")
Back to Journals » Integrated Blood Pressure Control » Volume 9
Clinical use of extended-release oral treprostinil in the treatment of pulmonary arterial hypertension
Authors Pugliese S, Bull T
Received 6 July 2015
Accepted for publication 15 October 2015
Published 25 January 2016 Volume 2016:9 Pages 1—7
DOI https://doi.org/10.2147/IBPC.S68230
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Steven Atlas
Steven C Pugliese,1 Todd M Bull1,2
1Division of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, 2UCD Pulmonary Vascular Disease Center, Division of Pulmonary Sciences and Critical Care Medicine and Cardiology, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
Abstract: The development of parenteral prostacyclin therapy marked a dramatic breakthrough in the treatment of pulmonary arterial hypertension (PAH). Intravenous (IV) epoprostenol was the first PAH specific therapy and to date, remains the only treatment to demonstrate a mortality benefit. Because of the inherent complexities and risks of treating patients with continuous infusion IV therapy, there is great interest in the development of an oral prostacyclin analog that could mimic the benefits of IV therapy. Herein, we highlight the development of oral prostacyclin therapy, focusing on oral treprostinil, the only US Food and Drug Administration approved oral prostacyclin. Recent Phase III clinical trials have shown the drug to improve exercise tolerance in treatment-naïve PAH patients, but not patients on background oral therapy. Oral treprostinil appears to be most efficacious at higher doses, but its side effect profile and complexities with dosing complicate its use. While oral treprostinil’s current therapeutic role in PAH remains unclear, ongoing studies of this class of medication should help clarify their role in the treatment of PAH.
Keywords: oral treprostinil, pulmonary arterial hypertension, selexipag
Introduction: management issues in pulmonary arterial hypertension
The first reported case of pulmonary arterial hypertension (PAH), published in 1891 by Romberg,1 described a patient who died suddenly, identifying both right ventricular and pulmonary artery sclerosis without apparent cause. It was not until 1951, following the development of cardiac catheterization, that Dresdale et al2 was able to describe the hemodynamic findings of PAH and coined the term “primary pulmonary hypertension”.2 Nearly 100 years later, the first specific treatment for PAH was the US Food and Drug Administration (FDA)-approved epoprostenol. Barst et al3 published their pivotal findings demonstrating that intravenous (IV) epoprostenol improved 6-minute walk distance (6MWD) and hemodynamics. Though the patient numbers were small and the study duration was short, this remains the only clinical trial in PAH to demonstrate a mortality benefit to date. Great progress has since been made with the development and approval of three separate classes of drugs for the treatment of PAH: prostacyclins, endothelin receptor antagonists (ERAs), and phosphodiesterase-5 (PDE-5) inhibitors/soluble guanylate cyclase stimulators. To date however, there is general agreement that by far the most effective therapy for patients with severe PAH is the parenteral or subcutaneously infused prostanoids. This class of medications is also however the most difficult to use. They require placement of a long-term tunneled vascular or subcutaneously positioned catheter, extensive patient training, and significant lifestyle modification to accommodate the preparation and administration of a continuously infused medication. Over the past two decades, considerable effort has been directed to the development of an effective and tolerable oral prostacyclin with the aim of both increasing the availability of this class of medication and perhaps ultimately eliminating the need for IV or subcutaneous (SubQ) prostanoid therapy. This review will highlight the development of oral prostacyclin therapies focusing specifically on the use and role of oral treprostinil.
Overview of prostacyclin pharmacology
Prostacyclin, or prostaglandin I2, was first described in 1976 by Moncada et al4 who isolated an enzyme from pig and rat aortas that inhibited platelet aggregation and caused arterial vasodilation. It is now known that prostacyclin is derived from the vascular endothelium and has potent pulmonary vasodilatory, antiplatelet, and antiproliferative properties.5,6 Patients with PAH have decreased prostacyclin production including a deficiency in prostacyclin synthase as well as increases in the vasoconstrictive and prothrombotic prostaglandin thromboxane.7,8 Prostacyclin exerts its effects primary through the IP receptor, a G-protein-coupled receptor that initially activates adenylate cyclase with subsequent downstream activation of protein kinase A mediating a host of effects including inhibition of platelet aggregation, relaxation of smooth muscle, and vasodilation of the pulmonary arteries.9 Prostacyclins also exert anti-inflammatory and antiproliferative effects on a variety of cell types.10–13 Epoprostenol is a synthetic prostacyclin with a half-life (T1/2) of 2–3 minutes and is stable at room temperature for only 8 hours.14 Due to its inherent instability, significant effort has been directed toward the development of other, more stable prostacyclin analogs including iloprost (T1/2=20–30 minutes), beraprost (T1/2=40–60 minutes), and treprostinil (T1/2=180–270 minutes).10,15,16 While all of the synthetic prostanoids primarily signal through the IP receptor, there are differences in both their selectivity and affinity to other prostaglandin receptors that may give them unique pharmacologic properties.10 For example, epoprostenol has some affinity for the vasoconstrictive prostaglandin E1 (EP1), prostaglandin E3 (EP3), and thromboxane (TP) receptors, while treprostinil can also bind the vasodilatory prostaglandin E2 (EP2) and prostaglandin D2 (DP1) receptors.10,17 Similarly, it appears that iloprost and treprostinil, but not epoprostenol, have potent venodilator effects in isolated human pulmonary veins which may be mediated through DP1 receptors.17 The additional anti-inflammatory and antiproliferative properties mediated through IP receptor activation can occur via cyclic adenosine monophosphate-dependent mechanisms (ie, inhibition of platelet-derived growth factor, mitogenic responses) as well as through cyclic adenosine monophosphate-independent mechanisms such as activation of the mitogen-activated protein kinases pathway.10 Prostacyclin and its analogs are also able to signal through nuclear receptors such as peroxisome proliferator-activated receptors (PPARs) with differences in their affinity for the three isoforms: PPARα, PPARβ, and PPARγ.10 While the PPARs have diverse cellular functions, with regard to prostacyclin activation, they appear to be important in vasorelaxation, vascular endothelial growth factor production, pulmonary artery endothelial survival, and inhibition of smooth muscle cell proliferation.10,11,18–20 Because none of these medications have been directly compared in clinical trials, the clinical implications of these differences remain unknown.
The development of oral prostanoid therapy
Enthusiasm for developing effective alternate routes of administration for prostacyclin therapy fueled a series of clinical studies published in 2002 involving SubQ, inhaled, and oral prostanoid formulations. Simonneau et al,21 in a Phase III randomized controlled trial (RCT), demonstrated that SubQ treprostinil improved exercise tolerance (median placebo-corrected 6MWD 16 m), dyspnea indices, and hemodynamics in PAH patients at 12 weeks compared to placebo. However, the medication required continuous infusion, treatment effect was modest compared to IV epoprostenol, and infusion-related site pain led to discontinuation of drug in a significant number of patients (8%). During the same year, Olschewski et al22 demonstrated (Phase III RCT) that inhaled iloprost led to an improvement in the combined end point of exercise capacity, New York Heart Association (NYHA) functional class (FC), and clinical deterioration in NYHA FC III/IV PAH/inoperable chronic thromboembolic pulmonary hypertension (PH) patients at 12 weeks. However, inhaled iloprost required administration every 2–4 hours and patients were often plagued by significant cough, further raising interest for an oral prostanoid formulation.
Beraprost sodium was the first chemically stable and orally active prostacyclin analog.23 The initial, large Phase III trial involving oral beraprost (Arterial Pulmonary Hypertension and Beraprost European Trial), also published in 2002, included 130 World Health Organization (WHO) FC II and III PAH patients randomized 1:1 to receive beraprost or placebo four times daily.23 There was a significant improvement in the primary outcome of placebo-corrected 6MWD (25.1 m, P=0.036) at 12 weeks with post hoc analysis suggesting that most of this occurred in patients with primary PH (53.9% of study population) versus associated PH. With regard to secondary outcomes, there was a significant improvement in Borg dyspnea index, but no change in FC, combined end point of hospitalization or death, or hemodynamics. However, Barst et al24 published a follow-up Phase III RCT of oral beraprost including 116 WHO FC II/III patients with the primary end point being disease progression (death, transplantation, initiation of prostanoid therapy, or a >25% decrease in peak Vo2 during exercise from baseline) at 12 months. The trial was stopped early due to sponsor-initiated accelerated data review, but 93% of the patients completed 12 months and 100% completed 9 months of therapy. There was no significant difference in the primary outcome of disease progression at 9 or 12 months or any of the secondary outcomes including change in WHO FC, placebo-corrected 6MWD, hemodynamics, Borg dyspnea score, or quality of life. Interestingly there were significant differences in disease progression at 6 months and placebo-corrected 6MWD at 3 and 6 months (similar to that in Arterial Pulmonary Hypertension and Beraprost European Trial) that waned by 9 and 12 months highlighting the importance of long-term studies in a chronic disease such as PAH. Additionally, beraprost use was associated with a 70% incidence of adverse events, mostly gastrointestinal side effects, flushing, headaches, and jaw pain.23,24 As a result, beraprost did not get FDA approval in the US, but is approved in Japan and South Korea.6
The pharmacology of oral treprostinil
Most recently, an oral formulation of treprostinil was developed, becoming the only other oral prostanoid to be tested in large clinical trials. The oral formulation known as treprostinil diolamine (previously UT-15C) is a salt-derived form of the original compound tricyclic benzidine treprostinil sodium, trade name Remodulin™ (United Therapeutics Corporation, Silver Spring, MD, USA).25 Oral treprostinil (Orenitram™, United Therapeutics Corporation) has a bioavailability of 17% compared to IV treprostinil with mean peak drug concentrations in approximately 4 hours, a plasma half-life of approximately 4.5 hours, and sustained plasma concentrations for 8–10 hours.26,27 There is a linear relationship between oral dosage and plasma concentrations (range =0.5–15 mg BID [two times a day]) in both BID and TID (three times a day) dosing indicating that peak plasma concentrations are mostly a function of tolerability of drug.27,28 Bioavailability is affected by food, and to ensure appropriate absorption, the drug should be taken with a minimum of 250 calories containing 30%–50% of fat.29 Metabolism occurs in the liver primarily through CYP2C8 and to a lesser extent through CYP2C9, where it is converted into six inactive metabolites.30 It is 96% protein bound and almost exclusively eliminated in the urine, but clearance is not affected by renal impairment or dialysis.27,31 Because it is metabolized in the liver, drug clearance is significantly impacted by liver impairment and is proportional to the degree of liver disease.30
With regard to drug–drug interactions between other PAH therapies, there is no conclusive evidence that interactions exist between PDE-5 inhibitors or ERAs. Pharmacokinetic (PK) data were obtained in healthy volunteers exposed to combinations of both treprostinil and sildenafil and treprostinil and bosentan without PK interaction for either agent.32,33
Oral treprostinil clinical trials
There have been three Phase III trials published to date to determine the efficacy and safety of oral treprostinil in patients with PAH (Table 1). The first trial to be published (Freedom-C) aimed at determining the efficacy and safety of oral treprostinil in patients receiving concomitant oral ERA and/or PDE-5 inhibitor therapy.34 They randomized 354 patients on background ERA, PDE-5 inhibitor, or both in a 1:1 manner to receive oral treprostinil or placebo. At 16 weeks, there was no significant difference in their primary end point of placebo-corrected change in 6MWD or the secondary end points of time to clinical worsening, mortality, or FC at 16 weeks. The only positive secondary end points were improvement in median dyspnea fatigue index score and combined 6MWD and Borg dyspnea score. Importantly, patients were initially started on 1 mg BID of drug, which was to be increased in 1 mg increments to 16 mg BID. Due to poor tolerability, the study protocol was amended mid-study to give patients access to 0.5 mg and later 0.25 mg doses of drug. The discontinuation rate due to adverse effects in patients starting with 1 mg tablets (tabs; n=51) was 25% and dropped to 12% in those with access to 0.5 mg tabs (n=100) and 0% in those with access to 0.25 mg tabs (n=23). When patients were stratified by having access to only 1 mg tabs versus those with access to all 3 doses, the former only had a 5 m change in placebo-corrected 6WMD, while the later had a 29.5 m change. Additionally, there appeared to be a dose response relationship to change in 6MWD distance: dose <1 mg BID +4 m (n=58), 1.25–3.25 mg BID +18 m (n=49), 3.5–16 mg BID +34 m (n=52). The median dose at 16 weeks for the overall population was 3.0 mg BID.
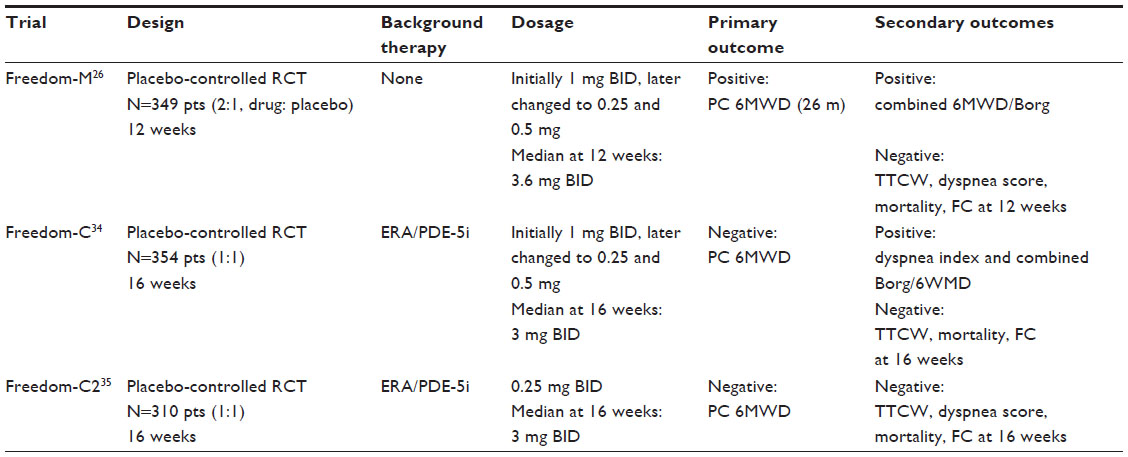 | Table 1 Comparison of Freedom trials determining the efficiency of oral treprostinil in pulmonary arterial hypertension |
In an attempt to resolve the problems with poor tolerance at higher doses, the follow-up Freedom-C2 trial was undertaken.35 This trial randomized 310 patients in a 1:1 manner with nearly identical design to Freedom-C, but gave all patients access to 0.25 mg tabs (all patients except those in the People’s Republic of China also had access to 0.125 mg tabs) from the start of the study. All patients were initiated on 0.25 mg BID and were able to increase the dose by 0.25 mg BID every 3 days. However, even using this approach, study participants achieved a median dose of only 3.1 mg BID at 16 weeks (very similar to the 3 mg BID achieved in Freedom-C). At study completion (16 weeks) there were no significant differences in the primary end point of placebo-corrected change in 6MWD or any of the secondary end points. Discontinuation of treprostinil because of adverse events occurred in 11% of patients in Freedom-C2 compared to 22% in Freedom-C. However, there were no discontinuations in the 23 patients in Freedom-C who had access to all three doses (0.25, 0.5, 1 mg).
The Freedom-M trial was designed to assess the safety and efficacy of oral treprostinil therapy in de novo PAH patients who were not concurrently receiving approved PAH therapy.26 Freedom-M enrolled 349 patients in a 2:1 fashion to receive drug (initially 1 mg tabs and later 0.25 or 0.5 mg tabs) or placebo for 12 weeks. Because of tolerability issues with the 1 mg doses also reported in Freedom-C, a modified intention to treat (mITT) group (n=228) that had access to the 0.25 or 0.5 mg tabs was analyzed separately from the entire group (ITT). Study drug doses achieved at week 12 were slightly higher in Freedom-M (3.4 in mITT and 3.6 mg in ITT) than in Freedom-C (3 mg) or Freedom-C2 (3.1 mg). There were significant differences in the primary outcome of placebo-corrected change in 6MWD at 12 weeks in both the mITT (23 m) and ITT (26 m) populations. There was also a significant difference in the combined 6MWD/Borg dyspnea score at week 12, but not in any of the other secondary outcomes such as time to clinical worsening or FC. Discontinuation of drug because of adverse effects was low at 10% for the ITT population and 4% in the mITT compared to 3% in those receiving placebo.
In summary, the Freedom trials suggest that oral treprostinil has beneficial effects on exercise tolerance in treatment-naïve PAH patients, but did not meet statistical significance in those on background oral PDE-5 inhibitor or ERA therapy. As a result of improved exercise tolerance in Freedom-M along with the established clinical efficacy of parenteral and inhaled treprostinil (modified formulation of previously approved medication), the FDA-approved oral treprostinil for the treatment of WHO group 1 PAH.
The future of oral treprostinil?
There was still some enthusiasm for oral treprostinil after Freedom-C because it appeared that it was well tolerated if titrated in smaller increments (0.25 mg tabs) and much more efficacious (34 m 6MWD difference) in the 52 patients that were able to tolerate higher doses in the range of 3.5–16 mg BID.34 This is not surprising as prior studies with IV and SubQ treprostinil as well as epoprostenol have all demonstrated both an efficacy and side effect profile that is dose dependent.3,21,36,37 With regard to tolerability, the initial 1 mg BID dosing used in Freedom-M and Freedom-C is equivalent to 8–12 ng/kg/min of IV or SubQ treprostinil, much higher than the typical 2 ng/kg/min starting dosages used for initiation of parental treprostinil therapy.27,34,35 The 0.25 mg BID dosage is equivalent to 2–3 ng/kg/min of IV or SubQ drug, the typical dosage increment used in parenteral titrations and understandably much better tolerated with regard to dose titration than the 0.5 or 1 mg tabs. Recently, White et al27 published PK data obtained from 70 patients enrolled in the Freedom trials showing that there is a linear relationship between the dose of oral treprostinil and maximum plasma concentrations. While the authors were not able to correlate plasma concentrations of drug to efficacy or adverse effect data, they did find that many of the patients were on an oral dose of treprostinil that produced plasma concentrations typically below what is considered a reasonable therapeutic dose for patients receiving parenteral therapy: 10–30 ng/kg/min.21,27,38 For example, in White et al’s27 cohort, most of the patients were either on 2 mg BID or 6 mg BID. In patients taking the 2 mg BID dose, they were above 10 ng/kg/min for less than 6 of the 12 hours studied, while it required a dose of 6 mg BID to be above this threshold for the entire 12-hour window, a dose that was nearly twice the median dosage in the Freedom trials (Figure 1). Importantly, drug clearance estimates and time to maximum plasma concentrations were similar between patients.
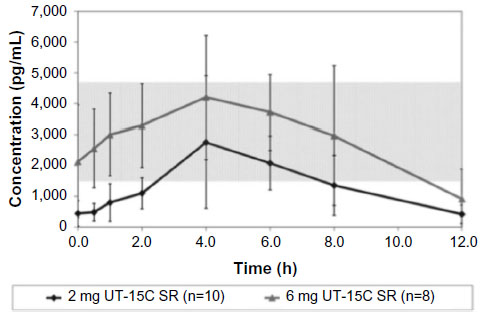 | Figure 1 Mean concentration–time profiles of treprostinil diolamine (UT-15C): 2 and 6 mg. |
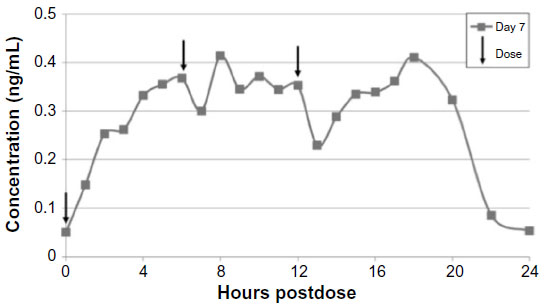 | Figure 2 Mean steady-state daily plasma treprostinil concentration after 0.5 mg TID regimen. |
It is also worth mentioning that concerns have been raised regarding potential drug interactions between oral treprostinil and patients on background PH therapy.38 White et al27 did find a trend (P<0.11) suggesting possible effects of background therapy (74% were taking an ERA) on the AUC0–12 of oral treprostinil. Furthermore, in the Freedom-C and Freedom-C2 trials 6MWDs were consistently higher in patients taking oral treprostinil and PDE-5 inhibitors versus oral treprostinil and ERAs.34,35 While intriguing, none of these data meet statistical significance and no firm conclusions can be made regarding potential synergy between treprostinil and PDE-5 inhibitors (or negative drug interaction with ERAs) as have been suggested between epoprostenol and PDE-5 inhibitors.39
Based on the PK data along with some of the efficacy data from Freedom-C, it is possible that oral treprostinil could be effective if titrated to higher doses or the dosing interval was reduced. PK data in healthy volunteers taking oral treprostinil TID revealed a sustained mean steady-state plasma concentration of drug over 24 hours without drug accumulation, an improvement over BID dosing28 (Figures 1 and 2). With regard to titrating to higher dosages, it remains unknown if the Freedom-C2 trial had been extended if patients would be able to tolerate further dose increases over time. It should be noted that despite better patient tolerability and less medication discontinuation owing to smaller dosage increments in Freedom-C2, median doses at week 16 were almost identical to those in Freedom-C. Results of the uncontrolled, open-label extension of Freedom-M may also reveal if patients are able to tolerate higher doses over time. Answers to the effectiveness of TID dosing and a longer duration of therapy will hopefully be answered in the ongoing FREEDOM EV (NCT01560624) trial. In this multicenter randomized placebo-controlled trial, oral treprostinil is being administered TID in patients with PAH on background ERA or PDE-5 inhibitor therapy (but not both) with the coprimary end points of 6MWD at 24 weeks and time to clinical worsening up to 4 years.
Oral prostacyclin receptor agonists (another approach)
Selexipag is an oral nonprostanoid selective IP receptor agonist. It is rapidly metabolized by hepatic microsomes into the active metabolite ACT-333679 (T1/2=9–12 hours) allowing for twice daily dosing.40 The reported advantages of selexipag over prostanoids are more favorable pharmacokinetics and greater selectivity for the IP receptor leading to less gastrointestinal intolerance.10,41,42 Simonneau et al,43 in a 43 patient randomized Phase II trial in patients with PAH (all oral background therapy), demonstrated a 30.3% change in pulmonary vascular resistance with a side effect profile similar to that of prostacyclin analogs. The recently completed GRIPHON trial, enrolled 1156 PAH patients (20% PAH treatment-naïve) randomized 1:1 to selexipag or placebo with the primary end point being time from randomization to first morbidity or mortality event with patients being treated up to 4.3 years.44 The final results of this trial are expected soon. A second oral prostacyclin agonist, ralinepag, or APD-811 (Arena Pharmaceuticals, San Diego, CA, USA), is currently in a Phase II clinical trial in PAH.
Conclusion
Treprostinil is currently the only FDA-approved oral prostacyclin for the treatment of PAH. While current evidence suggests that the drug is efficacious at improving exercise tolerance in treatment-naïve PAH patients, at this time, its place in a treatment paradigm remains unclear. The use of oral treprostinil is complicated, as it requires titration using a number of different formulation strengths and a TID dosing regimen. Additionally, its expected but significant side effect profile, high cost, and perceived modest drug effect without demonstrated efficacy in combination with other PAH medications compared have hindered its adoption in the therapeutic armamentarium for PAH. Still, despite these difficulties, complexities with administration and side effects of IV, SubQ and even inhaled prostanoid therapies have encouraged further investigation of its use. Long-term data with oral treprostinil will be required to determine whether it has effects on pulmonary vascular remodeling and whether this is mediated through activation of the IP receptor or through other pathways. While we are still without answers regarding the definitive role of oral prostacyclin therapy in PAH, the rapid pace of medication development and the potential benefits to patients with the expanding pool of pharmacotherapy offers great hope to clinicians and patients alike.
Disclosure
The authors report no conflicts of interest in this work.
References
Romberg E. Uebere Sklerose der Lungenarterien. Dtsch Arch Klin Med. 1891;48:197–206. German. | |
Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med. 1951;11(6):686–705. | |
Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med. 1996;334(5):296–301. | |
Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976; 263(5579):663–665. | |
Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655–1665. | |
Rich J, Hoeper MM. The search for an oral prostanoid to treat pulmonary arterial hypertension continues. Are we getting any closer? Int J Clin Pract. 2009;63:17–18. | |
Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75. | |
Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–1932. | |
Ruan C-H, Dixon RAF, Willerson JT, Ruan K-H. Prostacyclin therapy for pulmonary arterial hypertension. Tex Heart Inst J. 2010; 37(4):391–399. | |
Clapp LH, Gurung R. The mechanistic basis of prostacyclin and its stable analogs in pulmonary arterial hypertension: role of membrane versus nuclear receptors. Prostaglandins Other Lipid Mediat. 2015; 120:56–71. | |
Falcetti E, Hall SM, Phillips PG, et al. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182(9):1161–1170. | |
Raychaudhuri B, Malur A, Bonfield TL, et al. The prostacyclin analog treprostinil blocks NFkappaB nuclear translocation in human alveolar macrophages. J Biol Chem. 2002;277(36):33344–33348. | |
Zhou W, Hashimoto K, Goleniewska K, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol. 2007;178(2):702–710. | |
LeVarge BL. Prostanoid therapies in the management of pulmonary arterial hypertension. Ther Clin Risk Manag. 2015;11:535–547. | |
Demolis JL, Robert A, Mouren M, Funck-Brentano C, Jaillon P. Pharmacokinetics and platelet antiaggregating effects of beraprost, an oral stable prostacyclin analog, in healthy volunteers. J Cardiovasc Pharmacol. 1993;22(5):711–716. | |
Wade M, Baker FJ, Roscigno R, et al. Pharmacokinetics of treprostinil sodium administered by 28-day chronic continuous subcutaneous infusion. J Clin Pharmacol. 2004;44(5):503–509. | |
Benyahia C, Boukais K, Gomez I, et al. A comparative study of PGI2 mimetics used clinically on the vasorelaxation of human pulmonary arteries and veins, role of the DP-receptor. Prostaglandins Other Lipid Mediat. 2013;107:48–55. | |
Orie NN, Ledwozyw A, Williams DJ, Whittle BJ, Clapp LH. Differential actions of the prostacyclin analogs treprostinil and iloprost and the selexipag metabolite, MRE-269 (ACT-333679) in rat small pulmonary arteries and veins. Prostaglandins Other Lipid Mediat. 2013;106:1–7. | |
Biscetti F, Gaetani E, Flex A, et al. Peroxisome proliferator-activated receptor alpha is crucial for iloprost-induced in vivo angiogenesis and vascular endothelial growth factor upregulation. J Vasc Res. 2009; 46(2):103–108. | |
Piqueras L, Reynolds AR, Hodivala-Dilke KM, et al. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27(1):63–69. | |
Simonneau G, Barst RJ, Galiè N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analog, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800–804. | |
Olschewski H, Simonneau G, Galiè N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–329. | |
Galiè N, Humbert M, Vachiéry J-L, et al. Effects of beraprost sodium, an oral prostacyclin analog, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. JAC. 2002; 39(9):1496–1502. | |
Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41(12):2119–2125. | |
Hellawell JL, Bhattacharya S, Farber HW. Pharmacokinetic evaluation of treprostinil (oral) for the treatment of pulmonary arterial hypertension. Expert Opin Drug Metab Toxicol. 2014;10(10):1445–1453. | |
Jing Z-C, Parikh K, Pulido T, et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(5):624–633. | |
White RJ, Torres F, Allen R, et al. Pharmacokinetics of oral treprostinil sustained release tablets during chronic administration to patients with pulmonary arterial hypertension. J Cardiovasc Pharmacol. 2013; 61(6):474–481. | |
Jones A, Wang-Smith L, Pham T, Laliberte K. Pharmacokinetics of 3 times a day dosing of oral treprostinil in healthy volunteers. J Cardiovasc Pharmacol. 2014;63(3):227–232. | |
Lim A, Wang-Smith L, Kates J, Laurent A, Kumar P, Laliberte K. The effect of different meal compositions on the oral bioavailability of treprostinil diolamine in healthy volunteers. J Clin Pharm Ther. 2013; 38(6):450–455. | |
Peterson L, Marbury T, Marier J, Laliberte K. An evaluation of the pharmacokinetics of treprostinil diolamine in subjects with hepatic impairment. J Clin Pharm Ther. 2013;38(6):518–523. | |
Jenkins A, Wang-Smith L, Marbury T, Laliberte K. Pharmacokinetics of treprostinil diolamine in subjects with end-stage renal disease on or off dialysis. J Cardiovasc Pharmacol. 2013;61(4):272–276. | |
Gotzkowsky SK, Dingemanse J, Lai A, Mottola D, Laliberte K. Lack of a pharmacokinetic interaction between oral treprostinil and bosentan in healthy adult volunteers. J Clin Pharmacol. 2010;50(7):829–834. | |
Gotzkowsky SK, Kumar P, Mottola D, Laliberte K. Lack of a pharmacokinetic interaction between treprostinil diolamine and sildenafil in healthy adult volunteers. J Cardiovasc Pharmacol. 2013; 61(5):444–451. | |
Tapson VF. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients on background endothelin receptor antagonist and/or phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C study). Chest. 2012;142(6):1383–1388. | |
Tapson VF. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study). Chest. 2013;144(3):952–957. | |
Tapson VF. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension. Chest. 2006;129(3):683–688. | |
Hiremath J, Thanikachalam S, Parikh K, et al. Exercise improvement and plasma biomarker changes with intravenous treprostinil therapy for pulmonary arterial hypertension: a placebo-controlled trial. HEALUN. 2010;29(2):137–149. | |
de Jesus Perez VA. Understanding the pharmacokinetics of oral treprostinil in patients with pulmonary arterial hypertension. J Cardiovasc Pharmacol. 2013;61(6):471–473. | |
Simonneau G, Rubin LJ, Galiè N, et al. Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med. 2008; 149(8):521–530. | |
Kaufmann P, Okubo K, Bruderer S, et al. Pharmacokinetics and tolerability of the novel oral prostacyclin IP receptor agonist selexipag. Am J Cardiovasc Drugs. 2015;15(3):195–203. | |
Kuwano K, Hashino A, Asaki T, et al. 2-[4-[(5,6-Diphenylpyrazin-2-yl) (isopropyl)amino]butoxy]-N-(methylsulfonyl)acetamide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther. 2007;322(3):1181–1188. | |
Skoro-Sajer N, Lang IM. Selexipag for the treatment of pulmonary arterial hypertension. Expert Opin Pharmacother. 2014;15(3):429–436. | |
Simonneau G, Torbicki A, Hoeper MM, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–880. | |
McLaughlin VV, Channick R, Chin K, et al. Effect of selexipag on morbidity/mortality in pulmonary arterial hypertension: results of the GRIPHON study [abstract]. J Am Coll Cardiol. 2015; 65(10S):A1538. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.