")
Back to Journals » The Application of Clinical Genetics » Volume 7
Clinical problems of colorectal cancer and endometrial cancer cases with unknown cause of tumor mismatch repair deficiency (suspected Lynch syndrome)
Authors Buchanan D, Rosty C, Clendenning M, Spurdle A, Win AK
Received 17 May 2014
Accepted for publication 18 July 2014
Published 6 October 2014 Volume 2014:7 Pages 183—193
DOI https://doi.org/10.2147/TACG.S48625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Daniel D Buchanan,1,2 Christophe Rosty,1,3,4 Mark Clendenning,1 Amanda B Spurdle,5 Aung Ko Win2
1Oncogenomics Group, Genetic Epidemiology Laboratory, Department of Pathology, The University of Melbourne, Parkville, VIC, Australia; 2Centre for Epidemiology and Biostatistics, Melbourne School of Population and Global Health, The University of Melbourne, Parkville, VIC, Australia; 3Envoi Specialist Pathologists, Herston, QLD, Australia; 4School of Medicine, University of Queensland, Herston, QLD, Australia; 5Molecular Cancer Epidemiology Laboratory, Genetics and Computational Biology Division, QIMR Berghofer Medical Research Institute, Herston, QLD, Australia
Abstract: Carriers of a germline mutation in one of the DNA mismatch repair (MMR) genes have a high risk of developing numerous different cancers, predominantly colorectal cancer and endometrial cancer (known as Lynch syndrome). MMR gene mutation carriers develop tumors with MMR deficiency identified by tumor microsatellite instability or immunohistochemical loss of MMR protein expression. Tumor MMR deficiency is used to identify individuals most likely to carry an MMR gene mutation. However, MMR deficiency can also result from somatic inactivation, most commonly methylation of the MLH1 gene promoter. As tumor MMR testing of all incident colorectal and endometrial cancers (universal screening) is becoming increasingly adopted, a growing clinical problem is emerging for individuals who have tumors that show MMR deficiency who are subsequently found not to carry an MMR gene mutation after genetic testing using the current diagnostic approaches (Sanger sequencing and multiplex ligation-dependent probe amplification) and who also show no evidence of MLH1 methylation. The inability to determine the underlying cause of tumor MMR deficiency in these "Lynch-like" or "suspected Lynch syndrome" cases has significant implications on the clinical management of these individuals and their relatives. When the data from published studies are combined, 59% (95% confidence interval [CI]: 55% to 64%) of colorectal cancers and 52% (95% CI: 41% to 62%) of endometrial cancers with MMR deficiency were identified as suspected Lynch syndrome. Recent studies estimated that colorectal cancer risk for relatives of suspected Lynch syndrome cases is lower than for relatives of those with MMR gene mutations, but higher than for relatives of those with tumor MMR deficiency resulting from methylation of the MLH1 gene promoter. The cause of tumor MMR deficiency in suspected Lynch syndrome cases is likely due to either unidentified germline MMR gene mutations, somatic cell mosaicism, or biallelic somatic inactivation. Determining the underlying cause of tumor MMR deficiency in suspected Lynch syndrome cases is likely to reshape the current triaging schemes used to identify germline MMR gene mutations in cancer-affected individuals and their relatives.
Keywords: Lynch syndrome, cancer risk, screening, somatic mutation, germline mutation
Introduction
Lynch syndrome is the most common form of hereditary colorectal cancer and hereditary endometrial cancer, accounting for up to 6% of all colorectal and endometrial cancer cases.1,2 Lynch syndrome is an autosomal dominantly inherited disorder caused by germline mutations in one of the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2). Additionally, mutations in the gene upstream of MSH2 (EPCAM) have been shown to disrupt MSH2 expression and predispose to MSH2-deficient cancers.3 MMR gene mutation carriers have an increased risk of developing colorectal cancer and endometrial cancer, as well as cancers of the stomach, ovary, ureter, renal pelvis, brain, small bowel, pancreas, and biliary tract,4,5 and also second primary cancers.6–9 A 15-year controlled trial of MMR gene mutation carriers showed that colonoscopic screening and the removal of adenomas reduced both colorectal cancer incidence and mortality in carriers.10 Therefore, the identification of MMR gene mutation carriers is of critical importance for targeted colonoscopic screening to reduce the burden of disease in the community.
Loss of MMR function as a result of a germline mutation in one of the MMR genes can lead to the accumulation of errors in microsatellite repeats during DNA replication. As a result, tumors from MMR gene mutation carriers characteristically demonstrate MMR deficiency, defined as the presence of microsatellite instability (MSI) at high levels (MSI-H) and/or loss of MMR protein expression determined by immunohistochemistry (IHC).11 There have been several alternative recommendations for colorectal tumor testing for MMR deficiency by IHC and/or MSI testing using different ages of colorectal cancer diagnosis as a cut-off, regardless of family history; for example, age 50 years12,13 or 60 years14,15 or 70 years.16 However, several groups recommend that all cases of colorectal cancer should be tested for MMR deficiency regardless of their age at diagnosis or family history17–22 given that a proportion of colorectal cancers caused by MMR mutations occur at old age. This so-called “universal screening” has virtually complete sensitivity (100%; 95% confidence interval [CI]: 99.3% to 100%), as everyone is tested.22 The situation is similar for endometrial cancer, with a number of different strategies proposed based on age at diagnosis and/or family history,23,24 including diagnosis age <50 years21,22 and, more recently, <60 years,2 and also universal testing.25,26 A disadvantage of universal screening in colorectal or endometrial cancer cases is that it may be less cost-effective compared with having an age-of-diagnosis cut-off.2,13
While tumor MMR deficiency is a good predictor of carrying a germline MMR gene mutation, it can result from the two different mechanisms: “inherited” MMR deficiency, resulting from carrying a germline MMR gene mutation; or “sporadic” MMR deficiency, caused by somatic inactivation mechanisms in the tumor. The most common cause of somatic inactivation resulting in MSI identified to date is DNA methylation of the MLH1 gene promoter, specifically in the “C” region.2,27 In most cases, sporadic colorectal tumors with MSI exhibit extensive CpG island methylation phenotype (CIMP) and methylation of the MLH1 gene promoter. An association between sporadic MSI colorectal cancer and the presence of the BRAF V600E has been previously reported.22,23 No correlation, however, between MLH1 promoter methylation and BRAF V600E somatic mutations in endometrial cancer has been reported.22 MLH1 methylation (and BRAF V600E mutation in colorectal cancer) rarely occurs in colorectal and endometrial cancers from MMR gene mutation carriers.2,28 As a result, testing for tumor MMR deficiency incorporated with testing for MLH1 promoter methylation and/or BRAF V600E mutation in MLH1-deficient colorectal tumors is increasingly implemented in clinical diagnostics to triage individuals for subsequent germline MMR gene mutation testing.29 Figure 1 demonstrates a classical tumor testing approach used in triaging colorectal cancer cases for MMR gene mutation identification.
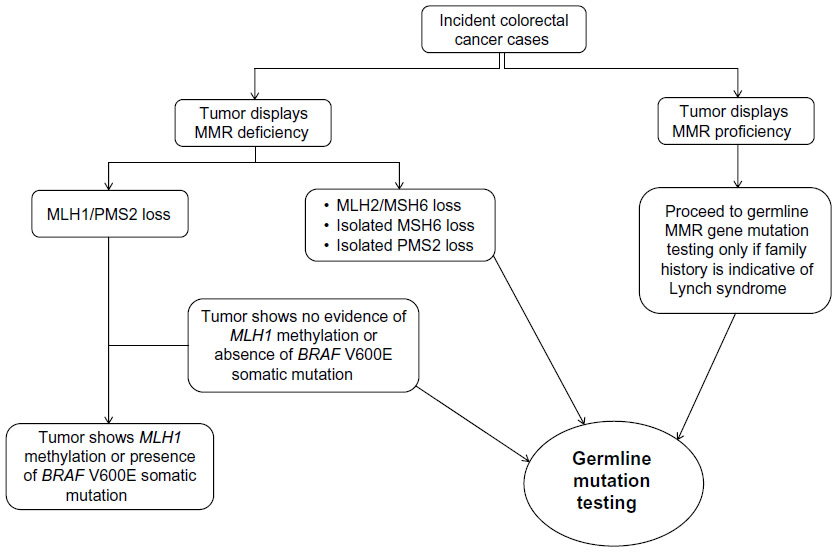 | Figure 1 Classic molecular testing approach for triaging colorectal cancer cases for germline mismatch repair (MMR) gene mutation testing. |
Differentiating individuals with inherited MMR deficiency from those with sporadic MMR deficiency is critical to defining optimal clinical management strategies. MMR gene mutation carriers and their carrier relatives have a high risk of developing cancers5 as well as second primary cancers.6–9,30,31 The same is not true for individuals, and relatives of individuals, with somatic MMR deficiency.32,33 Once diagnosed, MMR gene mutation carriers and their relatives are offered intensive clinical management including counseling, predictive mutation testing, increased surveillance, and prophylactic and chemotherapeutic treatment in order to reduce the development of cancer. Suspected Lynch syndrome (also called “Lynch-like”) cases refers to individuals in which no pathogenic germline MMR gene mutation has been identified, despite their tumor demonstrating MMR deficiency and there being no evidence for MLH1 promoter methylation in the tumor (Figure 2). Therefore, suspected Lynch syndrome cases cannot easily be assigned to either inherited or sporadic MMR deficiency categories.
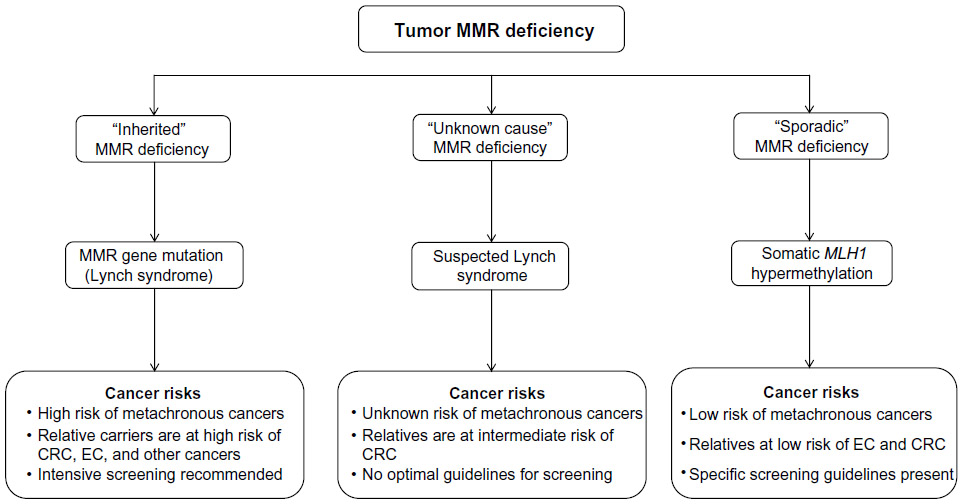 | Figure 2 The three groups of tumor mismatch repair (MMR) deficiency. |
The magnitude of the clinical problem of suspected Lynch syndrome cases
Across studies of differing design, up to 72% of colorectal cancer and 64% of endometrial cancer cases with tumor MMR deficiency do not have an MMR gene mutation identified by standard MMR gene testing approaches, nor do they show somatic MLH1 promoter methylation, depending on the gene and the population studied.2,19,26,33–37
The proportion of suspected Lynch syndrome cases in colorectal cancer cohorts varies among different studies (Table 1). In a large population-based study of the Colon Cancer Family Registry,38 3.8% (186/4,853) of all the colorectal cancer cases were confirmed MMR gene mutation carriers, whereas 5.6% (271/4,853) of all colorectal cancer cases were classified as suspected Lynch syndrome.33 Of the identified MMR-deficient colorectal cancers from this study (as evident by the loss of MMR protein expression in IHC) not related to MLH1 promoter methylation or the BRAF V600E somatic mutation, 56% (95% CI: 51% to 62%) were considered suspected Lynch syndrome. Similar proportions of suspected Lynch syndrome cases were reported in studies by Hampel et al at 69% (95% CI: 51% to 83%)17 and Rodríguez-Soler et al at 71% (95% CI: 58% to 83%).34 When data from these three studies are combined, the prevalence of suspected Lynch syndrome in MMR-deficient colorectal cancer cases with no MLH1 promoter methylation was estimated to be 59% (95% CI: 55% to 64%). Further, in a recent study of 102 early-onset colorectal cancer cases (diagnosed before age 50 years), Antelo et al identified that 13 of the 18 MMR-deficient colorectal cancer cases not related to MLH1 promoter methylation (72%; 95% CI: 47% to 90%) were considered suspected Lynch syndrome cases.35
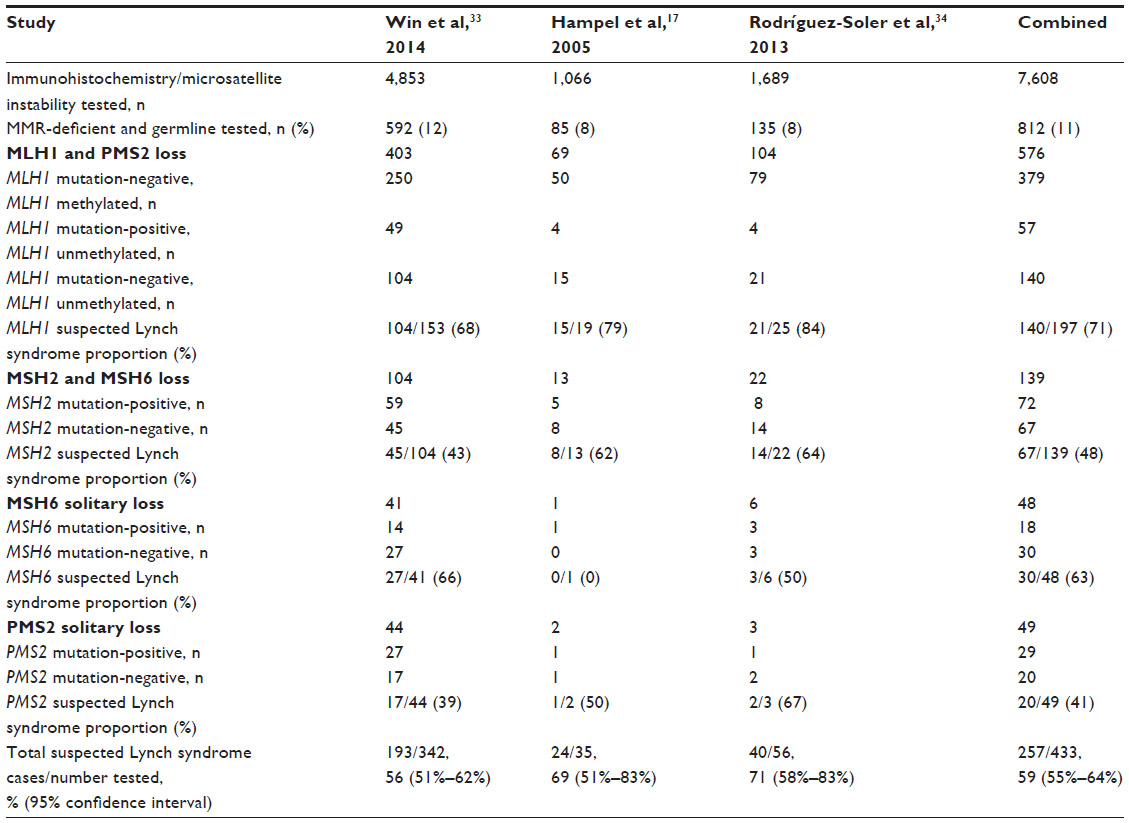 | Table 1 Summary of suspected Lynch syndrome cases from colorectal cancer cohorts |
Similar proportions of suspected Lynch syndrome cases are observed in endometrial cancer cohorts (Table 2). In a population-based endometrial cancer cohort from Australia, Buchanan et al tested 686 endometrial cancer cases for tumor MMR protein expression using IHC, tumor MLH1 promoter methylation, and germline MMR gene mutations. They found that, of the 55 endometrial cancer cases with MMR deficiency, only 45% (25/55) exhibited germline MMR gene mutations and 55% (95% CI: 41% to 68%) were identified as suspected Lynch syndrome.2 Of the 30 suspected Lynch syndrome–endometrial cancer cases, only one (3%) fulfilled the Amsterdam II criteria39 and 14 (47%) fulfilled the revised Bethesda Guidelines.4 Therefore, half of the suspected Lynch syndrome cases from this Australian endometrial cancer cohort had either no family history of cancer or a family history that did not meet criteria used for the identification of Lynch syndrome. Moline et al26 found a higher proportion of suspected Lynch syndrome cases at 64% (95% CI: 41% to 83%), while two other studies reported that around one-third of the MMR-deficient endometrial cancer cases were identified as suspected Lynch syndrome: 30% (95% CI: 7% to 65%) in Leenen et al37 and 36% (95% CI: 13% to 65%) in Hampel et al.19 When we combined the data from these four studies, suspected Lynch syndrome was identified in 52% (95% CI: 41% to 62%) of MMR-deficient endometrial cancer cases.
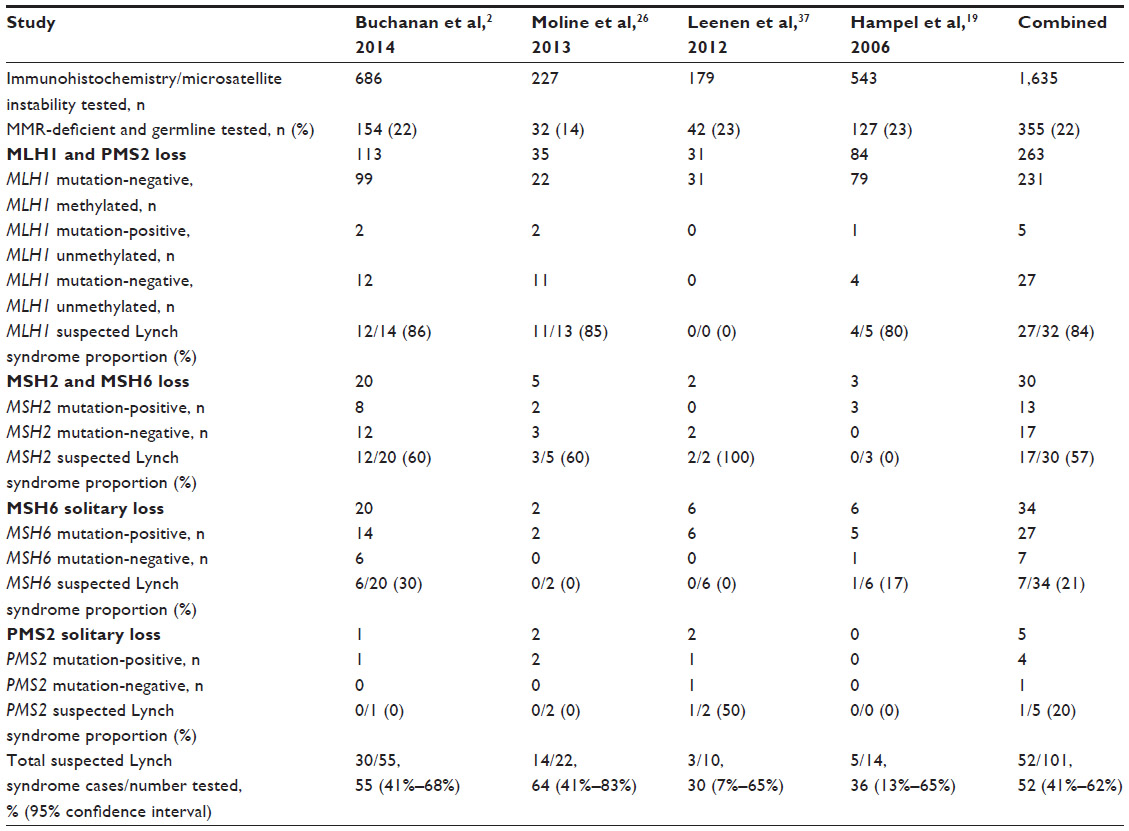 | Table 2 Summary of suspected Lynch syndrome cases from endometrial cancer cohorts |
These studies highlight a large proportion of colorectal and endometrial cancer cases that have no explanation for their tumor MMR deficiency. The move from MMR deficiency testing defined by age and/or family history criteria to universal screening is likely to increase the number of suspected Lynch syndrome cases identified.
Cancer risks in suspected Lynch syndrome
Little is known about the cancer risks for individuals with suspected Lynch syndrome. Only three studies have investigated cancer risks for suspected Lynch syndrome cases and their relatives (Table 3).33,34,40 Overbeek et al40 observed that 66% (50/75) of Lynch syndrome families from a Netherlands cohort fulfilled the Amsterdam II criteria;39 however, only 11% (2/18) of families with suspected Lynch syndrome fulfilled Amsterdam II (P<0.001). Rodríguez-Soler et al34 first quantified the risk of colorectal cancer for first-degree relatives of colorectal cancer cases with suspected Lynch syndrome and found the highest risk of colorectal cancer for first-degree relatives of MMR gene mutation carriers, an intermediate risk for those of individuals with suspected Lynch syndrome, and the lowest risk for those of MLH1-methylated MSI-H cases. A larger study confirmed the finding that first-degree relatives of colorectal cancer cases with suspected Lynch syndrome have an intermediate risk of colorectal cancer (between Lynch syndrome and sporadic MMR-deficient cases).33 The mean age at diagnosis of colorectal cancer of suspected Lynch syndrome was either similar to Lynch syndrome cases,34,40 or intermediate between Lynch syndrome and sporadic MMR-deficient cases.33 Currently, no studies have investigated the risk of cancers other than colorectal cancer for relatives of colorectal cancer cases with suspected Lynch syndrome. Further, no information is known about the risk of cancers for relatives of endometrial cancer cases with suspected Lynch syndrome.
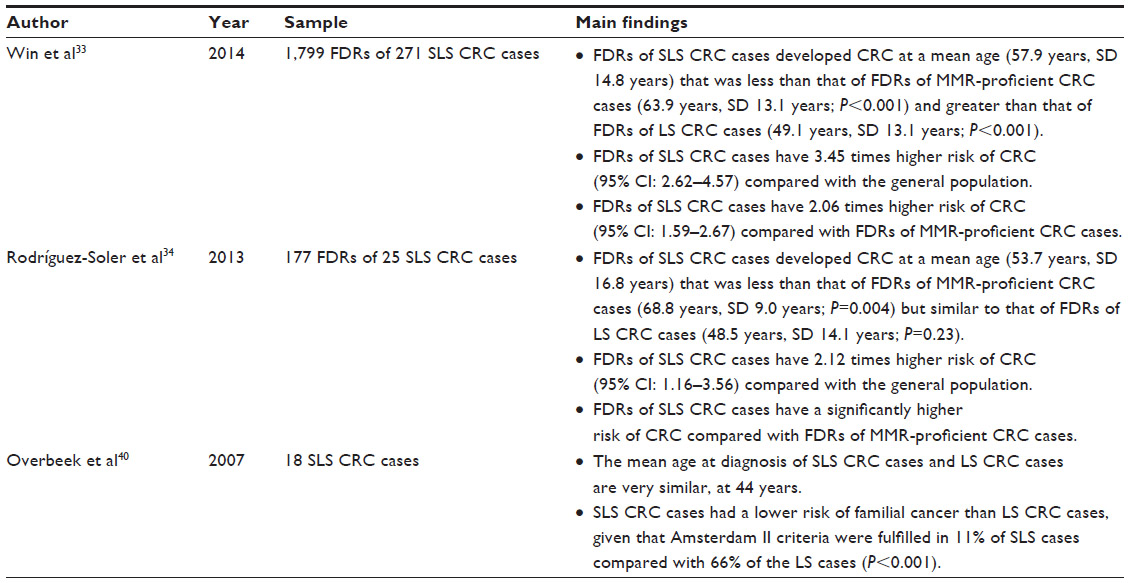 | Table 3 Estimates of cancer risks in SLS from previously published studies |
Current clinical management recommendation for suspected Lynch syndrome
The clinical management of individuals with suspected Lynch syndrome and their relatives in current practice is challenging, as there is insufficient information for defining evidence-based screening and treatment guidelines for them. As a consequence, individuals with suspected Lynch syndrome and their relatives are likely to receive varying degrees of screening, ranging from guidelines recommended for MMR gene mutation carriers to those recommended for low-risk individuals. Therefore, some individuals and their relatives are likely to be unnecessarily over-screened while others are likely to be under-screened, despite relatives of suspected Lynch syndrome cases having an increased risk of developing colorectal cancer. Such uncertainty in management is a great burden to both clinicians and families, as well as to the health care system.
Given that risk of colorectal cancer for first-degree relatives of colorectal cancer cases with suspected Lynch syndrome is intermediate (between that for first-degree relatives of Lynch syndrome and sporadic MMR-deficient cases), an intermediate screening strategy has been proposed, ie, a longer interval of colonoscopic screening for those without a strong family history of colorectal cancer (rather than 1- to 2-yearly colonoscopy given to MMR gene mutation carriers).34 However, optimal screening (including the age of starting colonoscopies) and clinical management strategies are yet to be defined, given that this group is likely to be heterogeneous with regards to family history and to the mechanism of MMR inactivation, as described in the next section.
Potential causes of tumor MMR deficiency in suspected Lynch syndrome
The potential underlying causes of tumor MMR deficiency in suspected Lynch syndrome cases may include: 1) germline inactivation via unidentified MMR gene mutations or genetic mutations in other MMR-related genes not attributed to Lynch syndrome that indirectly result in tumor MMR deficiency; and 2) somatic inactivation resulting from DNA hypermethylation or somatic mutations. In this section, we review evidence for each of these two mechanisms.
Germline inactivation
It is possible that a proportion of suspected Lynch syndrome cases are due to the existence of complex or cryptic mutations in MMR genes that are not readily identified by current Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA) techniques.3,41,42 It could also be argued that suspected Lynch syndrome–colorectal cancer cases carry undetected mutations associated with a more moderate penetrance of cancer compared with that of exonic and splice site mutations that are more readily detected.43
Unidentified germline MMR gene mutations in suspected Lynch syndrome cases
Unidentified mutations within regulatory/promoter regions of the MMR genes may also underlie suspected Lynch syndrome cases. The c.-93G>A single-nucleotide polymorphism (SNP) (rs1800734) within the MLH1 gene is associated with the risk of MSI-H colorectal cancer and endometrial cancer; however, the strength of the associations suggests the mutation is a low-penetrance variant.44,45 In contrast, a rare European haplotype containing the c.-27C>A variant in MLH1 has been shown to predispose individuals to dominantly inherited epimutation and silencing of the MLH1 gene.46 A deletion of the last exon of the EPCAM gene, which is located upstream of MSH2, results in a fusion transcript between EPCAM and MSH2 and promoter methylation-induced transcriptional silencing of MSH2.3 The 3’untranslated region (UTR) of most genes contain regulatory sequences controlling all aspects of mRNA processing, including message stability, and these regions are typically A/U-rich elements or U repeats. Germline mutations within 3′ UTR repetitive regulatory sequences could suppress MMR gene expression,47 as has been described for the MLH1 gene.48 Similarly, altered regulation of MMR gene expression by miRNA binding in the 3′ UTR, in particular by miR-21 and miR-155, has been demonstrated.49,50 These examples suggest that more extensive screening of the 5′ and 3′ UTR sequence of the MMR genes for germline mutations within suspected Lynch syndrome cases is warranted and may account for a proportion of these cases.
More extensive MMR gene mutation testing to include deeper intronic sequence may yet yield further germline mutations. An example of germline MMR gene mutations that would be undetected by current mutation testing approaches of Sanger sequencing and MLPA includes a mutation deep within intron 1 of the MSH2 gene (c.212-553_c.212-479) in a single family with multiple colorectal cancer-affected individuals that demonstrated loss of expression of MSH2 and MSH6 by IHC.41 RNA-based splicing detection revealed an intronic insertion of 75 nucleotides between exon 1 and 2. Germline sequencing revealed a T>G transversion at intronic position c.212-478, creating a canonical splice donor site at the 3′ end of the inserted sequence. The inserted sequence contains a stop codon at the 3′ end and is predicted to result in a truncated protein of just 94 amino acids. This newly identified mutation was not identified by standard Sanger sequencing testing due to its location deep within intron 1. Although next-generation sequencing could easily extend to capture intronic sequences, the high level of repetitive sequence within the introns of MMR genes means designing probes and aligning sequence to these regions will be difficult. For instance, 54% of the intronic regions for the MSH2 gene comprise interspersed repeats, where 33% of this sequence is Alu repeats (MLH1: 53% interspersed repeats with 20% Alu; MSH6: 48% interspersed repeats with 42% Alu; PMS2: 57% interspersed repeats with 45% Alu).
Complex structural variations involving MMR genes have been described, including: 1) an interstitial deletion on chromosome 3p21.3, which leads to an in-frame fusion of MLH1 (exons 1–11) with ITGA9 (integrin α9; exons 17–28);51 2) a paracentric inversion on chromosome 3p22.2, creating two new stable fusion transcripts between the MLH1 and LRRFIP2 genes;42 and 3) an inversion involving MSH2 exon 1–7,52,53 which is known to be a disease-causing mechanism in Lynch syndrome and was present in six out of the ten MSH2 suspected Lynch syndrome cases tested.54
Large-scale intragenic insertions and deletions in the MMR genes are a relatively common cause of MMR deficiency, particularly in the case of MSH2. Complications arising due to the presence of a large set of highly homologous pseudogenes have made the detection of such mutations very difficult for the PMS2 gene. Recently, the development of a new method, which integrates the findings from long-range polymerase chain reaction (PCR)-based sequencing with a modified MLPA panel, allows for accurate detection and interpretation of large-scale rearrangements in the 3′ end of the gene.36 A single large cohort study, which had previously identified a mutation in 78% of suspected PMS2 cases, revisited its mutation-negative samples and screened them using this new method.55 No additional mutations were identified in this subset of samples (n=16), suggesting that alternate mutational mechanisms also exist within the PMS2 locus. As such, other structural and complex mutations that disrupt MMR gene function through similar mechanisms are likely to exist and it is possible that the progression from Sanger sequencing and MLPA-based testing to targeted resequencing of the MMR genes will aid in the routine detection of such structural mutations. It should, however, be noted that current next-generation technologies are not compatible with the issues faced when screening the PMS2 gene and, as such, this gene is likely to be left in the shadows again when it comes to routine screening.
The mismatch repair pathway is comprised of genes other than MLH1, MSH2, MSH6, and PMS2, including the MSH3, MLH3, PMS1, and EXO1 genes, among others. Although rare variants that are putatively associated with colorectal cancer risk have been identified within these additional MMR genes, the literature provides little to no evidence for the presence of germline mutations in these minor MMR binding partners in individuals with tumor MMR deficiency: Loukola et al56 found no germline mutations in MLH3 in 52 MSI-H colorectal cancers; Wu et al57 and Liu et al58 found no loss of MMR expression or MSI in colorectal cancers from carriers of germline MLH3 variants; and Taylor et al59 found no loss of MMR expression or MSI in endometrial cancers from two carriers of germline MLH3 variants. Similarly, Plaschke et al60 found no loss of MMR protein expression in colorectal cancers from carriers of germline variants in MSH3. While germline variants in these additional MMR genes may predispose individuals to colorectal cancer, none to date has been shown to be associated with tumor MMR deficiency.
A number of studies61–64 have identified mutations within the heterodimer binding partner of the MutLα and MutSα complexes. For example, a MSH2 gene mutation was identified in a colorectal cancer-affected individual, where the tumor demonstrated isolated loss of MSH6 expression,63 suggesting that mutation screening of the heterodimer binding partner should be a high priority in cases with suspected Lynch syndrome. The most commonly reported of these scenarios (although still rare) are those with MLH1 mutations identified in cases demonstrating loss of PMS2 protein expression with normal or weak MLH1 staining in their tumor62,64 and may represent the occurrence of immunostable MLH1 mutations.65 Conversely, in a study of 80 colorectal cancer cases with loss of MLH1 and PMS2 protein expression, in which germline MLH1 mutations and MLH1 DNA hypermethylation had been excluded, no mutations were identified within the PMS2 gene.66 The increasing use of gene panel resequencing of colorectal cancer-associated genes will inadvertently address issues of mutations in heterodimer binding partners for the MMR genes.67
Mosaicism
Mosaicism has been implicated in more than 30 monogenic disorders that show variable expressivity;68 however, it has not been well studied in suspected Lynch syndrome cases, with only a few cases identified from a small series of cases.69,70 MMR gene mutations might be overlooked as a result of somatic cell mosaicism in lymphocyte-derived DNA, accounting for the failure to detect mutations using standard mutation testing approaches. Mosaic mutation detection could be facilitated by the increased sensitivity resulting from the high read depth provided by massively parallel sequencing. Testing of multiple DNA sources per individual, including blood-derived DNA as well as normal mucosa- and tumor tissue-derived DNA, would be necessary along with confirmation of the mutation in blood relatives.
Somatic inactivation
A recent meta-analysis has shown that somatic mutations in the MLH1 and MSH2 genes are more common in tumors from individuals carrying a germline mutation within the same MMR gene.71 This supports the concept that a somatic MMR gene mutation is the “second hit” that contributes to tumor MMR deficiency in Lynch syndrome.71 The prevalence of biallelic somatic mutations in MMR genes in colorectal cancers has been estimated to be around 10%.71 Biallelic mutations identified as “double somatic mutation” are likely in trans (one on each allele), and may explain the inactivation of the MMR gene involved and the MMR-deficient status of the tumor.70,72 In a study of 25 suspected Lynch syndrome cases, Mensenkamp et al found that 13 (52%; 8/18 in MLH1 and 5/7 in MSH2) had biallelic somatic mutations in the form of point mutations and loss of heterozygosity.73 Biallelic somatic inactivation for at least some of these suspected Lynch syndrome cases would explain why the risk of colorectal cancer for first-degree relatives of suspected Lynch syndrome cases is reduced compared with Lynch syndrome cases. Therefore, additional studies aimed at confirming the high proportion of biallelic somatic mutations as a cause of MMR deficiency in suspected Lynch syndrome is necessary.
One study has reported on a small number of cases where reduced or absent MSH6 protein expression can be attributed to somatic frameshift mutations within the (C)8 tract in exon 5 of the MSH6 gene, secondary to the loss of mismatch repair function in MLH1/PMS2-deficient colorectal cancers.74 The same study also reported on non-genetic mechanisms as a further cause of MMR deficiency. Preoperative chemotherapy or chemoradiation therapy was shown to be associated with reduced MSH6 protein expression in the absence of germline or somatic mutations in otherwise MMR-proficient colorectal cancers,74 presumably as a result of hypoxia or cytotoxic stress secondary to treatment.
Epigenetic mechanisms of MMR gene inactivation
MMR gene inactivation as a result of promoter hypermethylation has been previously demonstrated in both the germline (epimutations) and tumor tissue (somatic mutations) for the MLH1 gene.2,27,32,75 For MSH2, MSH6, and PMS2 genes, however, much less is known regarding their inactivation by DNA hypermethylation. Rare cases of germline MSH2 methylation have been reported,3,76 the majority of which were attributable to 3′ EPCAM (TACSTD1) deletions. One study found somatic MSH2 hypermethylation in 24% (11/46) of MSH2-deficient tumors without a germline MSH2 mutation, and no MSH2 hypermethylation in sporadic colorectal cancers with normal expression of the MMR genes.77 For seven of the eleven cases with somatic MSH2 hypermethylation, a germline mutation in the MSH2 gene was present, suggesting methylation of the wild-type allele acted as the second hit in these MSH2-deficient colorectal cancers. However, nothing is known about somatic MSH2 or MSH6 hypermethylation in endometrial cancers with suspected Lynch syndrome. Furthermore, somatic PMS2 hypermethylation has not been previously investigated in colorectal or endometrial cancer cases.
Future directions
It is clear from the few studies of suspected Lynch syndrome cases conducted to date that the underlying cause of MMR deficiency is likely to be heterogeneous, comprising both cases with inherited MMR deficiency due to unidentified mutations in the MMR genes and cases with sporadic MMR deficiency resulting from biallelic somatic mutations or MMR gene promoter hypermethylation. More comprehensive studies will be needed to determine: 1) the full spectrum of genetic mutations in the MMR genes; and 2) the proportion of cases attributable to genetic, epigenetic, or somatic inactivation mechanisms. Strategies exploiting the power of massively parallel sequencing are likely to aid in the identification of germline noncoding and structural variation mutations that were beyond previous testing feasibility. The recently reported biallelic somatic mutation frequency of 50% in suspected Lynch syndrome cases, if confirmed by others, suggests triaging strategies designed to identify germline MMR gene mutation carriers within colorectal- and endometrial cancer-affected populations will need to be revised to accommodate additional tumor testing steps prior to germline MMR gene testing. Similarly, as suspected Lynch syndrome cases are apportioned into inherited or sporadic MMR deficiency groups, the cancer risks for first-degree relatives will need to be reevaluated. Translation of new knowledge regarding MMR gene somatic inactivation into clinical practice is essential for a large number of families who have previously tested negative for MMR gene mutations but exhibited tumor MMR deficiency. Findings will be instrumental for partitioning these individuals and their relatives as low-risk or high-risk, and then more intensive screening and surveillance can be targeted to those individuals at high risk. To provide optimal care, the underlying causes of tumor MMR deficiency in suspected Lynch syndrome cases will need to be identified in order to provide effective and targeted clinical management for the affected individuals and their relatives, thereby reducing the burden on the health system.
Acknowledgments
AKW is an Australian National Health and Medical Research Council (NHMRC) Early Career Fellow. ABS is an NHMRC Senior Research Fellow. CR is the Jass Pathology Fellow. DDB is a Senior Research Fellow funded by Research at Melbourne University Accelerator Program (R@MAP).
Disclosure
The authors report no conflicts of interest in this work.
References
Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525–2538. | |
Buchanan DD, Tan YY, Walsh MD, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol. 2014;32(2):90–100. | |
Ligtenberg MJ, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41(1):112–117. | |
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–268. | |
Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30(9):958–964. | |
Win AK, Lindor NM, Young JP, et al. Risks of primary extracolonic cancers following colorectal cancer in Lynch syndrome. J Natl Cancer Inst. 2012;104(18):1363–1372. | |
Win AK, Lindor NM, Winship I, et al. Risks of colorectal and other cancers after endometrial cancer for women with Lynch syndrome. J Natl Cancer Inst. 2013;105(4):274–279. | |
Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut. 2011;60(7):950–957. | |
Win AK, Parry S, Parry B, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Ann Surg Oncol. 2013;20(6):1829–1836. | |
Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829–834. | |
Win AK, Macinnis RJ, Dowty JG, Jenkins MA. Criteria and prediction models for mismatch repair gene mutations: a review. J Med Genet. 2013;50(12):785–793. | |
Southey MC, Jenkins MA, Mead L, et al. Use of molecular tumor characteristics to prioritize mismatch repair gene testing in early-onset colorectal cancer. J Clin Oncol. 2005;23(27):6524–6532. | |
Jenkins MA, Dowty JG, Hopper JL, Southey MC. Molecular screening of all colorectal tumors diagnosed before age 50 years followed by genetic testing efficiently identifies Lynch syndrome cases. Int J Cancer. 2009;124(5):x–xi. | |
Schofield L, Watson N, Grieu F, et al. Population-based detection of Lynch syndrome in young colorectal cancer patients using microsatellite instability as the initial test. Int J Cancer. 2009;124(5):1097–1102. | |
Schofield L, Grieu F, Goldblatt J, Amanuel B, Iacopetta B. A state-wide population-based program for detection of lynch syndrome based upon immunohistochemical and molecular testing of colorectal tumours. Fam Cancer. 2012;11(1):1–6. | |
Boland CR, Shike M. Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology. 2010;139(6):2197. e1–e7. | |
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352(18):1851–1860. | |
Piñol V, Castells A, Andreu M, et al; Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA. 2005;293(16):1986–1994. | |
Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7817. | |
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–5788. | |
Morrison J, Bronner M, Leach BH, Downs-Kelly E, Goldblum JR, Liu X. Lynch syndrome screening in newly diagnosed colorectal cancer in general pathology practice: from the revised Bethesda guidelines to a universal approach. Scand J Gastroenterol. 2011;46(11):1340–1348. | |
Moreira L, Balaguer F, Lindor N, et al; EPICOLON Consortium. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012;308(15):1555–1565. | |
Lancaster JM, Powell CB, Kauff ND, et al; Society of Gynecologic Oncologists Education Committee. Society of Gynecologic Oncologists Education Committee statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol. 2007;107(2):159–162. | |
Tan YY, McGaughran J, Ferguson K, et al; ANECS Group. Improving identification of lynch syndrome patients: a comparison of research data with clinical records. Int J Cancer. 2013;132(12):2876–2883. | |
Clarke BA, Cooper K. Identifying lynch syndrome in patients with endometrial carcinoma: shortcomings of morphologic and clinical schemas. Adv Anat Pathol. 2012;19(4):231–238. | |
Moline J, Mahdi H, Yang B, et al. Implementation of tumor testing for lynch syndrome in endometrial cancers at a large academic medical center. Gynecol Oncol. 2013;130(1):121–126. | |
Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95(12):6870–6875. | |
Parsons MT, Buchanan DD, Thompson B, Young JP, Spurdle AB. Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J Med Genet. 2012;49(3):151–157. | |
Heald B, Plesec T, Liu X, et al. Implementation of universal microsatellite instability and immunohistochemistry screening for diagnosing lynch syndrome in a large academic medical center. J Clin Oncol. 2013;31(10):1336–1340. | |
Win AK, Lindor N, Jenkins M. Risk of breast cancer in Lynch syndrome: a systematic review. Breast Cancer Res. 2013;15(2):R27. | |
Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23(3):437–449. | |
Levine AJ, Win AK, Buchanan DD, et al. Cancer risks for the relatives of colorectal cancer cases with a methylated MLH1 promoter region: data from the Colorectal Cancer Family Registry. Cancer Prev Res (Phila). 2012;5(2):328–335. | |
Win AK, Buchanan DD, Rosty C, et al. Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut. Epub March 10, 2014. | |
Rodríguez-Soler M, Pérez-Carbonell L, Guarinos C, et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013;144(5):926–932. e1; quiz e13–e14. | |
Antelo M, Milito D, Shia J, et al. High frequency of Lynch-like syndrome in early-onset non-familial colorectal cancer. 17th Annual Meeting of the CGA-ICC; October 6–8, 2013; Anaheim, CA. | |
Vaughn CP, Baker CL, Samowitz WS, Swensen JJ. The frequency of previously undetectable deletions involving 3′ Exons of the PMS2 gene. Genes Chromosomes Cancer. 2013;52(1):107–112. | |
Leenen CH, van Lier MG, van Doorn HC, et al. Prospective evaluation of molecular screening for Lynch syndrome in patients with endometrial cancer ≤70 years. Gynecol Oncol. 2012;125(2):414–420. | |
Newcomb PA, Baron J, Cotterchio M, et al; Colon Cancer Family Registry. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomarkers Prev. 2007;16(11):2331–2343. | |
Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology. 1999;116(6):1453–1456. | |
Overbeek LI, Kets CM, Hebeda KM, et al. Patients with an unexplained microsatellite instable tumour have a low risk of familial cancer. Br J Cancer. 2007;96(10):1605–1612. | |
Clendenning M, Buchanan DD, Walsh MD, et al. Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam Cancer. 2011;10(2):297–301. | |
Morak M, Koehler U, Schackert HK, et al; German HNPCC consortium. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J Med Genet. 2011;48(8):513–519. | |
Dowty JG, Win AK, Buchanan DD, et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat. 2013;34(3):490–497. | |
Raptis S, Mrkonjic M, Green RC, et al. MLH1-93G>A promoter polymorphism and the risk of microsatellite-unstable colorectal cancer. J Natl Cancer Inst. 2007;99(6):463–474. | |
Whiffin N, Broderick P, Lubbe SJ, et al. MLH1-93G > A is a risk factor for MSI colorectal cancer. Carcinogenesis. 2011;32(8):1157–1161. | |
Kwok CT, Vogelaar IP, van Zelst-Stams WA, et al. The MLH1 c.-27C>A and c.85G>T variants are linked to dominantly inherited MLH1 epimutation and are borne on a European ancestral haplotype. Eur J Hum Genet. 2014;22(5):617–624. | |
Wilding JL, McGowan S, Liu Y, Bodmer WF. Replication error deficient and proficient colorectal cancer gene expression differences caused by 3′UTR polyT sequence deletions. Proc Natl Acad Sci U S A. 2010;107(49):21058–21063. | |
Mao G, Pan X, Gu L. Evidence that a mutation in the MLH1 3′-untranslated region confers a mutator phenotype and mismatch repair deficiency in patients with relapsed leukemia. J Biol Chem. 2008;283(6):3211–3216. | |
Valeri N, Gasparini P, Braconi C, et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc Natl Acad Sci U S A. 2010;107(49):21098–21103. | |
Valeri N, Gasparini P, Fabbri M, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A. 2010;107(15):6982–6987. | |
Meyer C, Brieger A, Plotz G, et al. An interstitial deletion at 3p21.3 results in the genetic fusion of MLH1 and ITGA9 in a Lynch syndrome family. Clin Cancer Res. 2009;15(3):762–769. | |
Wagner A, van der Klift H, Franken P, et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivates MSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosomes Cancer. 2002;35(1):49–57. | |
Chen JM. The 10-Mb paracentric inversion of chromosome arm 2p in activating MSH2 and causing hereditary nonpolyposis colorectal cancer: re-annotation and mutational mechanisms. Genes Chromosomes Cancer. 2008;47(6):543–545. | |
Rhees J, Arnold M, Boland CR. Inversion of exons 1–7 of the MSH2 gene is a frequent cause of unexplained Lynch syndrome in one local population. Fam Cancer. 2014;13(2):219–225. | |
Clendenning M, Walsh MD, Gelpi JB, et al. Detection of large scale 3′ deletions in the PMS2 gene amongst Colon-CFR participants: have we been missing anything? Fam Cancer. 2013;12(3):563–566. | |
Loukola A, Vilkki S, Singh J, Launonen V, Aaltonen LA. Germline and somatic mutation analysis of MLH3 in MSI-positive colorectal cancer. Am J Pathol. 2000;157(2):347–352. | |
Wu Y, Berends MJ, Sijmons RH, et al. A role for MLH3 in hereditary nonpolyposis colorectal cancer. Nat Genet. 2001;29(2):137–138. | |
Liu HX, Zhou XL, Liu T, et al. The role of hMLH3 in familial colorectal cancer. Cancer Res. 2003;63(8):1894–1899. | |
Taylor NP, Powell MA, Gibb RK, et al. MLH3 mutation in endometrial cancer. Cancer Res. 2006;66(15):7502–7508. | |
Plaschke J, Preussler M, Ziegler A, Schackert HK. Aberrant protein expression and frequent allelic loss of MSH3 in colorectal cancer with low-level microsatellite instability. Int J Colorectal Dis. 2012;27(7):911–919. | |
Niessen RC, Kleibeuker JH, Westers H, et al. PMS2 involvement in patients suspected of Lynch syndrome. Genes Chromosomes Cancer. 2009;48(4):322–329. | |
Halvarsson B, Lindblom A, Rambech E, Lagerstedt K, Nilbert M. The added value of PMS2 immunostaining in the diagnosis of hereditary nonpolyposis colorectal cancer. Fam Cancer. 2006;5(4):353–358. | |
Loconte DC, Patruno M, Lastella P, et al. A rare MSH2 mutation causes defective binding to hMSH6, normal hMSH2 staining, and loss of hMSH6 at advanced cancer stage. Hum Pathol. Epub June 30, 2014. | |
de Jong AE, van Puijenbroek M, Hendriks Y, et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(3):972–980. | |
Mangold E, Pagenstecher C, Friedl W, et al. Tumours from MSH2 mutation carriers show loss of MSH2 expression but many tumours from MLH1 mutation carriers exhibit weak positive MLH1 staining. J Pathol. 2005;207(4):385–395. | |
Clendenning M, Macrae FA, Walsh MD, et al. Absence of PMS2 mutations in colon-CFR participants whose colorectal cancers demonstrate unexplained loss of MLH1 expression. Clin Genet. 2013;83(6):591–593. | |
Pritchard CC, Smith C, Salipante SJ, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn. 2012;14(4):357–366. | |
Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3(10):748–758. | |
Pastrello C, Fornasarig M, Pin E, Berto E, Pivetta B, Viel A. Somatic mosaicism in a patient with Lynch syndrome. Am J Med Genet A. 2009;149A(2):212–215. | |
Sourrouille I, Coulet F, Lefevre JH, et al. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer. 2013;12(1):27–33. | |
Zhang R, Qin W, Xu GL, Zeng FF, Li CX. A meta-analysis of the prevalence of somatic mutations in the hMLH1 and hMSH2 genes in colorectal cancer. Colorectal Dis. 2012;14(3):e80–e89. | |
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–2087. e3. | |
Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146(3):643–646. e8. | |
Shia J, Zhang L, Shike M, et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1/PMS2 deficiency. Mod Pathol. 2013;26(1):131–138. | |
Hitchins M, Williams R, Cheong K, et al. MLH1 germline epimutations as a factor in hereditary nonpolyposis colorectal cancer. Gastroenterology. 2005;129(5):1392–1399. | |
Rumilla K, Schowalter KV, Lindor NM, et al. Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated Lynch syndrome cases. J Mol Diagn. 2011;13(1):93–99. | |
Nagasaka T, Rhees J, Kloor M, et al. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer Res. 2010;70(8):3098–3108. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.