")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 8
Clinical use and applications of histone deacetylase inhibitors in multiple myeloma
Authors Tandon N, Ramakrishnan V, Kumar S
Received 24 December 2015
Accepted for publication 25 February 2016
Published 6 May 2016 Volume 2016:8 Pages 35—44
DOI https://doi.org/10.2147/CPAA.S94021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Nidhi Tandon, Vijay Ramakrishnan, Shaji K Kumar
Division of Hematology, Mayo Clinic, Rochester, MN, USA
Abstract: The incorporation of various novel therapies has resulted in a significant survival benefit in newly diagnosed and relapsed patients with multiple myeloma (MM) over the past decade. Despite these advances, resistance to therapy leads to eventual relapse and fatal outcomes in the vast majority of patients. Hence, there is an unmet need for new safe and efficacious therapies for continued improvement in outcomes. Given the role of epigenetic aberrations in the pathogenesis and progression of MM and the success of histone deacetylase inhibitors (HDACi) in other malignancies, many HDACi have been tried in MM. Various preclinical studies helped us to understand the antimyeloma activity of different HDACi in MM as a single agent or in combination with conventional, novel, and immune therapies. The early clinical trials of HDACi depicted only modest single-agent activity, but recent studies have revealed encouraging clinical response rates in combination with other antimyeloma agents, especially proteasome inhibitors. This led to the approval of the combination of panobinostat and bortezomib for the treatment of relapsed/refractory MM patients with two prior lines of treatment by the US Food and Drug Administration. However, it remains yet to be defined how we can incorporate HDACi in the current therapeutic paradigms for MM that will help to achieve longer disease control and significant survival benefits. In addition, isoform-selective and/or class-selective HDAC inhibition to reduce unfavorable side effects needs further evaluation.
Keywords: HDAC inhibitors, Panobinostat, epigenetics, myeloma, relapse
Introduction
Multiple myeloma (MM) is a plasma cell malignancy, characterized by an accumulation of high levels of monoclonal immunoglobulins or paraproteins in blood and/or urine and end organ damage, including anemia, renal failure, hypercalcemia, and bony lesions.1 MM is the second most commonly diagnosed hematologic malignancy representing 1.6% of all new cancer cases in the US. The outcomes of these patients have not been satisfactory, and the 5-year survival is 46.6% according to surveillance, epidemiology, and end results analysis.2
Over the last two decades, the treatment paradigm for MM has changed with the use of autologous stem cell transplantation (ASCT) and novel therapeutic options including proteasome inhibitors (PIs) and immunomodulatory drugs (IMiDs).3 The incorporation of novel drugs, particularly thalidomide, lenalidomide (Len), and bortezomib (Btz), has resulted in a significant prolongation of overall survival (OS) in newly diagnosed and relapsed patients.4 Despite these advances, acquired or intrinsic resistance to therapy leads to eventual relapse and fatal outcomes in vast majority of patients. In an analysis of 286 patients with relapsed MM, who were refractory to Btz and had relapsed following, or were refractory to or ineligible to receive an IMiD, OS and event-free survival were 9 and 5 months, respectively. These findings indicate the poor outcome of patients, once they become refractory to current modalities and an unmet need for safe and efficacious novel therapies.5
MM is a biologically complex disease, with great heterogeneity in terms of genetic alterations, thereby giving rise to individual differences in overall response and survival of patients receiving the same treatment. In addition to genetic alterations such as point mutations, deletions, or translocations, epigenetic alterations and abnormal microRNA expression also contribute to the pathogenesis of MM.6–9
Epigenetic aberrations are heritable changes in gene expression that occur independent of changes in the primary DNA sequence. Most of the epigenetic mechanisms occur at the level of chromatin. Chromatin is built up by nucleosomes that contain ~146 bp of DNA wrapped around an octamer consisting of four core histones (H3–H4 tetramer and two H2A–H2B dimers). The modifications of these nucleosomes play an important role in the transition of chromatin between open and closed states.10 The N-terminal tail regions of histones undergo a wide variety of enzyme modifications, including acetylation, methylation, sumoylation, phosphorylation, and ubiquitination, which are crucial in modulating gene expression. This phenomenon is referred to as the “histone code” and has a significant effect on gene expression and chromatin structure.11 Histone acetylation is controlled by histone acetyl transferases, which transfer acetyl groups to the side chain of lysine residues on N-terminal, and histone deacetylases (HDACs), which deacetylate and counterbalance activity of histone acetyl transferases.12,13
The HDAC family of enzymes regulates the acetylation level of histones in chromatin and various nonhistone substrates, including many proteins involved in tumor progression, cell cycle control, apoptosis, angiogenesis, and cell invasion.14 The HDAC family consists of 18 genes that are grouped into classes I, II, III, and IV based on their homology to respective yeast orthologs Rpd3, HdaI, and Sir2.15 Various classes of HDAC and their localization and function have been shown in Table 1. HDAC inhibitors (HDACi) typically target the “classical” classes I, II, and IV HDACs (which contain a Zn2þ catalytic ion in their active site) and not class III HDACs that use NAD+ as the essential cofactor.14,16
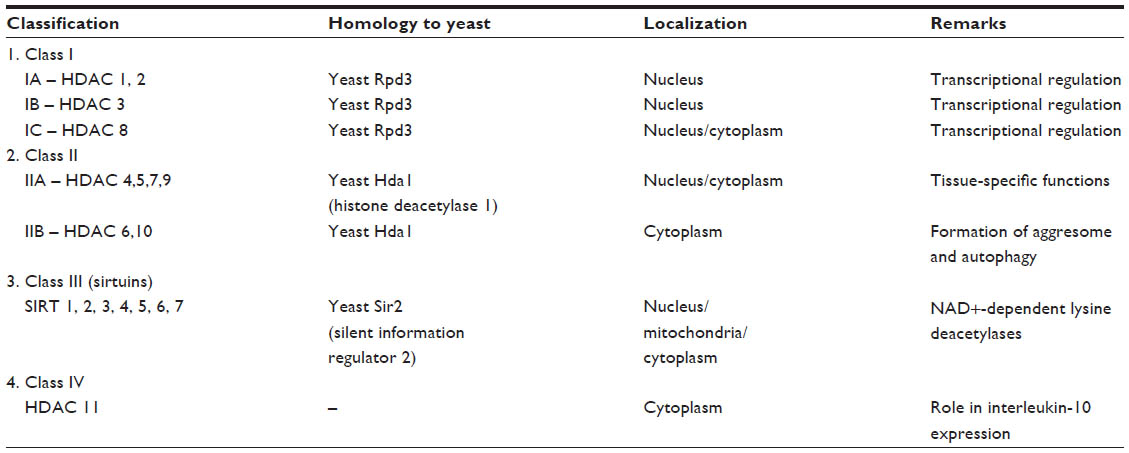 | Table 1 Various classes of HDAC |
This review focuses on the antimyeloma activity of different HDACi in preclinical and clinical settings.
Epigenetic changes in MM
Epigenetic aberrations play an important role in the initiation and progression of most of the malignancies, including MM, and this is largely attributed to alterations in the expression of histone-modifying enzymes.17,18
In cancer, global DNA hypomethylation of repetitive sequences (such as long interspersed nuclear element 1 [LINE-1] and Alu repeats), gene bodies, and intergenic regions has been observed. This contributes to genomic instability, transposon activation, proto-oncogene activation, and loss of normal imprinting patterns. In addition, site-specific CpG island hypermethylation of gene promoters such as tumor suppressor genes results in gene silencing.19 Even in MM, there is increased global hypomethylation of the LINE-1 and Alu repetitive elements compared to normal control subjects.20 This seems to be an early event in the pathogenesis, and the global methylation levels of repetitive elements decrease as the disease progresses from monoclonal gammopathy of unknown significance to MM.20,21
Epigenetic alterations also increase the vulnerability to genomic instability. LINE-1 hypomethylation is found to be associated with translocations of chromosome 14 and deletion of chromosome 13q.21 Also, t(4;14) translocation showed more frequent hypermethylation that may underline the poor prognosis associated with its presence.22
The most characteristic documentation of aberrations of histone modifications is in t(4;14) MM, which leads to overexpression of multiple myeloma SET domain containing protein (MMSET) (NSD-2), a histone methyltransferase. MMSET regulates genes involved in the p53 pathway, nuclear factor kappa B pathway, apoptosis, cell cycle regulation, DNA repair, and adhesion, and its upregulation enhances survival and adhesion of MM cells.23,24
In addition, epigenetic alterations may result in dysregulation of critical oncogenic pathways such as cyclin dependent kinase/retinoblastoma (CDK/Rb), Wnt/β-catenin, Janus kinase/signal transducer and activator of transcription protein (JAK/STAT), death associated protein kinase-1/p14-ARF/p53 (DAPK-1/ p14 ARF/p53) pathways, which contribute to the pathogenesis of MM.8
HDACs in MM
Overexpression of HDAC proteins, especially class I HDACs, has been observed in both solid and hematological malignancies.25–29 In the majority of tumors, HDAC expression is associated with a poor prognosis.30–32 However, HDAC expression is correlated with a better prognosis in breast cancer, acute lymphoblastic leukemia, and chronic lymphocytic leukemia.33–35 Patients with MM with high transcript levels of HDACs 1, 2, 4, 6, and 11 show a shorter progression-free survival (PFS) than those expressing lower levels. However, when HDAC protein levels were examined, it was found that only increased HDAC1 expression correlated with poor PFS and OS.36
HDACi in MM
Butyrate and trichostatin A were among the initial molecules identified as HDACi. Since then, various natural and synthetic HDACi have been developed and evaluated as anticancer agents in the preclinical and clinical settings. Major HDACi can be divided into five categories on the basis of their chemical structure (Table 2). The direct impact of HDAC inhibition on chromatin is hyperacetylation of histone proteins, which alters the chromatin structure and results in up- or downregulation of gene expression involved in cell cycle regulation, apoptosis, cytokine signaling, adhesion and migration, proteasomal degradation, drug resistance, and DNA damage.37–39
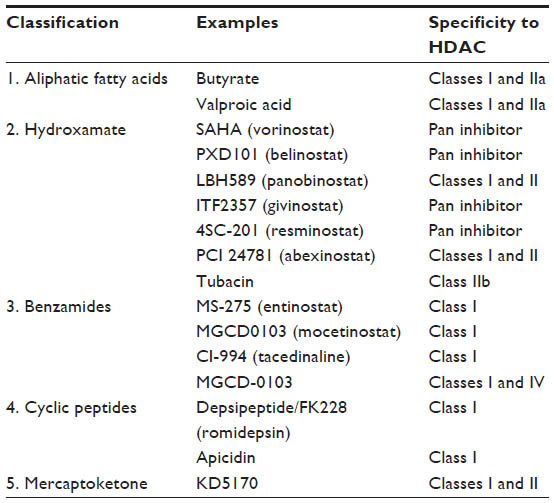 | Table 2 Classification of HDAC inhibitors |
Preclinical activity of HDACi in MM
As a single agent
Microarray analysis has shown that HDACi induce transcriptional modulation of 7%–10% of the genes in myeloma and human lymphoid cell lines by acetylation of histones and nonhistone proteins.38,40 The pattern of gene alteration is quite similar across different HDACi in the same cell line.41,42 HDACi such as valproate, FK228, and ITF2357 affect the viability of interleukin (IL)-6-dependent and -independent MM cell lines, indicating that the antimyeloma activity of HDACi is not influenced by IL-6.43–45 Moreover, coculturing MM cells with bone marrow stromal cells (BMSCs) do not protect them from death induced by LAQ824, ITF2357, LBH589, or KD5170, suggesting that HDACi could overcome the protective effect of the BMSCs.45–48 The various possible mechanisms of the anti-myeloma activity of HDACi have been described later.
Cell cycle arrest
Almost all HDACi induce G0/SG1 arrest due to increase in histone acetylation and upregulation of cyclin-dependent kinase (CDK) inhibitor CDKN1A by p53-dependent and -independent ways, as observed in MM cell lines treated with valproate, NVP-LAQ824, LBH589, NaB, SAHA, and ITF2357.43,45,46,49–51 Other effects include upregulation of other CDK inhibitors such as p27 and p19 and/or the decrease of cyclins D1 and D2.43,52,53
Apoptotic pathway
HDACi upregulate expression of proapoptotic Bcl-2 family proteins (Bax, Bak, and Bim) and downregulate antiapoptotic proteins (Bcl-2, Bcl-xL, MCL1, and XIAP). Overall, this triggers increased mitochondrial permeability and cytosolic release of cytochrome C and Smac followed by activation of intrinsic apoptotic pathway, as seen in MM cell lines treated with depsipeptide, ITF2357, LBH589, SAHA, and KD5170.44,45,47,50,51 Extrinsic apoptotic pathway is activated by upregulation of death receptors and ligands, caspase-8 cleavage, and downregulation of Flice-like inhibitory protein (caspase-8 inhibitor) as seen in MM cells after valproate, suberoylanilide hydroxamic acid (SAHA), and LBH589 treatment.50,51,54 Autophagy is a catabolic process involving the degradation of long-lived proteins or cytoplasmic organelles through the lysosomal machinery, which plays a role in valporate-induced cytotoxicity in human myeloma cell lines.54
DNA damage and oxidative stress
HDACi interfere with the function of DNA-repair proteins such as Ku70, RAD51, RAD50, DNA-PKcs, BRCA1, and BRCA2, thus inducing double-stranded breaks in DNA.55,56 The HDACi PDX-101 and KD-5170 phosphorylate H2AX on ser139 and induce DNA damage.57 Another HDACi, SDNX-275, could enhance the DNA damage response induced by the alkylating agent melphalan in MM cell lines.58 Moreover, HDACi-induced chromatin hyperacetylation makes DNA more sensitive to drugs, radiation, and reactive oxygen species.59 The production of reactive oxygen species observed after HDAC inhibition seems crucial as evidenced by the upregulation of several antioxidant genes such as glutathione S-transferase, glutathione reductase, and superoxide dismutase 1 and 2 on the treatment of U937 leukemic cells with vorinostat.60
Ubiquitin–proteasome system
HDACi decrease activity of 20S proteasome and downregulate genes encoding 26S proteasome and ubiquitin conjugating enzymes in MM cells.38,46 Also, tubacin or pan HDACi such as SAHA or LBH589, hyperacetylate α-tubulin, accumulate polyubiquitinated proteins, leading to apoptosis subsequently.48 HDAC inhibition enhances the cytotoxic effects of Btz both in vitro and in vivo,38,47,48,61,62 which will be discussed later.
BMSC interaction
Multiple cytokines such as IL-6, IL-1, insulin-like growth factor-1 (IGF-1), tumor necrosis factor-α, vascular endothelial growth factor (VEGF), Dickkopf-related protein 1, and secreted frizzled-related protein are secreted at high levels by either the malignant plasma cells or the BMSCs. This then causes activation of signaling pathways in the MM cells and further promotes their interaction with cells in the tumor microenvironment such as BMSCs, endothelial cells, osteoblasts, and osteoclasts. The net result of such interaction is increased tumor growth, angiogenesis, bone disease, and drug resistance.63,64
HDACi downregulate the expression of genes involved in cytokine signaling such as IGF-1, IGF-1 receptor, and IL-6 receptor.38 Mitsiades et al38,50 showed that vorinostat not only suppresses the expression of receptor genes involved in MM cell proliferation, survival, and/or migration such as IGF-1R, IL-6R, TNF-R, CD138 (syndecan-1), and CXCR-4.55 but also reduces the autocrine IGF-1 and paracrine IL-6 secretion of BMSC.
Antiangiogenesis
HDACi induce alteration of numerous pro- and antiangiogenic genes (angiopoietin, TIE2, eNOS, p53, pVHL, and thrombospondin 1), thereby targeting increased angiogenesis in MM.65–67 Valporate decreases VEGF secretion and VEGF receptor expression, resulting in inhibition of the vascular tubule formation of endothelial cells in cocultures with MM cells.43,68,69
In combination with PIs
HDACi have been tried in combination with a variety of agents for MM, but the most synergistic effects are seen with Btz. The precise mechanisms causing this synergy are not yet completely defined. The best understood mechanism is dual inhibition of the proteasomal and aggresomal protein degradation pathways, targeted by Btz and HDACi, respectively. Btz inhibits proteasome and causes accumulation of polyubiquitinated proteins that form an aggresome by a process dependent on the interaction of HDAC6 with tubulin and dynein complex. HDAC6 inhibition leads to increased hyperacetylation of tubulin and upregulation of polyubiquitinated proteins, resulting in apoptosis.70,71 In accordance with the above mentioned dual inhibition phenomenon, non-selective HDACi like vorinostat as well as selective HDAC6i like tubacin and ACY-1215 have been found to inhibit aggresome formation and induce caspase-mediated apoptosis in MM when combined with Btz.72–74
In addition, HDAC1 overexpression causes resistance to Btz both in vitro and in vivo, which is reversed by the class I HDACi romidepsin. Moreover, Btz downregulates the expression of class I HDACs and enhances HDACi cytotoxicity.75 Taken together, Btz and HDACi combination appears to be a promising therapeutic strategy that can overcome drug resistance.
In combination with other agents
Preclinical studies have shown that addition of vorinostat or panobinostat to MM cell lines and tumor cells derived from patients resistant to conventional therapies increases their susceptibility to IMiDs (such as pomalidomide or Len) and dexamethasone.38,76 Moreover, treatment of MM cells with vorinostat increases their sensitivity to DNA-damaging agents, such as doxorubicin or melphalan.38,77
Treatment of MM cell line with sodium butyrate in combination with DNA methyltransferase inhibitor, decitabine, resulted in increased expression of p16 gene and G1 arrest, a phenomenon not seen with either agent alone.78 Furthermore, mTORC1 inhibitor RAD001 caused potent G0/G1 arrest, while LBH589 induced pronounced apoptosis, both of which were enhanced when the drugs were used in combination.79
In addition, additive effects of HDACi have been seen in conjunction with RSK2 (Ser227) inhibitor BI-D1870 and heat shock protein-90 (alpha/beta) inhibitor NVP-AUY922 in preclinical studies.80,81 Also, HDACi-inducible Bim is primarily neutralized by Bcl-2 and Bcl-xL, thus providing a mechanistic framework by which Bcl-2 antagonists potentiate the lethality of HDACi.82 Also, SAHA and trichostatin A induce G1 arrest by upregulating p21 and p27 and inhibiting E2F transcriptional activity. The tumor necrosis factor-related apoptosis-inducing ligand effect can be enhanced after HDACi pretreatment and is found to be consistent with the upregulation of proapoptotic Bim, Bak, Bax, Noxa, and p53 upregulated modulator of apoptosis (PUMA) and downregulation of antiapoptotic Bcl-2 and Bcl-xL.83
In combination with immune therapies
In addition to all the above-mentioned combination therapies, HDACi enhance MHC classes I and II expression and tumor-associated antigens on tumor cells, inducing cell death mediated by natural killer cells and cytotoxic T-cells.84–89 Also, vorinostat induces the secretion of adenosine triphosphate and high mobility group box 1 protein (HMGB-1) and expression of calreticulin on the tumor cell surface, which are important mediators of recognition and phagocytosis by dendritic cells.88–90 However, the effect of HDACi on the immune cells is far from clear and has been reviewed in detail elsewhere. Overall, it appears that the HDACi could promote or inhibit the functions of regulatory T-cells, myeloid-derived suppressor cells, and tumor-associated macrophages.91
HDACi have shown favorable responses in combination with immune therapies in preclinical settings. Christiansen et al88 observed synergistic responses when vorinostat or panobinostat was used in combination with anti-CD40 and anti-CD137 antibodies in solid tumors. He also noted an important role for CD8+ cytotoxic T-cells and natural killer cells for the synergy observed.88 In another study, LAQ824 induced synergistic cell death in combination with adoptive transfer of tumor-specific T-cells in melanoma.92 However, the effect of this combination remains unexplored in MM. One preclinical study showed that LBH589 impairs the phenotype and function of dendritic cells by downregulating dendritic cell maturation, antigen presentation, and T-cell costimulation markers on immature and mature dendritic cells.93 Thus, it is important to examine the immune status of patients with MM before and after HDACi treatment. Such studies will help us not only to better understand the effects of HDACi on immune cells but also identify potential combinations of HDACi with immune therapies.
Clinical trials using HDACi in MM
HDAC represents a very interesting clinical target for the development of novel antimyeloma therapy. The early clinical trials of different HDACi have revealed only modest single-agent activity, but encouraging clinical response rates have been reported in combination with other antimyeloma agents such as PIs, IMiDs, dexamethasone, and conventional cytotoxic therapy.
Vorinostat
Vorinostat (SAHA) is a potent nonselective HDACi with a hydroxamic acid moiety, which causes reversible inhibition of classes I and II HDACs. It was the first epigenetic agent used therapeutically in malignancy and was approved by the US Food and Drug Administration (FDA) for the treatment of cutaneous T-cell lymphoma in 2006.94
In the initial dose-escalating Phase I trial of vorinostat in relapsed/refractory MM (RRMM), 13 patients with a median of three prior lines of therapy were included. The most common drug-related adverse effects (AEs) included fatigue, anorexia, dehydration, diarrhea, and nausea and were mostly grade ≤2. Among the ten evaluable patients, one had a minimal response and nine had stable disease (SD).95
Based on the synergy with PIs depicted in preclinical studies, a Phase I trial evaluated vorinostat in combination with Btz in patients with RRMM. The 23 patients enrolled in the study had received a median of seven prior regimens with 20 patients post ASCT and 19 patients with prior Btz (nine of whom were Btz refractory). The dose-limiting toxicity was prolonged QT interval seen in two patients. The most common toxicities were myelosuppression, diarrhea, and fatigue. The overall response rate (ORR) was 42%, with two patients having very good partial response (VGPR) and seven patients having PR, including three patients who were Btz refractory.96
VANTAGE 095 was a multicenter, open-label Phase IIB study in which 143 patients with RRMM (Btz refractory) received vorinostat in combination with Btz till progressive disease, unacceptable toxicities, or patient withdrawal. The ORR was 11%, while 47% of patients had SD. The median duration of response (DOR) was 7.0 months, and the median OS was 11.1 months. However, serious AEs were reported in 65% of patients, resulting in treatment discontinuations in 11% of patients.97
On the basis of these encouraging responses, a multicenter, randomized, double-blind Phase III study, VANTAGE 088 trial, was conducted. They enrolled 637 patients with RRMM who had progressive disease after one to three prior antimyeloma treatments (but were Btz sensitive) and randomized them to receive Btz with vorinostat or placebo. The addition of vorinostat to Btz significantly improved the ORR (56% vs 41%) and clinical benefit rates (CBRs) (71% vs 53%). The median PFS also increased from 6.83 to 7.63 months, but the median OS was not significantly different between the two groups. More patients in the vorinostat group developed high-grade AEs, especially fatigue, myelosuppression, and gastrointestinal disorders compared to the placebo group. The authors concluded that though the study achieved the primary end point of prolonging the PFS, the clinical value of adding vorinostat to Btz needed further evaluation with regard to optimizing the dose of vorinostat to minimize toxicity.98
Vorinostat has also been used in combination with carfilzomib in compassionate use setting for patients with RRMM and was well tolerated.99 A Phase I dose-escalation trial of vorinostat with Len/dexamethasone in RRMM demonstrated an ORR of 47%. Serious AEs were reported in 45% of the patients and were considered to be study drug related in 22%.100 Hence, this combination seems to be effective and needs further evaluation.
Panobinostat
Panobinostat (LBH589) is a cinnamic hydroxamic acid analog that exhibits tenfold higher inhibitory activity against classes I, II, and IV HDACs than vorinostat. A Phase II multicenter study of oral panobinostat in 38 heavily pretreated patients with RRMM showed that it was well tolerated and the most common AEs were nausea and fatigue. But the ORR was lower than what was seen in the preclinical studies with VGPR in one patient, mixed response (MR) in one patient, and SD in three patients.101
In view of poor results with its use as monotherapy and preclinical data depicting synergy with Btz, a Phase Ib trial studied the use of panobinostat in combination with Btz in RRMM. Among the 47 patients enrolled in the dose-escalation phase, 76% of patients had ≥MR with responses seen in ten of 15 Btz refractory patients. Out of the 12 evaluable patients enrolled in the dose-expansion phase, MR was seen in 75% of patients.102
PANORAMA 2 is a Phase II trial of panobinostat in combination with Btz and dexamethasone in patients with relapsed and Btz refractory MM with at least two prior lines of therapy. Fifty-five heavily pretreated patients with a median of four prior regimens were enrolled. The ORR was 34.5%, and the CBR was 52.7%. Median PFS was 5.4 months, and the median DOR was 6.0 months. Common grade 3/4 AEs included thrombocytopenia (63.6%), fatigue (20.0%), and diarrhea (20.0%).103
PANORAMA 1 is a multicenter double-blind Phase III trial of patients with RRMM after one to three previous treatment regimens. Approximately 768 eligible patients were randomized to receive Btz and dexamethasone with panobinostat or placebo. It was demonstrated that though the ORR (60.7% vs 54.6%) was similar, the proportion of patients achieving complete response (CR) or near CR (27.6% vs 16.7%) was significantly higher with panobinostat compared to placebo. The addition of panobinostat prolonged the median DOR (13.14 vs 10.87 months), median PFS (11·99 vs 8·08 months), and median OS (33.6 vs 30.4 months). Serious AEs were higher in the panobinostat group (60% vs 42%). Common grade 3–4 AEs were thrombocytopenia, lymphopenia, diarrhea, asthenia, and peripheral neuropathy.104 Recent subgroup analysis of PANORAMA 1 trial demonstrated a clear PFS benefit of 7.8 months for panobinostat–Btz–Dex among patients who received two or more prior regimens, including Btz and IMiD, a population with poorer prognosis and limited treatment options.105
Collectively, the results of PANORAMA 1 and 2 show that the combination of panobinostat and Btz appears promising and has recently been approved by the FDA for the treatment of RRMM in patients with two prior treatments, including Btz and IMiDs.
Romidepsin
Romidepsin (FR901228 or FK228) is a depsipeptide derived from the bacterium Chromobacterium violaceum with activity mainly against class I HDAC. It was approved by the FDA for the treatment of relapsed cutaneous T-cell lymphoma in 2009.106
A Phase II study evaluated the activity of romidepsin in heavily pretreated patients with MM who were refractory to therapies, including ASCT, Btz, and IMiDs. Although no objective responses were achieved, ~30% of patients exhibited stabilization of M-protein, resolution of hypercalcemia, and improvement in bone pain. The most common AEs were grade 1/2 and included nausea, fatigue, taste alteration, and clinically insignificant electrocardiographic abnormalities.107
A Phase II trial used romidepsin with Btz and dexamethasone based on preclinical synergy. The incidence of grade 3 anemia and neutropenia was similar to that reported in previous trials using Btz–dexamethasone. PR was seen in 52% (VGPR in 28%) and CR was seen in 8% of the 25 patients enrolled. The median time to progression was 7.2 months, and the median OS was > 36 months.108
A Phase I/II trial is evaluating the combination of romidepsin and Len in patients with relapsed/refractory lymphoma and myeloma. The study is ongoing, but the Phase I results suggest that the combination is well tolerated up to standard single-agent doses of each drug.109
ACY-1215
ACY-1215 is an oral small molecule targeted against HDAC6. In view of responses seen in xenograft severe combined immunodeficiency mouse models,60 a Phase I trial is evaluating ACY-1215 alone (part 1, Phase Ia) and in combination with Btz (part 2, Phase Ib) in patients with RRMM after at least two lines of treatment. In Phase Ia, no maximal tolerated dose was identified and AEs reported were elevated creatinine, fatigue, hypercalcemia, and upper respiratory tract infection (not attributed to ACY-1215). In Phase Ib, grade 3 or 4 gastrointestinal AEs were rare and hematologic AEs were manageable. The ORR was 25%, and the CBR was 60% in this heavily pretreated patient population.110
Another ongoing trial is exploring the combination of ACY-1215 with Len/dexamethasone. ACY-1215 is found to be well tolerated, and no dose-limiting toxicity has been observed so far. The most common AEs, mainly grades 1/2, were fatigue, upper respiratory tract infections, and neutropenia. At the interim analysis, the ORR was 81%, including one CR and three VGPR.111
Belinostat
Belinostat (PXD101) is a nonselective HDACi of hydroxamic acid class. A Phase II study enrolled 24 patients with RRMM who received belinostat as monotherapy and in combination with high dose of dexamethasone. This treatment was well tolerated, with minimal side effects, obtaining one MR and five SD.112
Givinostat
Givinostat (ITF2357) is an orally active HDACi. In a Phase II trial, givinostat (alone or combined with dexamethasone) proved tolerable but showed only a modest clinical benefit. Only five of the 19 patients with advanced MM achieved SD. All patients experienced grade 3/4 thrombocytopenia, three had grade 3/4 gastrointestinal toxicity, and three had transient electrocardiographic abnormalities.113
Conclusion
Epigenetic aberrations have now been recognized to contribute to the development and progression of various types of cancer, including MM. HDACi regulate the acetylation status of various histone and nonhistone proteins required for cellular processes, including gene expression, protein recycling, cell proliferation, and apoptosis, that are important for myeloma cell growth and survival. Preclinical evidence from studies of HDACi, alone or in combination with other antimyeloma agents, provides a strong scientific rationale for the evaluation of these regimens in the clinical setting. Results from early-stage clinical trials demonstrate that though HDACi show only modest activity as single agent, using them in combination with other anti-MM agents, especially Btz, show significant clinical responses. It must be noted that most of these trials were performed in patients relapsed on or refractory to Btz, and perhaps their utilization earlier in therapy, likely in combination with Btz, would be more effective. Hence, their precise role in the armamentarium of therapy for MM is yet to be defined. In addition, isoform-selective and/or class-selective HDAC inhibition needs further evaluation to reduce unfavorable side effects.
Disclosure
SKK has received research support from Novartis for clinical trials. The authors report no other conflicts of interest in this work.
References
Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004; 351(18):1860–1873. | |
National Cancer Institute [webpage on the Internet]. SEER Stat Fact Sheets: Myeloma. 2016. Available from: http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed March 7, 2016. | |
Kristinsson SY, Landgren O, Dickman PW, Derolf AR, Bjorkholm M. Patterns of survival in multiple myeloma: a population-based study of patients diagnosed in Sweden from 1973 to 2003. J Clin Oncol. 2007;25(15):1993–1999. | |
Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. | |
Kumar SK, Lee JH, Lahuerta JJ, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012;26:149–157. | |
Sharma A, Heuck CJ, Fazzari MJ, et al. DNA methylation alterations in multiple myeloma as a model for epigenetic changes in cancer. Wiley Interdiscip Rev Syst Biol Med. 2010;2:654–669. | |
Chapman MA, Lawrence MS, Keats JJ, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. | |
Dimopoulos K, Gimsing P, Gronbaek K. The role of epigenetics in the biology of multiple myeloma. Blood Cancer J. 2014;4:e207. | |
Dimopoulos K, Gimsing P, Grønbaek K. Aberrant microRNA expression in multiple myeloma. Eur J Haematol. 2013;91:95–105. | |
Bentley GA, Finch JT, Lewit-Bentley A, Roth M. The crystal structure of the nucleosome core particle by contrast variation. Basic Life Sci. 1984;27:105–117. | |
Moniot S, Weyand M, Steegborn C. Structures, substrates, and regulators of mammalian sirtuins – opportunities and challenges for drug development. Front Pharmacol. 2012;3:16. | |
Khan O, La Thangue NB. Drug insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat Clin Pract Oncol. 2008;5:714–726. | |
Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85–94. | |
Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett. 2009;277:8–21. | |
Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. | |
Liu T, Liu PY, Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009;69:1702–1705. | |
Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. | |
Smith EM, Boyd K, Davies FE. The potential role of epigenetic therapy in multiple myeloma. Br J Haematol. 2009;148:702–713. | |
Maes K, Menu E, Valckenborgh EV, Riet IV, Vanderkerken K, Bruyn ED. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers. 2013;5:430–461. | |
Bollati V, Fabris S, Pegoraro V, et al. Differential repetitive DNA methylation in multiple myeloma molecular subgroups. Carcinogenesis. 2009;30:1330–1335. | |
Aoki Y, Nojima M, Suzuki H, et al. Genomic vulnerability to LINE-1 hypomethylation is a potential determinant of the clinicogenetic features of multiple myeloma. Genome Med. 2012;4:101. | |
Walker BA, Wardell CP, Chiecchio L, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2011;117:553–562. | |
Martinez-Garcia E, Popovic R, Min DJ, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–220. | |
Brito JL, Walker B, Jenner M, et al. MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells. Haematologica. 2009;94:78–86. | |
Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280(2):168–176. | |
Marquard L, Gjerdrum LM, Christensen IJ, Jensen PB, Sehested M, Ralfkiaer E. Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology. 2008;53(3):267–277. | |
Skov V, Larsen TS, Thomassen M, et al. Increased gene expression of histone deacetylases in patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leuk Lymphoma. 2012;53(1):123–129. | |
Wang JC, Chen C, Dumlao T, et al. Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk Lymphoma. 2008; 49(12):2321–2327. | |
Marquard L, Poulsen CB, Gjerdrum LM, et al. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology. 2009;54(6):688–698. | |
Weichert W, Röske A, Gekeler V, et al. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 2008;9(2):139–148. | |
Weichert W, Röske A, Niesporek S, et al. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14(6):1669–1677. | |
Weichert W, Denkert C, Noske A, et al. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia. 2008;10(9):1021–1027. | |
Zhang Z, Yamashita H, Toyama T, et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res Treat. 2005;94(1):11–16. | |
Moreno DA, Scrideli CA, Cortez MA, et al. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br J Haematol. 2010;150(6):665–673. | |
Van Damme M, Crompot E, Meuleman N, et al. HDAC isoenzyme expression is deregulated in chronic lymphocytic leukemia B-cells and has a complex prognostic significance. Epigenetics. 2012; 7(12):1403–1412. | |
Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics. 2014;9(11):1511–1520. | |
Heller G, Schmidt WM, Ziegler B, et al. Genome-wide transcriptional response to 5-aza-2’-deoxycytidine and trichostatin a in multiple myeloma cells. Cancer Res. 2008;68:44–54. | |
Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci U S A. 2004; 101:540–545. | |
Neri P, Tagliaferri P, di Martino MT, et al. In vivo anti-myeloma activity and modulation of gene expression profile induced by valproic acid, a histone deacetylase inhibitor. Br J Haematol. 2008;143:520–531. | |
Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–253. | |
Gray SG, Qian CN, Furge K, Guo X, Teh BT. Microarray profiling of the effects of histone deacetylase inhibitors on gene expression in cancer cell lines. Int J Oncol. 2004;24:773–795. | |
Peart MJ, Smyth GK, van Laar RK, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:3697–3702. | |
Kaiser M, Zavrski I, Sterz J, et al. The effects of the histone deacetylase inhibitor valproic acid on cell cycle, growth suppression and apoptosis in multiple myeloma. Haematologica. 2006;91:248–251. | |
Khan SB, Maududi T, Barton K, Ayers J, Alkan S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br J Haematol. 2004;125:156–161. | |
Golay J, Cuppini L, Leoni F, et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia. 2007;21:1892–1900. | |
Catley L, Weisberg E, Tai YT, et al. NVPLAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–2622. | |
Feng R, Ma H, Hassig CA, et al. KD5170, a novel mercaptoketone-based histone deacetylase inhibitor, exerts antimyeloma effects by DNA damage and mitochondrial signaling. Mol Cancer Ther. 2008;7:1494–1505. | |
Catley L, Weisberg E, Kiziltepe T, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood. 2006;108:3441–3449. | |
Lavelle D, Chen YH, Hankewych M, De-Simone J. Histone deacetylase inhibitors increase p21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am J Hematol. 2001;68:170–178. | |
Mitsiades N, Mitsiades CS, Richardson PG, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–4062. | |
Maiso P, Carvajal-Vergara X, Ocio EM, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66:5781–5789. | |
Deleu S, Lemaire M, Arts J, et al. The effects of JNJ-26481585, a novel hydroxamate-based histone deacetylase inhibitor, on the development of multiple myeloma in the 5T2MM and 5T33MM murine models. Leukemia. 2009;23:1894–1903. | |
Bai LY, Omar HA, Chiu CF, Chi ZP, Hu JL, Weng JR. Antitumor effects of (S)-HDAC42, a phenylbutyrate-derived histone deacetylase inhibitor, in multiple myeloma cells. Cancer Chemother Pharmacol. 2010;35:373–379. | |
Schwartz C, Palissot V, Aouali N, et al. Valproic acid induces non-apoptotic cell death mechanisms in multiple myeloma cell lines. Int J Oncol. 2007;30:573–582. | |
Rosato RR, Almenara JA, Maggio SC, et al. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther. 2008;7(10):3285–3297. | |
Chen CS, Wang YC, Yang HC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67(11):5318–5327. | |
Feng R, Oton A, Mapara MY, Anderson G, Belani C, Lentzsch S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol. 2007;139:385–397. | |
Lee CK, Wang S, Huang X, Ryder J, Liu B. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010;296:233–240. | |
Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. | |
Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011;25(2):226–235. | |
Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–3852. | |
Deleu S, Lemaire M, Arts J, et al. Bortezomib alone or in combination with the histone deacetylase inhibitor JNJ-26481585: effect on myeloma bone disease in the 5T2MM murine model of myeloma. Cancer Res. 2009;69:5307–5311. | |
Maes K, Menu E, Van Valckenborgh E, Van Riet I, Vanderkerken K, De Bruyne E. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers (Basel). 2013;5(2):430–461. | |
Lemaire M, Deleu S, de Bruyne E, van Valckenborgh E, Menu E, Vanderkerken K. The microenvironment and molecular biology of the multiple myeloma tumor. Adv Cancer Res. 2012;110:19–42. | |
Qian DZ, Wang X, Kachhap SK, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64(18):6626–6634. | |
Ellis L, Hammers H, Pili R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009;280(2):145–153. | |
Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32(3):157–165. | |
Dong XF, Song Q, Li LZ, Zhao CL, Wang LQ. Histone deacetylase inhibitor valproic acid inhibits proliferation and induces apoptosis in KM3 cells via downregulating VEGF receptor. Neuro Endocrinol Lett. 2007;28:775–780. | |
Kitazoe K, Abe M, Hiasa M, et al. Valproic acid exerts anti-tumor as well as antiangiogenic effects on myeloma. Int J Hematol. 2009;89:45–57. | |
Davenport EL, Moore HE, Dunlop AS, et al. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–2649. | |
Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90. J Biol Chem. 2005;280(267):29–34. | |
Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 2009;280:201–210. | |
Hideshima T, Bradner JE, Wong J, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A. 2005;102:8567–8572. | |
Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589. | |
Kikuchi J, Wada T, Shimizu R, et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 2010;116:406–417. | |
Ocio EM, Vilanova D, Atadja P, et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95:794–803. | |
Campbell RA, Sanchez E, Steinberg J, et al. Vorinostat enhances the antimyeloma effects of melphalan and bortezomib. Eur J Haematol. 2010;84:201–211. | |
Du HL, Ren LM, Chen H, Zhu Y, Qi Y. Re-expression of p16 gene in the myeloma cell line U266 induced by synergy of sodium butyrate and 5-Aza-2’-deoxycytidine. Di Yi Jun Yi Da Xue Xue Bao. 2002;22:981–984. | |
Ramakrishnan V, Kimlinger T, Timm M, Haug J, Rajkumar SV, Kumar S. Multiple mechanisms contribute to the synergistic anti-myeloma activity of the pan-histone deacetylase inhibitor LBH589 and the rapalog RAD001. Leuk Res. 2014;38:1358–1366. | |
Shimura Y, Kuroda J, Ri M, et al. RSK2(Ser227) at N-terminal kinase domain is a potential therapeutic target for multiple myeloma. Mol Cancer Ther. 2012;11(12):2600–2609. | |
Kaiser M, Lamottke B, Mieth M, et al. Synergistic action of the novel HSP90 inhibitor NVP-AUY922 with histone deacetylase inhibitors, melphalan, or doxorubicin in multiple myeloma. Eur J Haematol. 2010;84:337–344. | |
Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol Cell Biol. 2009;29:6149–6169. | |
Fandy TE, Shankar S, Ross DD, Sausville E, Srivastava RK. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia. 2005;7(7):646–657. | |
Magner WJ, Kazim AL, Stewart C, et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000;165(12):7017–7024. | |
Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65(23):11136–11145. | |
Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008;57(5):647–654. | |
Manning J, Indrova M, Lubyova B, et al. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology. 2008;123(2):218–227. | |
Christiansen AJ, West A, Banks KM, et al. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc Natl Acad Sci U S A. 2011;108(10):4141–4146. | |
West AC, Mattarollo SR, Shortt J, et al. An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res. 2013;73(24):7265–7276. | |
Sonnemann J, Gressmann S, Becker S, Wittig S, Schmudde M, Beck JF. The histone deacetylase inhibitor vorinostat induces calreticulin exposure in childhood brain tumour cells in vitro. Cancer Chemother Pharmacol. 2010;66(3):611–616. | |
Kroesen M, Gielen P, Brok IC, Armandari I, Hoogerbrugge PM, Adema GJ. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget. 2014;5(16):6558–6572. | |
Vo DD, Prins RM, Begley JL, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009;69(22):8693–8699. | |
Song W, Tai YT, Tian Z, et al. HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia. 2011;25(1):161–168. | |
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–1252. | |
Richardson P, Mitsiades C, Colson K, et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma. 2008;49(3):502–507. | |
Badros A, Burger AM, Philip S, et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009;15(16):5250–5257. | |
Siegel DS, Dimopoulos MA, Yoon S-S, et al. Vantage 095: Vorinostat in Combination with Bortezomib in Salvage Multiple Myeloma Patients: Final Study Results of a Global Phase 2b Trial. ASH; San Diego; 2011. | |
Dimopoulos MA, Siegel DS, Lonial S, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 2013;14(11):1129–1140. | |
Alsayed Y, Nair BP, Kauffman M, et al. Carfilzomib (CFZ) in combination with DEX and other agents (Dox, Thal, DDP, vorinostat) for far advanced and refractory multiple myeloma (FARMM). J Clin Oncol. 2010;28:e18504. ASCO Annual Meeting Abstracts. | |
Siegel DS, Richardson P, Dimopoulos M, et al. Vorinostat in combination with lenalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood Cancer J. 2014;4:e182. | |
Wolf LW, Siegel D, Matous J, et al. A phase II study of oral panobinostat (LBH589) in adult patients with advanced refractory multiple myeloma. Blood. 2008(11):112. ASH Annual Meeting Abstracts. | |
San-Miguel JF, Richardson PGG, Sezer O, et al. A phase lb study of oral panobinostat and IV bortezomib in relapsed or relapsed and refractory multiple myeloma. J Clin Oncol. 2011;29(15):abstr8075. ASCO Annual Meeting Proceedings. | |
Richardson PG, Schlossman RL, Alsina M, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood. 2013;122:2331–2337. | |
San-Miguel JF, Hungria VTM, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15:1195–1206. | |
Richardson PG, Hungria VTM, Yoon SS, et al. Panobinostat plus bortezomib and dexamethasone in relapsed/relapsed and refractory myeloma: outcomes by prior treatment. Blood. 2016;127(6):713–721. | |
Bertinoa EM, Ottersona GA. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin Investig Drugs. 2011;20(8):1151–1158. | |
Niesvizky R, Ely S, Mark T, et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 2011;117(2):336–342. | |
Harrison SJ, Quach H, Link E, et al. A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood. 2011;118(24):6274–6283. | |
Lunning MA, Ruan J, Nair S, et al. A phase I/II trial of the combination of romidepsin and lenalidomide in patients with relapsed/refractory lymphoma and myeloma: phase I results. J Clin Oncol. 2014; 32(5 suppl):abstr8582. | |
Raje N, Vogl DT, Hari PN, et al. ACY-1215, a Selective Histone Deacetylase (HDAC) 6 Inhibitor: Interim Results Of Combination Therapy With Bortezomib In Patients With Multiple Myeloma (MM). ASH 2013 Annual Meeting Abstract 759 (Oral Presentation). New Orleans, LA; 2013. | |
Yee A, Vorhees P, Bensinger WI, et al. ACY-1215, a Selective Histone Deacetylase (HDAC) 6 Inhibitor, In Combination With Lenalidomide and Dexamethasone (dex), Is Well Tolerated Without Dose Limiting Toxicity (DLT) In Patients (Pts) With Multiple Myeloma (MM) At Doses Demonstrating Biologic Activity: Interim Results Of a Phase 1b Trial. ASH 2013 Annual Meeting Abstract 3190 (Poster Presentation). New Orleans, LA; 2013. | |
Sullivan DSS, Schuster M, Berenson J, et al. A phase II study of PXD 101 in advanced multiple myeloma. Blood. 2006(11):108. (ASH Annual Meeting Abstracts). | |
Galli M, Salmoiraghi S, Golay J, et al. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. 2010;89(2):185–190. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.