ma Y, Lin L, Fu P, Xiao H, Guo QN ![]()
Received 11 February 2020
Accepted for publication 19 April 2020
Published 8 May 2020 Volume 2020:13 Pages 3943—3952
DOI https://doi.org/10.2147/OTT.S249317
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Ying Zeng,1,2 Xiangfeng Zhu,2 Yali Wang,1 Bo Liu,1 Xin Yang,2 Qiushi Wang,2 Juan Du,2 Yu Ma,2 Li Lin,2 Ping Fu,2 Hualiang Xiao,2 Qiao-Nan Guo1
1Department of Pathology, Second Affiliated Hospital, Amy Medical University (Third Military Medical University), Chongqing 400037, People’s Republic of China; 2Department of Pathology, Daping Hospital, Amy Medical University (Third Military Medical University), Chongqing 400042, People’s Republic of China
Correspondence: Qiao-Nan Guo; Hualiang Xiao Email [email protected]; [email protected]
Purpose: To observe the clinicopathological, immunohistochemical, and molecular genetic features of epithelioid glioblastoma (E-GBM), and identify tumor-associated prognostic factors.
Patients and Methods: The clinical and radiological data of fifteen cases of E-GBM were collected, and their pathological, immunohistochemical, and molecular features were examined. A 1p/19q analysis via FISH, MGMT promoter methylation by MS-PCR, and IDH1 and BRAF V600E mutation analysis by HRM-PCR were performed. The level of EZH2 expression was valuated by immunohistochemistry in 15 E-GBM cases, and the prognostic factors were analyzed in E-GBM patients. Fifteen non-E-GBM cases were used as a control.
Results: The fifteen cases of E-GBM included twelve males and three females, with fourteen cases supratentorially located. Headache was the main symptom. Microscopy revealed that the tumors were composed of epithelioid cells and some rhabdoid cells. The epithelioid and rhabdoid cells displayed focal discohesion, scant intervening neuropil, a distinct cell membrane, eosinophilic cytoplasm, and a laterally positioned nucleus. Most tumors showed high mitosis, zonal necrosis, and microvascular hyperplasia. Immunohistochemical findings included epithelioid cells positive for GFAP, vimentin, nestin, S-100, and INI-1. The molecular findings included no deletions of 1p/19q, EGFR amplifications, or IDH1 mutations in any case, a methylated MGMT promoter in 46.7% (7/15) cases, and a BRAFV600E mutation in 46.7% (7/15) cases. EZH2 overexpression occurred in 60.0% (9/15) of E-GBM cases. E-GBM patients with OS (≤ 12 months) exhibited extensive necrosis (6/6), EZH2 overexpression (6/6), MGMT promoter unmethylation (5/6), BRAFV600E mutation (3/6), and treatment (surgery4/6). E-GBM patients with OS (> 12 months) exhibited focal or limited necrosis, low or negative EZH2 expression, MGMT promoter methylation (2/3), BRAFV600E mutation (3/3), and treatment (surgery+radiotherapy/chemo-radiotherapy, 2/3).
Conclusion: E-GBM was a rare variant of glioblastoma, with histological epithelioid features and poor prognosis. Extensive necrosis, MGMT promoter unmethylation, EZH2 overexpression, and lack of adjuvant chemo-radiotherapy may indicate a poor prognosis.
Keywords: central nervous system tumor, epithelioid glioblastoma, immunohistochemistry, clinicopathological features, molecular genetics, differential diagnosis
Introduction
Epithelioid glioblastoma (E-GBM) is a new variant of glioblastoma (GB) that was recently formally recognized by the World Health Organization (WHO) Classification of Tumours of the Central Nervous System (2016, revised 4th edition). E-GBM is defined as a high-grade diffuse astrocytic tumor variant with a dominant population of closely packed epithelioid cells, some rhabdoid cells, mitotic activity, microvascular proliferation, and necrosis. E-GBMs predominantly occur in young adults and children, and are preferentially located in the cerebrum or diencephalon. Moreover, E-GBMs are aggressive tumors associated with short survival, particularly in children compared to typical GBM.1 Since E-GBM shares morphological features with other tumors and lacks specific radiological2,3 and molecular hallmarks, it poses a diagnostic challenge. Although the prognosis is dismal in a majority of cases, a relatively longer survival has been noted in a small subset of patients.4–7 Thus, multi-institutional studies with a close follow-up are necessary for determining the actual frequency, prognostic evaluation, and appropriate therapeutic intervention for E-GBM. In addition, there is considerable molecular and clinical heterogeneity within the E-GBM category, making it difficult to identify significant prognostic parameters.8 Therefore, it is essential to accumulate additional cases to understand the various tumor characteristics and explore relevant prognostic factors. In this study, we report fifteen cases of E-GBM and the associated clinical, pathological, immunohistochemical, and molecular features, detect the expression of EZH2 in E-GBM. We found that extensive necrosis, unmethylated MGMT promoter, EZH2 overexpression, and lack of adjuvant chemo-radiotherapy may indicate a poor prognosis. We further discuss the clinical features of the tumor, pathological diagnosis, and molecular features based on these cases and those in a related literature review.
Materials and Methods
Data Collection and Histopathological Review
This study performed a retrospective review of the data from 15 E-GBM cases that were treated between 2010 and 2019 were obtained from the Department of Pathology at Third Affiliated Hospital and Second Affiliated Hospital, Amy Medical University. The data from 15 non-E-GBM cases were used as a control. The pathological materials were subjected to a blinded review by two pathologists (Y.Z and H.L.X) according to the definition of E-GBM in the WHO Classification of Tumors of the Central Nervous System. This study was conducted following approval by the Research Ethics Committee of Daping Hospital of The Amy Military Medical University (approval number: 2018–125) and conformed to the 1975 Declaration of Helsinki. All patients provided written informed consent.
Immunohistochemistry
An immunohistochemical (IHC) analysis was performed on 3-μm-thick representative sections following deparaffinization in xylene and rehydration in alcohol. All IHC procedures were performed using a Roche BENCHMARK XT (Roche, Ventana, USA) according to routine laboratory practices and the manufacturer’s protocol. EZH2 staining was graded as “−”, “+”, and “++”, when the percentage of positive nuclei was <5%, 5–25%, and >25%, respectively.
Molecular Study
Analysis of 1p/19q and epidermal growth factor receptor (EGFR) gene status was conducted by fluorescence in situ hybridization (FISH) using Vysis probes (Abbott Laboratories, Abbott Park, IL, USA). O-6-methylguanine-DNA methyltransferase (MGMT) methylation-specific PCR (MSP), HRM-PCR of isocitrate dehydrogenase 1 (IDH1) R132, and BRAF V600E were performed. For DNA extraction, the tumor areas were manually microdissected from 6-μm unstained histological sections obtained from formalin-fixed, paraffin-embedded (FFPE) tissues. DNA was isolated from the target tissues using a DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) in accordance with the manufacturer’s instructions.
Statistical Analysis
The patient characteristics were summarized based on the medians and standard deviations or ranges for continuous data, as well as the frequencies and percentages for the categorical data. Patient characteristics were compared between the two groups using a chi-squared and Fisher’s exact tests, as appropriate. Overall survival (OS) was defined as the time between the diagnosis and the last follow up or death. Survival curves were calculated using the Kaplan–Meier method. Differences between the curves were assessed using a log-rank analysis. A p-value <0.05 was considered to be statistically significant. Statistical analyses were performed using SPSS version 20.0™ (SPSS, Inc, USA).
Results
Patient Characteristics
Epithelioid glioblastoma (n = 15) accounted for 3% of glioblastoma (n = 498) during the same period. The mean age of the 12 male patients and 3 female patients was 39.6 years (range: 18–77 years). The median age at diagnosis was 34 years. Nine patients experienced headaches for up to eight months, and six patients had experienced dizziness and vomiting, left limb weakness, and progressive memory loss was observed in one patient.
One patient had a history of anaplastic astrocytoma for five years. A tumor location in the temporal lobe accounted for 53.3% (8/15) of cases, frontal lobe accounted for 46.7% (7/15) of cases, and two lobes occurred in 33.3% (5/15) of cases. Tumor sizes ranged from 2.7 × 1.7 × 1.6 cm to 9.2 × 9.0 × 2.2cm (Table 1). Radiologically, a well-circumscribed enhancing mass was observed in ten cases, an ill-circumscribed enhancing mass was observed in three cases, a cystic and solid space mass in two cases, and dura mater attachment was observed in two cases. T2-weighted images revealed peritumoral edema in all 15 cases. Neuroradiological findings for E-GBM case #2 showed a heterogeneous lesion with necrosis and perilesional edema on T1 in the left temporal lobe, 5.3 cm × 4.3 cm in size (Figure 1A), a heterogeneous lesion with perilesional edema on T2 (Figure 1B), and a rim-enhancing mass with perilesional edema on T1-weighted enhanced (Figure 1C)
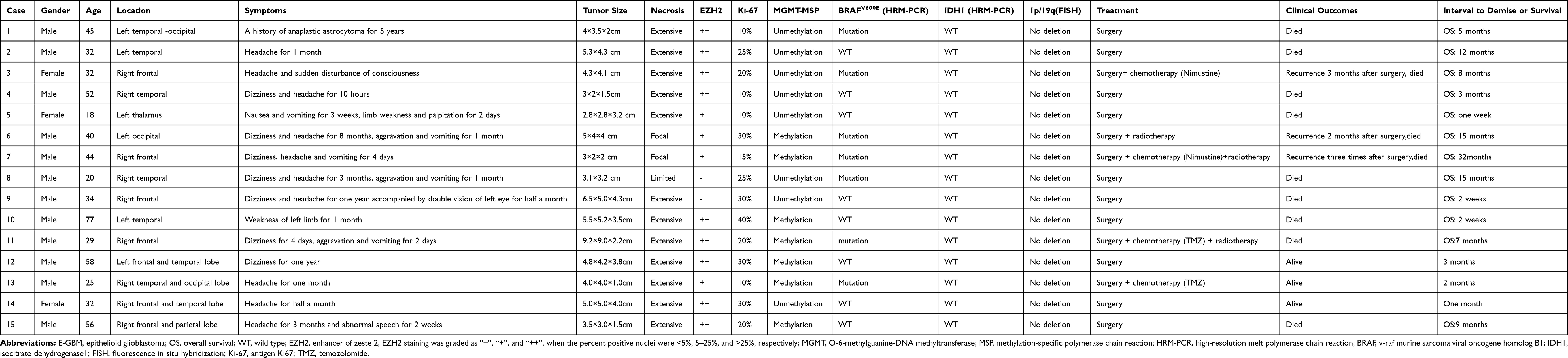 |
Table 1 Summary of the Clinical Parameters, Immunohistochemistry and Molecular Findings of 15 E-GBM Patients |
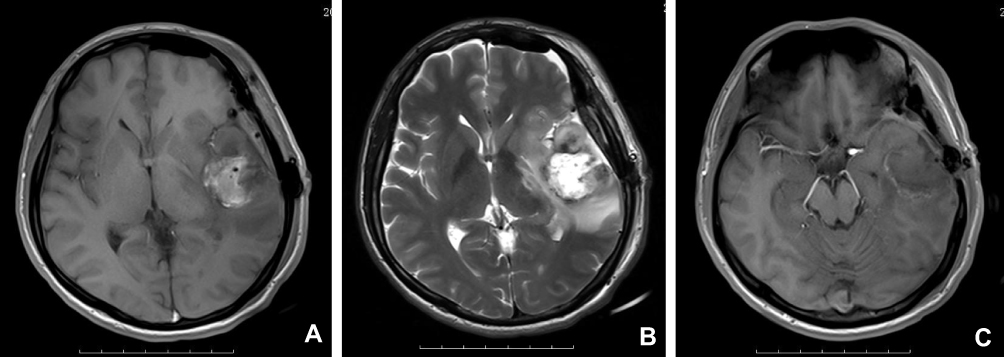 |
Figure 1 Neuroradiological findings for E-GBM case #2. (A) A heterogeneous lesion with necrosis and perilesional edema on T1 in the left temporal lobe, 5.3 cm × 4.3 cm in size. (B) A heterogeneous lesion with perilesional edema (T2). (C) A rim-enhancing mass with perilesional edema (T1-weighted enhanced). Abbreviation: E-GBM, epithelioid glioblastoma. |
All patients underwent complete surgical resection. Two patients were treated with combined chemotherapy and radiation therapy after surgery, one patient was treated only with chemotherapy (Nimustine) after surgery, one patient was treated with chemotherapy (TMZ) after surgery, one patient was treated with radiation therapy after surgery, and five patients were treated with neither chemotherapy nor radiation therapy after surgery. The follow-up time for two patients was short, and no post-operative chemoradiotherapy had been performed. Three patients died of post-operative complications. The follow-up time ranged from 1 to 48 months. For the three patients who relapsed (one within 3 months, one within 2 months and one within 3, 5, and 15 months), salvage management, including a second, third, fourth operations, was performed. By the time of the last follow-up, 12 patients had died, one patient died one week after surgery, and two patients died two weeks after surgery due to respiratory failure. The OS of nine patients was 3–32 months, the mean OS was 10.8 months, the median OS was 8.0 months, and the one-year survival rate was 27.00% (95% CI: 0.1042–0.6998). Three patients remained alive at 1, 2, and 3 months after diagnosis. The clinical findings, including radiologic features, treatment modalities, and follow-up data are summarized in Table 1.
Pathological Findings
Macroscopically, the tumor was classified as soft in fourteen cases and moderate in one case. The results of the pathological findings are shown in Figure 2. Tumors were composed of both closely arranged epithelioid and rhabdoid cells (Figure 2A). Tumors were rich in vasculature and vascular proliferation was observed, including thin-walled and thick-walled vessels with hyaline degeneration, microvascular proliferation, and glomerulus-like vasculature. Epithelioid cells were grown around the vascular wall and formed a papillary structure in one case. The tumors had zonal necrosis (Figure 2B) in fourteen cases, and palisading necrosis was observed in case #5. The epithelioid cells and some of the rhabdoid cells were round or oval, and contained a laterally positioned nucleus and abundant, homogeneous eosinophilic cytoplasm. In addition, a high mitotic rate was observed (Figure 2C). Three cases exhibited a large amount of hemorrhaging and two cases displayed focal calcification.
 |
Figure 2 Histological, immunohistochemical, and molecular findings of E-GBM. (A) Epithelioid and rhabdoid cells were arranged in patches or were in close proximity with rich vasculature with thin walls (×100). (B) The tumor exhibited zonal necrosis (×100). (C) Epithelioid and rhabdoid cells were round or oval, containing laterally located oval to pleomorphically shaped nuclei and abundant and homogeneously eosinophilic cytoplasm. Mitoses were easily observed (×400). (D) Epithelioid cells were positive for GFAP, INI-1 (E) and EZH2 (F) (×200). (G) MGMT-MSP revealed MGMT methylation in case 6. (H) HRM-PCR revealed a BRAF V600E mutation in case 1. Curve 1 shows the positive control, curve 2 shows the tumor specimen, and curve 3 shows the negative control. (I) EGFR FISH revealed a high level of polysomy in the tumor in case 7. Abbreviations: E-GBM, epithelioid glioblastoma; GFAP, glial fibrillary acidic protein; SMARCB1/INI-1, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily b, member 1; EZH2, enhancer of zeste 2; MGMT, O-6-methylguanine-DNA methyltransferase; MSP, methylation-specific polymerase chain reaction; HRM-PCR, high-resolution melt polymerase chain reaction; BRAF, v-raf murine sarcoma viral oncogene homolog B1; EGFR, epithelial growth factor receptor; FISH, fluorescence in situ hybridization. |
Immunohistochemical Analysis
The results of the immunohistochemical analysis are summarized and presented in Figure 2D–F. These epithelioid cells were immunoreactive for GFAP (Figure 2D) in nine cases and focally in six cases. The epithelioid cells were immunoreactive for vimentin, nestin, c-Met, INI1 (Figure 2E), ATRX, Nanog, MDM2, and S-100 in all 15 cases. The epithelioid cells were immunoreactive for P53 in seven cases. A small population of cells also reacted with EMA in six cases. The epithelioid cells were immunoreactive focally for EGFR in six cases. The Ki-67 proliferation index was 10–40%. The epithelioid cells were negative for NeuN, NF, IDH1, P16, myogenin, MyoD1, SMA, desmin, CK, LCA, CD117, HMB45, CD68, Syn, CD34, and PTEN. EZH2 expression occurred in 86.7% (13/15) of E-GBM cases and EZH2 overexpression occurred in 60.0% (9/15) of E-GBM cases (Figure 2F).
FISH and MGMT Methylation-Specific PCR
Molecular analysis by FISH, HRM-PCR, and MGMT-MSP is summarized in Table 1 and the results are presented in Figure 2G–I. MS-PCR showed that the MGMT promoter was methylated in 46.7% (7/15) cases (Figure 2G). The BRAFV600E mutation was detected in 46.7% (7/15) cases (Figure 2H). Neither an IDH1 mutation by HRM-PCR nor the 1p/19q co-deletion by FISH was detected in any case. EGFR FISH revealed no amplification in any case, a low polysomy in five cases, and high polysomy in one case (Figure 2I).
Outcome Analyses
Follow-up data were accepted from nine E-GBM patients. E-GBM patients (n = 6) with an OS≤12 months exhibited extensive necrosis (6/6), EZH2 overexpression (6/6), unmethylation of the MGMT promoter (5/6), BRAFV600E mutation (3/6), treatment (surgery only, 4/6), E-GBM patients (3 cases) with OS > 12 months exhibited focal or limited necrosis (3/3), low expression or negative EZH2 expression (3/3), MGMT promoter methylation (2/3), BRAFV600E mutation (3/3), and treatment (surgery+ radiotherapy/chemoradiotherapy, 2/3). These findings show extensive necrosis, MGMT promoter unmethylation, EZH2 overexpression, and a lack of adjuvant chemo-radiotherapy may indicate a poor prognosis.
There were significant differences in the median age (p = 0.002) of E-GBM and non-E-GBM. There was no difference in gender (p = 1.000) or median OS (p = 0.079). Histologically, the tumor cells were dominated by homogeneous epithelioid and rhabdoid cells, and necrosis was dominated by zonal necrosis in E-GBM, whereas the tumor cells were characterized by heterogeneous polymorphic cells, palisading necrosis was the main necrosis pattern in non-E-GBM patients. EGFR was expressed focally in E-GBM (6/15) and diffuse expression was observed in non-E-GBM patients (10/15). There was no difference in EZH2 overexpression (p = 0.713), MGMT promoter methylation (p = 0.715), IDH mutation (p = 1.000), and 1p/19q LOH (p = 1.000). The BRAF mutations (p = 0.01) differed between the E-GBM and non-E-GBM patients. The data are summarized in Table 2.
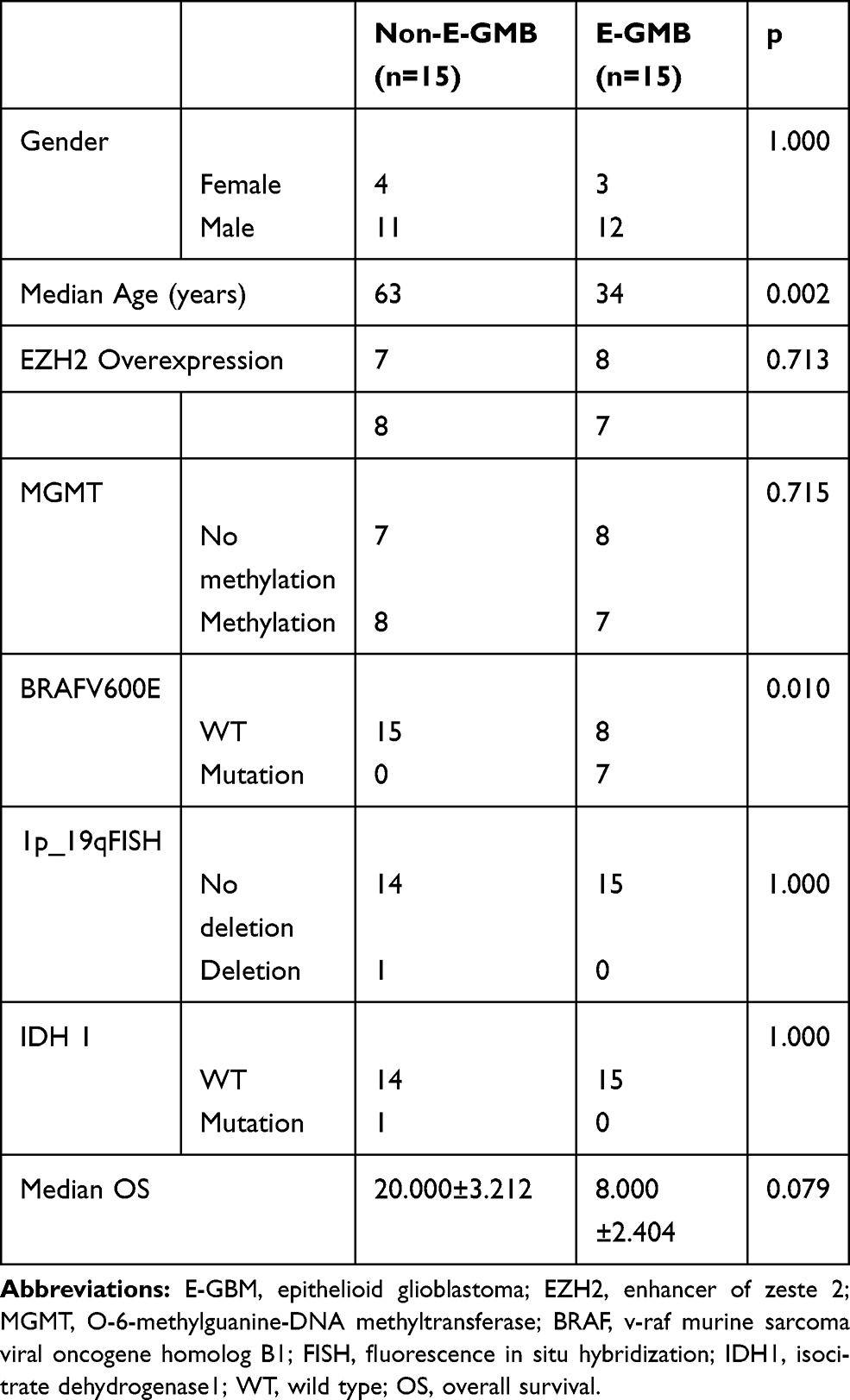 |
Table 2 The Clinical Parameters, Immunohistochemistry and Molecular Findings of 15 non-E-GMB and E-GMB Patients |
Discussion
E-GBM is a rare variant of GBM that has recently been formally recognized by the WHO.1 Approximately 135 cases of E-GBM have been reported in the literature. Tumors predominantly arise in the cerebral hemisphere, although examples in the lateral ventricles, deep nuclei, and posterior fossa have been described. The median age of the reported patients at diagnosis was 30 years, with 54% male patients,9 25 years, with 70% male patients.8 These cases also predominantly occurred in the cerebrum, males were predominantly affected, and the median age paralleled that reported in the literature.9 The clinical manifestation of E-GBM in this panel parallels that of non-E-GBMs, with the majority of patients presenting symptoms and signs of raised intracranial pressure. Only one patient had transformed from anaplastic astrocytoma in this study, suggesting that most E-GBM cases were primary GBM. E-GBM has been reported to demonstrate leptomeningeal spread10 and pulmonary metastasis,11 and we only observed microscopic meningeal spread in one case.
The histological characteristics of pediatric E-GBM are similar to those in adults. E-GBM tumors are dominated by a relatively uniform population of epithelioid cells, exhibiting focal discohension, scant intervening neuropil, a distinct cell membrane, eosinophilic cytoplasm, and a laterally positioned nucleus. Osteoclast-like giant cell-rich E-GBM,12 E-GBM with a subependymal giant cell astrocytoma (SEGA)-like morphology13 was reported. By definition, squamous nests, granular formation, and adenoid features were absent from E-GBM patients. We did not observe squamous nests, granular formation, or adenoid features in these cases. Tumors are rich in vasculature, including thin-walled and thick-walled vessels with hyaline degeneration and microvascular proliferation, as well as glomerulus-like vasculature. In addition, we observed extensive zonal necrosis in most cases, focal zonal necrosis in a few cases and palisading necrosis as an unusual necrosis pattern in one pediatric E-GBM case. Although tumor cells are typically positive for S100 and GFAP, the latter may be present only focally. Staining for cytokeratins and epithelial membrane antigen (EMA) may also be observed. SMARCB1 (INI1) and BRG1 staining is universally intact, and both myogenic and melanocytic markers are routinely negative.14 ATRX was also retained in the tumor cells. Our findings are consistent with those described in the literature.
E-GBM and APXA are closely related.15 However, E-GBM and APXA are two rare entities associated with different prognoses. The ratio of p53 mutation, EZH2 overexpression, and CDKN2A homozygous deletion was higher in E-GBM than in APXA.16 The detected EZH2 overexpression rate (60.0%, 9/15) in E-GBM was similar to that reported in the literature (69.2%, 9/13).16 The PXA feature was not observed in this panel.
E-GBM is associated with a particularly poor prognosis. Lu et al found that the median OS and PFS were estimated to be 11.0 months (range: 6.5–13.0 months) and 7.0 months (range: 3.0–10.0 months), respectively,9 whereas Korshunov et al found the median OS and PFS was 23.0 months and 11.0 months, respectively.8 The median OS in this study was similar to that of Lu et al,9 and more cases are required to further assess the median OS and PFS of E-GBM in the future. Based on methylation patterns, copy number alterations, and mutation analysis data in combination with clinical findings, Korshunov et al disclosed three different, well-established tumor subtypes: 1) PXA-like tumors with favorable prognosis, predominantly in children and young adults; 2) IDH wt GBM-like tumors with a poor prognosis, which primarily occurs in older adults, albeit with more frequent BRAF mutations; and 3) RTK1 pediatric GBM-like neoplasms of intermediate prognosis in children and young adults, which are associated with chromothripsis and frequent PDGFRA amplifications. The authors concluded that histopathologically defined E-GBM does not represent a single diagnostic entity, but rather at least three molecularly and biologically distinct categories. They focused on prognostic factors in E-GBM patients, the univariate OS analysis revealed that older age, presence of any oncogene amplification, 10q deletion, and tumors from either the RTK1 pediatric GBM or those adult IDH wt GBM clusters were associated with a significantly poor survival.8 In contrast, the multivariate analysis did not identify any significant prognostic parameter.8 The surgical extent of resection, radiation therapy, and chemotherapy all predicted a superior OS and PFS in the multivariate analysis.9 Another study demonstrated that an abundance of epithelioid cells, extensive necrosis, EZH2 overexpression, and BRAF V600E mutations were significantly associated with decreased OS in the E-GBM patients.16 We also found that extensive necrosis, unmethylation of the MGMT promoter, EZH2 overexpression indicated a shorter OS, and lack of adjuvant chemo-radiotherapy also indicated a shorter OS. However, we found that E-GBM patients (3 cases) with an OS > 12 months harbored a BRAF V600E mutation, and the role of a BRAF V600E mutation in E-GBM prognosis requires further evaluation. Therefore, in view of molecular heterogeneity and different prognosis in E-GBM, additional molecular testing using genome-wide molecular profiling is recommended to further stratify these rare cases.8
We observed two patients with BRAF V600E mutations and MGMT promoter methylation had a survival time of 15 and 32 months for patients treated with chemotherapy/radiotherapy following surgical resection based on the standard therapeutic course for conventional GBM. As mentioned above, treatment interventions, including maximal surgical resection, radiation therapy, and chemotherapy were all significant independent predictors of better survival outcomes.9 Therefore, E-GBM patients also benefit from standard chemoradiotherapy for typical GBM. In addition to standard chemoradiotherapy for typical GBM, the use of other alternative agents (eg, BRAF inhibitors) has proven to be effective in some patients.10,17-19 However, this would not be restricted to only E-GBM, but rather all glioblastoma that tested positive for the BRAF mutation. We anticipate outcomes based on the BRAF-mutation status and BRAF-inhibitor effect,20 as well as more well-defined prognostic relevance of a BRAF-mutant E-GBM subtype in the future.
The BRAF gene is located on chromosome 7q34 and encodes a cytoplasmic serine-threonine kinase. The mutated BRAF protein constitutively activates the MAPK signaling pathway, which results in increased cell proliferation, apoptosis resistance, and tumor progression. BRAF V600E mutations may be detected using VE1 mutation-specific BRAF V600E antibodies and show good correlation between mutational status and BRAF VE1 IHC.7,21 The BRAF V600E mutation was detected in approximately half of the cases in this panel, which was consistent with those described in the literature.5,6,21 Similar findings have been reported to be as high as 63%9 and 93%21 of cases. In contrast, there is a low frequency of this mutation in non-E-GBM patients. Moreover, E-GBMs rarely have EGFR amplifications,22 and we only observed focal epithelioid cells that were immunopositive for EGFR in six E-GBM cases and no EGFR amplification was observed in any case. Methylation of the MGMT promoter was observed in about half of adult E-GBMs and the MGMT promoter was unmethylated in two cases of pediatric E-GBM. A total of 100% (37/37) cases were IDH1-wild-type,9 no IDH1 mutations were detected in our study, which suggested that these cases of E-GBM are IDH1-wildtype, supporting the notion that most E-GBMs are primary GBM. In addition, a TERT mutation was observed in 40% of cases. H3F3A mutations15,22 and PTEN deletions22 were not observed in any of the cases.
EZH2 is the core protein of the polycomb group (PcG), which is comprised of epigenetic chromatin modifiers involved in tumorigenesis and development; these enzymes function by catalyzing the trimethylation of histone3 lysine27 (H3K27). The rate of EZH2 positivity increased with an increasing WHO tumor grade. This EZH2 expression was significantly correlated with a worse overall and progression-free survival.23 EZH2 overexpression has been reported to occur in 69.2% (9/13) of E-GBM cases,16 and occurred in 60.0% (9/15) of E-GBM cases in our study. The BRAF V600E mutation was only detected in E-GBM colocalized with BRAF intact low-grade diffuse astrocytoma.24 Several studies have found identical BRAF V600E mutation patterns in both low- and high-grade lesions in the biphasic EGBM cases,16,25-27 suggesting an identical origin for these lesions and that the acquisition of this mutation might be an early event. However, given that strong EZH2 expression was identified in epithelioid cells but not in low-grade lesions may indicate that EZH2-positive cells are related to the intratumoral heterogeneity and malignant tumor progression.16 Intratumoral heterogeneity in E-GBM has been found in some studies.21 Intratumoral heterogeneity in E-GBM needs to be investigated in the future, and requires a thorough histomorphological and genetic workup to fully recognize E-GBM pathogenesis.
In summary, we observed the clinicopathologic features and detected the EZH2 status, MGMT, and BRAF V600E in 15 E-GBM cases and 15 non-E-GBM cases. The results demonstrate extensive necrosis, unmethylation of the MGMT promoter, EZH2 overexpression, and lack of adjuvant chemo-radiotherapy indicated a poor prognosis of E-GBM. These results suggest that testing for EZH2 expression, MGMT, and BRAF V600E, as well as assessing the extent of necrosis might be helpful for evaluating E-GBM prognosis. Moreover, adjuvant chemo-radiotherapy may also improve the prognosis. E-GBM and non-E-GBM patients share common characteristics, including a benefit from adjuvant chemo-radiotherapy. Thus, a comprehensive understanding of the characteristics of E-GBM can better guide clinical treatment.
Acknowledgments
We would like to thank the native English-speaking scientists of Elixigen Company (Huntington Beach, Califonia) for editing our manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Louis DN, Ohgaki H, Wiestler OD, et al. WHO Classification of Tumours of the Central Nervous System.
2. Huang QL, Cao X, Chai X, et al. The radiological imaging features of easily misdiagnosed epithelioid glioblastoma in seven patients. World Neurosurg. 2019;125:546. doi:10.1016/j.wneu.2019.02.023
3. Gasco J, Franklin B, Fuller GN, Salinas P, Prabhu S. Multifocal epithelioid glioblastoma mimicking cerebral metastasis: case report. Neurocirugia. 2009;20(6):550–554. doi:10.1016/s1130-1473(09)70133-2
4. Alexandrescu S, Korshunov A, Lai SH, et al. Epithelioid glioblastomas and anaplastic epithelioid pleomorphic xanthoastrocytomas–same entity or first cousins? Brain Pathol. 2016;26(2):215–223. doi:10.1111/bpa.12295
5. Broniscer A, Tatevossian RG, Sabin ND, et al. Clinical, radiological, histological and molecular characteristics of paediatric epithelioid glioblastoma. Neuropathol Appl Neurobiol. 2014;40(3):327–336. doi:10.1111/nan.12093
6. Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol. 2013;37(5):685–698. doi:10.1097/PAS.0b013e31827f9c5e
7. Kleinschmidt-DeMasters BK, Aisner DL, Foreman NK. BRAF VE1 immunoreactivity patterns in epithelioid glioblastomas positive for BRAF V600E mutation. Am J Surg Pathol. 2015;39(4):528–540. doi:10.1097/PAS.0000000000000363
8. Korshunov A, Chavez L, Sharma T, et al. Epithelioid glioblastomas stratify into established diagnostic subsets upon integrated molecular analysis. Brain Pathol. 2018;28(5):656–662. doi:10.1111/bpa.12566
9. Lu VM, George ND, Brown DA, et al. Confirming diagnosis and effective treatment for rare epithelioid glioblastoma variant: an integrated survival analysis of the literature. World Neurosurg. 2019;131:243–51 e2. doi:10.1016/j.wneu.2019.08.007
10. Sugimoto K, Ideguchi M, Kimura T, et al. Epithelioid/rhabdoid glioblastoma: a highly aggressive subtype of glioblastoma. Brain Tumor Pathol. 2016;33(2):137–146. doi:10.1007/s10014-015-0243-3
11. He XY, Xu L, Li CL, Xiang H. Brain epithelioid glioblastoma with pulmonary metastasis: report of a case. Zhonghua Bing li Xue Za Zhi. 2019;48(5):418–420. doi:10.3760/cma.j.issn.0529-5807.2019.05.022
12. Funata N, Nobusawa S, Yamada R, Shinoura N. A case of osteoclast-like giant cell-rich epithelioid glioblastoma with BRAF V600E mutation. Brain Tumor Pathol. 2016;33(1):57–62. doi:10.1007/s10014-015-0239-z
13. Georgescu MM, Li Y, Islam MZ, et al. Mutations of the MAPK/TSC/mTOR pathway characterize periventricular glioblastoma with epithelioid SEGA-like morphology-morphological and therapeutic implications. Oncotarget. 2019;10(40):4038–4052. doi:10.18632/oncotarget.27005
14. Meredith DM. Advances in diagnostic immunohistochemistry for primary tumors of the central nervous system. Adv Anat Pathol. 2019. doi:10.1097/PAP.0000000000000225
15. Furuta T, Miyoshi H, Komaki S, et al. Clinicopathological and genetic association between epithelioid glioblastoma and pleomorphic xanthoastrocytoma. Neuropathology. 2018;38(3):218–227. doi:10.1111/neup.12459
16. Wang J, Liu Z, Cui Y, et al. Evaluation of EZH2 expression, BRAF V600E mutation, and CDKN2A/B deletions in epithelioid glioblastoma and anaplastic pleomorphic xanthoastrocytoma. J Neurooncol. 2019;144(1):137–146. doi:10.1007/s11060-019-03212-0
17. Li J, Ling X, Lai M, Hu Q, Shan C, Cai L. Clinicopathological features for epithelioid glioblastoma: a newly defined tumor by the 2016 World Health Organization classification of tumors of the central nervous system. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2018;43(4):398–402. doi:10.11817/j.issn.1672-7347.2018.04.010
18. Ceccon G, Werner JM, Dunkl V, et al. Dabrafenib treatment in a patient with an epithelioid glioblastoma and BRAF V600E mutation. Int J Mol Sci. 2018;19(4):1090. doi:10.3390/ijms19041090
19. Kanemaru Y, Natsumeda M, Okada M, et al. Dramatic response of BRAF V600E-mutant epithelioid glioblastoma to combination therapy with BRAF and MEK inhibitor: establishment and xenograft of a cell line to predict clinical efficacy. Acta Neuropathol Commun. 2019;7(1):119. doi:10.1186/s40478-019-0774-7
20. Kaley T, Touat M, Subbiah V, et al. BRAF inhibition in BRAF(V600)-mutant gliomas: results from the VE-BASKET study. J Clin Oncol. 2018;36(35):3477. doi:10.1200/JCO.2018.78.9990
21. Nakajima N, Nobusawa S, Nakata S, et al. BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions are frequent in epithelioid glioblastomas: a histological and molecular analysis focusing on intratumoral heterogeneity. Brain Pathol. 2018;28(5):663–673. doi:10.1111/bpa.12572
22. Khanna G, Pathak P, Suri V, et al. Immunohistochemical and molecular genetic study on epithelioid glioblastoma: series of seven cases with review of literature. Pathol Res Pract. 2018;214(5):679–685. doi:10.1016/j.prp.2018.03.019
23. Pyo JS, Kang DW. Prognostic role of EZH2 in gliomas: a meta-analysis. Int J Biol Markers. 2018;33(1):62–67. doi:10.5301/ijbm.5000293
24. Kuroda J, Nobusawa S, Nakamura H, et al. A case of an epithelioid glioblastoma with the BRAF V600E mutation colocalized with BRAF intact low-grade diffuse astrocytoma. Neuropathology. 2016;36(2):181–186. doi:10.1111/neup.12258
25. Nobusawa S, Hirato J, Kurihara H, et al. Intratumoral heterogeneity of genomic imbalance in a case of epithelioid glioblastoma with BRAF V600E mutation. Brain Pathol. 2014;24(3):239–246. doi:10.1111/bpa.12114
26. Tanaka S, Nakada M, Nobusawa S, et al. Epithelioid glioblastoma arising from pleomorphic xanthoastrocytoma with the BRAF V600E mutation. Brain Tumor Pathol. 2014;31(3):172–176. doi:10.1007/s10014-014-0192-2
27. Matsumura N, Nakajima N, Yamazaki T, et al. Concurrent TERT promoter and BRAF V600E mutation in epithelioid glioblastoma and concomitant low-grade astrocytoma. Neuropathology. 2017;37(1):58–63. doi:10.1111/neup.12318
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.