")
Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 4
Clinical potential of eliglustat tartrate in the treatment of type 1 Gaucher disease
Authors Kaplan P
Received 5 November 2013
Accepted for publication 9 January 2014
Published 14 May 2014 Volume 2014:4 Pages 1—8
DOI https://doi.org/10.2147/RRED.S36669
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Paige Kaplan
Lysosomal Disorders Center, Section of Metabolic Diseases, Children's Hospital of Philadelphia, Philadelphia, PA, USA
Abstract: Nonneuropathic type 1 Gaucher disease is an autosomal recessive inherited disease caused by the deficiency or absence of beta glucocerebrosidase (beta glucosidase). The highest prevalence of type 1 is in Ashkenazi Jews, but it affects all ethnic groups. It manifests at any age but is seen predominantly in the first two decades. The phenotype is characterized by painless splenomegaly and secondary hypersplenism (low hemoglobin concentration and low platelet and white blood cell counts). Symptoms and signs include splenomegaly; chronic fatigue, frequent nose bleeds, prolonged bleeding, and/or bruising; hepatomegaly; bone pain, bone destruction and low bone density; and poor growth in childhood and delayed pubertal development. Current treatment with intravenous enzyme replacement has been generally successful. However, oral treatments have been developed because enzyme replacement is time-consuming and invasive, and intravenous infusions are not universally available for patients who live far from medical centers or home infusion nurses. Furthermore, it may become difficult to access veins after repeated infusions. Orally administered substrate reduction is a newer treatment approach. The aim is to limit the synthesis of the substrate, glucosylceramide. The residual intrinsic enzyme, acting alone or with recombinant enzyme, can then completely catabolize the smaller amounts of glucosylceramide that are transported into lysosomes. Eliglustat tartrate is a new specific inhibitor of glucosylceramide synthase. Phase III trials in humans have been completed. Eliglustat tartrate has been shown to be efficacious and safe in adult humans. The results are as good or better compared with intravenous replacement with regard to reductions in spleen and liver enlargement and improvements in hemoglobin concentrations, platelet counts, and bone density, as well as decreases in biomarkers of Gaucher disease activity. Few adverse events, none of which was serious, have been reported. Eliglustat tartrate has the clinical potential to enable a larger number of patients with type 1 Gaucher disease to be treated successfully.
Keywords: type 1 Gaucher disease, substrate reduction therapy, eliglustat tartrate
Introduction
Eliglustat tartrate is a novel, oral treatment for nonneuropathic (type 1) Gaucher disease. Gaucher disease is the most common lysosomal storage disease and is caused by deficiency or absence of beta glucocerebrosidase (beta glucosidase).1 This results in accumulation of an undegraded glycosphingolipid glucosylceramide, in lysosomes in macrophages, mainly in the spleen, liver, bone marrow, and in some cases, the brain and lungs. These macrophages have a distinctive microscopic appearance and are named “Gaucher cells.”
Gaucher disease is inherited in an autosomal recessive manner. There is a spectrum of clinical manifestations, designated as type 1 (nonneuropathic), type 2 (acute neuropathic), and type 3 (chronic neuropathic). Type 1 is the most common form in the Western world, with a high prevalence in Ashkenazi Jews, but it affects people of all origins. The neuropathic forms are more prevalent than type 1 in other parts of the world.2 Type 1 can manifest at any age, but more than half the patients with symptomatic type 1 Gaucher disease manifest in the first two decades.3 The hallmarks of type 1 disease are painless significant splenomegaly with secondary hypersplenism (low hemoglobin concentration and low platelet and white blood cell counts), hepatomegaly, acute bone pain (Gaucher bone crisis), bone destruction (particularly in hips, knees, vertebrae, and shoulders), low bone density, poor growth in childhood, and delayed pubertal development.1,3,4 Chronic fatigue, frequent nose bleeds, prolonged bleeding, and/or bruising, and joint and bone pains are common complaints. The signs and symptoms can vary, even among affected members within the same family. Many untreated patients have been severely incapacitated and crippled. Patients with long-standing untreated type 1 Gaucher disease have a higher incidence of cholecystitis, pulmonary hypertension, hematologic malignancies, and possibly hepatocellular carcinoma compared with the general population.
Until 1991, treatment was based on alleviating the symptoms of the disease because there was no therapy that could prevent damage or restore function. Alglucerase (Ceredase; Genzyme Corporation, Cambridge, MA, USA), a mannose-terminated enzyme, given intravenously, was the first specific treatment. This was approved by the US Food and Drug Administration in 1991. Subsequently, recombinant mannose-terminated enzymes imiglucerase (Cerezyme; Genzyme Corporation, Cambridge, MA, USA), velaglucerase alfa (VPRIV; Shire Pharmaceuticals, St Helier, Jersey, United Kingdom), and taliglucerase alfa (Elylyso; Protalix BioTherapeutics, Carmiel, Israel, and Pfizer Inc., New York, NY, USA) were introduced. They have been effective in reducing spleen and liver enlargement; reversing or preventing anemia, thrombocytopenia and leukopenia, bone disease, and lung disease; and improving growth. However, because the enzyme does not penetrate the blood–brain barrier, it is not effective for the neuropathic manifestations of types 2 and 3.
Different modalities, including chaperones and gene therapies, have been tried in efforts to penetrate the blood–brain barrier. Most have not been as successful as enzyme replacement in treating the nonneurologic aspects of the disease.
Although enzyme replacement therapy has been generally successful, other treatment modalities have been studied because enzyme replacement must be administered indefinitely by intravenous infusions every 2–4 weeks. This is time-consuming, invasive, and not universally available for people living far from medical centers or home infusion nurses. Furthermore, it may become difficult to access veins after repeated infusions. Also, enzyme replacement has caused anaphylactic reactions in a small number of cases.
A potential therapy that could overcome these problems in affected individuals with residual enzyme activity would be an oral treatment that would limit the amount of glucosylceramide that is synthesized. The residual intrinsic enzyme, alone or with recombinant enzyme, then could completely catabolize the smaller amounts of glucosylceramide transported into lysosomes.
This concept, called substrate reduction, was suggested in 1972 by Radin (cited by Shayman),5,6 who synthesized a compound, PDMP, (2-decanoylamino-3-morpholino-1-phenylpropanol) that inhibited glycosphingolipid synthesis. Iminosugar N-butyldeoxynojirimycin, a nonspecific inhibitor of glucosylceramidase, the initial and rate-limiting enzyme in glycosphingolipid synthesis, was developed as an oral drug called miglustat (Zavesca; Actelion Pharmaceuticals, Allschwill, Switzerland). This can reduce the load of accumulated glucosylceramide and other glycosphingolipids in lysosomes. However, its action is nonspecific, and it also inhibits other glycosidases, including intestinal disaccharidases, which caused peripheral neuropathy and diarrhea in a large number of patients.7
A promising new, orally active, specific inhibitor of glucosylceramide synthase, eliglustat tartrate ((1R,2R)-octanoic acid [2-(2′,3′-dihydro-benzo[1,4] dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide-L-tartaric acid salt) was developed.5 Clinical trials in humans have just been completed.10–12,14–17
Development of eliglustat tartrate
Shayman5,6 refined the compound developed by Radin, which contained three primary functional groups: a cyclic amine, an aromatic group, and a fatty acid in amide linkage. The authors identified the pharmacophore, the chemical structure that defines the activity of the inhibitor, by sequentially substituting each of these groups to identify potential glycolipid synthesis inhibitors with higher activity and greater specificity. They determined that cyclic amines were required for activity and that a five-membered ring (pyrrolidine group) was optimal. Increasing the carbon chain length resulted in measurable improvements in the concentration at which the drug achieves 50% of enzyme inhibition of the inhibitor. Decreasing the fatty acyl chain length to eight carbons resulted in a drug that had a half-life of 5 hours with only a modestly higher concentration at which the drug achieves 50% of enzyme inhibition against the glucosylceramide synthase. The substitution of the aromatic phenyl group with an aromatic 1,2-ethylenedioxy-phenyl group resulted in a more than 1,000-fold increase in inhibitory activity against the glucosylceramide synthase. Tartrate salt provided optimal bioavailability and stability. This was the ultimate configuration of eliglustat tartrate (Figure 1). Eliglustat tartrate is influenced by cytochrome P450 2D6,6 encoded by the CYP2D6 gene, that controls the metabolism and elimination of approximately 25% of drugs; accordingly, some people metabolize it quickly and others slowly.
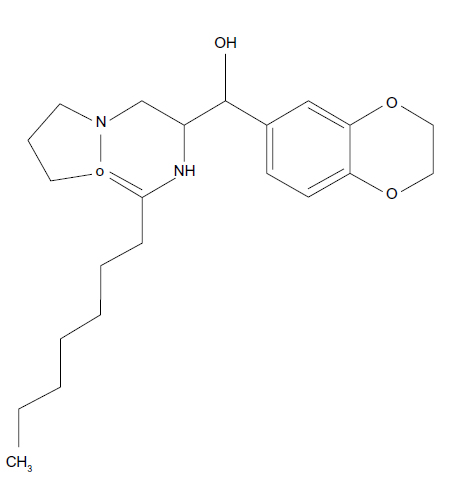 | Figure 1 Free base of eliglustat tartrate. |
In vitro studies showed that eliglustat tartrate was effective in decreasing GM1 (mono-sialo-tetra-hexosyl-ganglioside) and GM3 gangliosides (glycosphingolipids) in cultured human erythroleukemia cells and murine melanoma cells.
Animal studies of eliglustat tartrate
In vivo, eliglustat tartrate was administered to genetically engineered mice with a severe Gaucher genotype, D409H/null (Asp409Val/null).8 Eliglustat tartrate reduced the load of glycosphingolipids and numbers of Gaucher cells in older mice (7 months) and prevented the ongoing development of Gaucher cells, followed by depletion of Gaucher cells and glycosphingolipid accumulation in young, 10-week-old mice.8 In another cohort of these mice, treated with eliglustat tartrate after substrate clearance by imiglucerase, the reduction of glucosylceramide in the visceral organs was greater than with either treatment alone.9
Human trials of eliglustat tartrate
The safety and tolerability of eliglustat tartrate were demonstrated in a Phase I trial with 20 healthy volunteers.10 In a Phase Ib clinical trial in healthy volunteers, plasma glucocerebroside (GL1) concentrations decreased after administration of eliglustat tartrate.10 Single daily doses of 20 mg/kg or less were well tolerated without adverse effects. Multiple increasing doses given twice daily were tolerated up to 200 mg/day, although there was vomiting or nausea in a few volunteers taking doses higher than 100 mg/day. Plasma therapeutic levels of 6–14 ng/mL were achieved with doses of 50 mg twice daily.
A Phase II, single-group, open-label, multicenter study of efficacy, safety, and pharmacologic kinetics was completed in 2 years.11,12 In the first part, 26 adults (mean age, 34 years) with moderate or severe type 1 Gaucher disease with intact spleens who had not been treated in the 12 months before enrollment with miglustat (Zavesca), recombinant (glucosidase) enzyme, bisphosphonates, or vitamin B12 were enrolled. Four patients were excluded during the study because two had ventricular tachycardia during the first dose and two became pregnant during the study.
Inclusion criteria were spleen volume more than 10 multiples of normal, platelet counts 40–100 K, and hemoglobin concentrations lower than 10 g/dL in women and lower than 10.5 g/dL in men. Spleen volumes were measured with magnetic resonance imaging (MRI) or spiral computed tomography; normal volumes were calculated as two times weight (kg) and recorded as milliliters. Primary endpoints were decreases in spleen volumes and increases in platelet counts and hemoglobin concentrations. Biomarkers (chitotriosidase and chemokine [C-C motif] ligand 18 [CCL18]), genotyping of CYP2D6 to evaluate slow and fast metabolizers, bone density (dual energy absorptiometry scans [DXA]), and pharmacokinetic data were studied. The initial dose was 50 mg twice daily. The calculated therapeutic level was 6–14 ng/mL; accordingly, on day 20, the dose was adjusted to 100 mg twice daily for 18 patients whose trough concentrations were less than 5 ng/mL after taking eliglustat tartrate for 10 days.
In the 22 patients completing the 52-week study, spleen and liver volumes decreased, bone mineral density improved, abnormal bone marrow signal with MRI resolved, and GL1 and GM3 concentrations normalized (Figure 2, Figure 3A).11 The hemoglobin concentrations and platelet counts increased statistically significantly by weeks 13 and 26, respectively; by week 52, mean hemoglobin concentration increased by 1.62 mg/dL (P<0.001) and mean platelet count increased by 40.3% (P<0.001). Spleen and liver volumes decreased significantly by week 26 and by week 52 (by 38.5% [P<0.001] and additional 17.0% [P<0.001], respectively). In type 1 Gaucher disease, the increase in spleen volume is much greater than in the liver, so this was considered to be an excellent response.
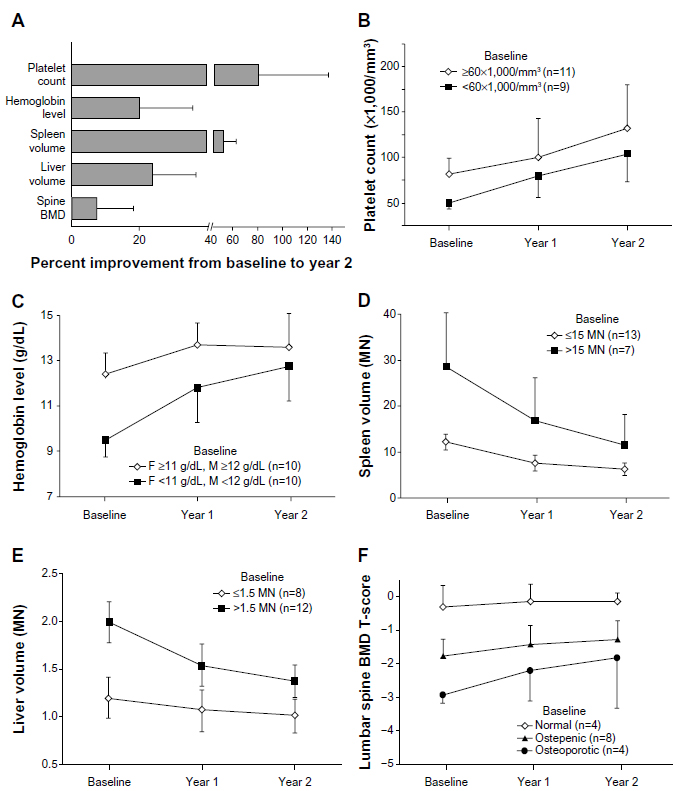 | Figure 2 Improvements in hematological, visceral, and skeletal parameters: overall and stratified by baseline disease severity. |
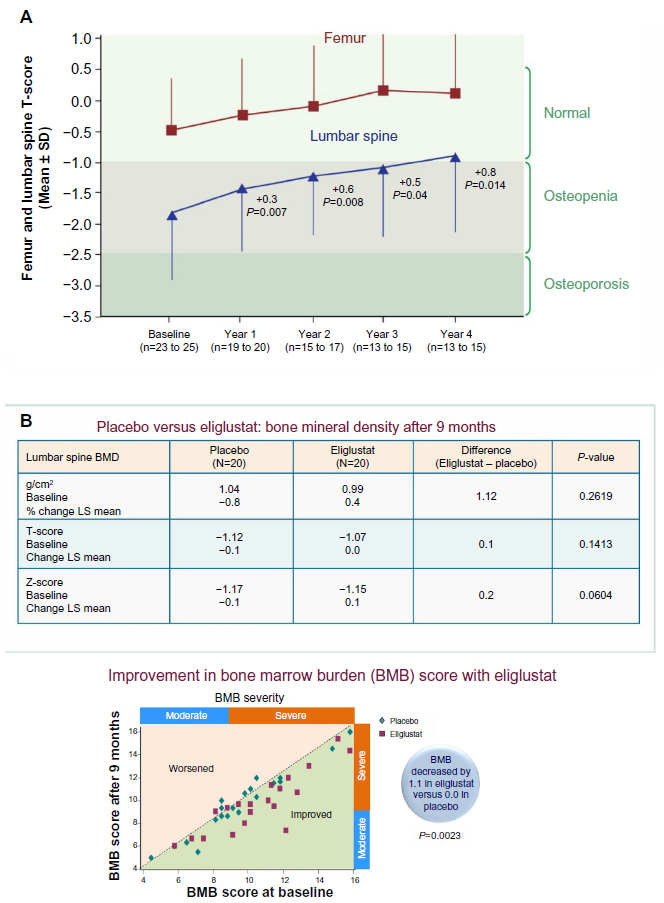 | Figure 3 (A) Long-term effect of eliglustat tartrate on bone in the phase II trial in 19 patients. (B) Improved bone mineral density and bone marrow burden (BMB) in patients treated with eliglustat tartrate compared with placebo. |
There were important improvements in bone by week 52.11 Bone density, measured by dual energy absorptiometry, improved by 7.8% (0.31 Z-score), particularly in those who were osteopenic or osteoporotic.11 This was a greater improvement than had occurred in 342 adult patients with similar baseline scores who were treated with varying doses of imiglucerase.13 Infiltrates of Gaucher cells into bone marrow, measured by MRI, decreased or stabilized.11 There were no new lytic lesions or bone crises, but there was worsening of preexisting osteonecrosis in one participant who withdrew from the trial after completing 1 year of treatment.
Improvements in all parameters were comparable to or better than those obtained with imiglucerase therapy. Plasma biomarkers, chitotriosidase, CCL18, angiotensin-converting enzyme, and tartrate-resistant acid phosphatase are secreted by Gaucher cells and reflect disease activity; these were elevated at baseline, but by week 52, the median levels declined by 35%–50%. Concentrations of GL1 and GM3 normalized.
Twenty participants continued participation in an extension of the Phase II trial for a second year and showed continuing, although slower, improvement (Figure 2).12 This trend of rapid initial improvement, followed by a slower rate of improvement, is similar to that with enzyme replacement. Eighty-five percent of the patients achieved therapeutic goals for three or all of the four parameters. Lumbar spine bone mineral density increased 7.8% ±10.6% (P<0.01), and T-score increased 0.6±0.8 (P<0.012), especially in those with low initial density. Bone marrow infiltration, assessed by MRI, decreased in eight of 18 patients and remained stable in ten individuals. Bone lysis, seen in nine patients at baseline, remained stable without new lesions. In seven patients with baseline bone infarcts, one improved and six remained stable. No bone crises or pathologic fractures occurred. In 12 of 15 patients who continued taking eliglustat tartrate for another 4 years and who had osteopenia or osteoporosis at baseline, the mean bone density T-score (standard deviation [SD]) increased from −1.6 (SD, 1.1; osteopenia) to −0.9 (SD,1.3; normal).14
Three Phase III trials to confirm the efficacy of eliglustat tartrate, determine optimal doses, monitor adverse effects, and compare it with recombinant enzyme therapy in adults with type 1 Gaucher have just been completed. One trial (Study of Eliglustat Tartrate [Genz-112638] in Patients With Gaucher Disease [ENGAGE]) was a randomized, double-blind, placebo-controlled study in treatment-naïve adults; the second (Study of Eliglustat Tartrate [Genz-112638] in Patients With Gaucher Disease Who Have Reached Therapeutic Goals With Enzyme Replacement Therapy [ENCORE]) tested whether eliglustat tartrate can maintain the stability achieved with imiglucerase in adults; and the third (Study of Eliglustat Tartrate [Genz-112638] in Patients With Gaucher Disease to Evaluate Once Daily Versus Twice Daily Dosing [EDGE]) tested the efficacy of two different doses (50 and 100 mg) given once or twice daily in patients stabilized on enzyme therapy. Preliminary results from the ENGAGE and ENCORE trials have been reported at national and international conferences.14–17
ENGAGE, a prospective, randomized, double-blind, placebo-controlled trial of eliglustat in treatment-naïve adults with type 1 Gaucher disease, enrolled 20 subjects in each group.14,15 After 9 months, there were statistically significant improvements in the treated group in spleen and liver volumes (Figure 4A and B), platelet counts (Figure 4C), hemoglobin concentrations (Figure 4D), bone marrow burden (BMB), biomarkers (chitotriosidase, CCL18, angiotensin-converting enzyme, and tartrate-resistant acid phosphatase), and self-administered quality of life surveys. Spleen volume in patients treated with eliglustat tartrate decreased significantly from baseline by a mean of 28% compared with a mean increase of 2% in placebo patients, for an absolute difference of 30% (P<0.0001). Platelet levels increased from baseline by an absolute difference of 41% compared with placebo (P<0.0001). Hemoglobin concentrations increased from baseline by an absolute difference of 1.2 g/dL compared with placebo (P<0.0006). Liver volume decreased from baseline by an absolute difference of 7% compared with placebo (P<0.0072). At baseline, 80% of subjects treated with eliglustat tartrate and 75% of subjects given placebo had moderately to severe disease. The total BMB (a semiquantitative measure by MRI of marrow infiltration in the lumbar spine and femur) decreased in the group treated with eliglustat tartrate (Figure 3B), but not in the placebo group (eliglustat tartrate compared with placebo (BMB scores: total body [−1.1 versus 0.0], spine [−0.6 versus 0.1], and femur [−0.3 versus 0.0]). All other markers of bone disease showed trends toward improvement (Figure 3B).
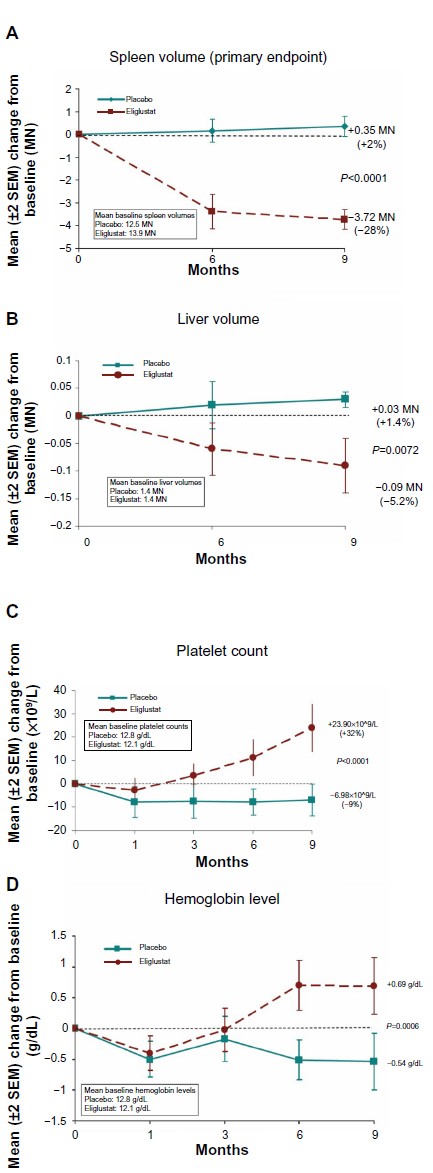 | Figure 4 (A–D) Type 1 Gaucher disease in untreated adults; response to treatment with eliglustat compared with placebo treatment, showing significant improvement in most parameters with eliglustat tartrate. |
ENCORE, a multinational, randomized, controlled, open-label, 1-year study, determined whether eliglustat tartrate could maintain improvements achieved in patients treated for at least 3 years with imiglucerase (Cerezyme) and who had reached therapeutic goals.16 One hundred sixty patients were randomized in a ratio of 2:1 to receive either eliglustat tartrate or imiglucerase for 1 year. Participants treated with eliglustat tartrate maintained stability for most criteria: 94% for spleen volume, 96% for liver volume, 95% for hemoglobin concentrations, and 93% for platelet counts. There were no significant bone symptoms, pathologic fractures, or bone crises.
Adverse events
An analysis of 393 subjects in these clinical trials totaling 535 patient-years of eliglustat tartrate exposure shows it was well-tolerated.17 Most adverse events were mild to moderate in severity and were considered by the treating physicians to be unrelated to eliglustat tartrate. These included headaches, arthralgia, and diarrhea. Most serious adverse events were unrelated to eliglustat tartrate: pregnancy, myocardial infarction (three patients with risk factors), and aspects of Gaucher disease (femur fracture, joint dislocation, one case of hepatocellular carcinoma retrospectively noted on baseline MRI, and cholecystitis [n=2]). Adverse events probably related to eliglustat tartrate occurred in two patients (abdominal pain, lethargy, and exfoliative rash). In the Phase II study, the only adverse event considered to be possibly treatment-related was in an asymptomatic 41-year-old man who showed decreased bilateral sural nerve amplitude and unilateral sural nerve conduction velocity testing at 18 months. Previously, during miglustat treatment, he had experienced symptomatic peripheral neuropathy that resolved when miglustat was discontinued. Mild sural nerve abnormalities were also detected by nerve conduction velocity study in a 50-year-old man during the first year of eliglustat tartrate treatment, but these were not considered treatment-related. Both men have remained asymptomatic, with normal neurologic examinations after 3 years of treatment. Peripheral neuropathy, often subclinical, may possibly be a manifestation of type 1 Gaucher disease.18 Twelve patients with Gaucher disease (3%) stopped participating because of adverse effects.
Different doses of eliglustat tartrate, depending on CYP2D6 status (fast or slow metabolizers) and body weight, were needed to achieve therapeutic goals.
Conclusion
Eliglustat tartrate, with potent effects on type 1 Gaucher disease, and possibly other glycosphingolipidoses, appear likely to fulfill the high expectations for clinical efficacy. Phase II and III trials of the oral drug, eliglustat tartrate, in type 1 nonneuropathic Gaucher disease have shown that in both enzyme-treated and treatment-naïve adults, the responses to substrate depletion in organ volume, hematologic parameters, and bone have been as good or better than with enzyme replacement, with minimal severe adverse events. Depending on whether a patient is a fast or slow metabolizer, because of CYP2D6 status and body weight, different doses of eliglustat may be needed to achieve therapeutic goals. The CYP2D6 genotype may have to be determined before treatment is started to individualize optimal therapeutic doses. Many more people affected with this debilitating disease will benefit because of increased access to treatment with an orally administered drug.
It is not known whether eliglustat tartrate will be safe to use in children, whose nervous systems are still developing. Although eliglustat tartrate is a small molecule, which should permit it to cross the blood–brain barrier, brain penetration is limited by its attachment to a P-glycoprotein transporter that is highly expressed at the blood–brain barrier. This may prevent interference with myelin and neuronal function in children. It also makes eliglustat ineffective for the neurologic treatment of types 2 and 3 Gaucher disease. However, an analog of eliglustat has been devised that penetrates the blood–brain barrier and is being tested in mice.19 This may prove to be useful in adults with type 1 Gaucher disease, which is associated with a higher incidence of Parkinsonism than that seen in the general population, as well as the neuropathic forms of Gaucher disease, and also offers hope for effective treatment of other lysosomal disorders.
Disclosure
Paige Kaplan is an advisor for the International Collaborative Gaucher Group Gaucher Registry and Elylyso Gaucher registry, has received honoraria from Genzyme Corporation (a Sanofi company) and Pfizer, and owns Pfizer shares. She had received an unrestricted educational grant from Genzyme Corporation.
References
Grabowski GA, Kolodny EH, Weinreb NJ, Rosenbloom BE, Prakash-Cheng A. Gaucher disease: Phenotypic and genetic variation. In: Scriver CR, Beaudet A, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Diseases. 9th ed. New York: McGraw-Hill; 2006:3635–3658. | |
Ida H, Rennert OM, Iwasawa K, Kobayashi M, Eto Y. Clinical and genetic studies of Japanese homozygotes for the Gaucher disease L444P mutation. Hum Genet. 1999;105(1–2):120–126. | |
Charrow J, Andersson HC, Kaplan P, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr. 2004;144(1):112–120. | |
Kaplan P, Andersson HC, Kacena KA, Yee JD. The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160(6):603–608. | |
Shayman JA. ELIGLUSTAT TARTRATE: Glucosylceramide Synthase Inhibitor Treatment of Type 1 Gaucher Disease. Drugs Future. 2010;35(8):613–620. | |
Shayman JA. The design and clinical development of inhibitors of glycosphingolipid synthesis: will invention be the mother of necessity? Trans Am Clin Climatol Assoc. 2013;124:46–60. | |
Hollak CE, Hughes D, van Schaik IN, Schwierin B, Bembi B. Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. Pharmacoepidemiol Drug Saf. 2009;18(9):770–777. | |
McEachern KA, Fung J, Komarnitsky S, et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol Genet Metab. 2007;91(3):259–267. | |
Marshall J, McEachern KA, Chuang WL, et al. Improved management of lysosomal glucosylceramide levels in a mouse model of type 1 Gaucher disease using enzyme and substrate reduction therapy. J Inherit Metab Dis. 2010;33(3):281–289. | |
Peterschmitt MJ, Burke A, Blankstein L, et al. Safety, tolerability, and pharmacokinetics of eliglustat tartrate (Genz-112638) after single doses, multiple doses, and food in healthy volunteers. J Clin Pharmacol. 2011;51(5):695–705. | |
Lukina E, Watman N, Arreguin EA, et al. A phase II study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood. 2010;116(6):893–899. | |
Lukina E, Watman N, Arreguin EA, et al. Improvement in hematological, visceral, and skeletal manifestations of Gaucher disease type 1 with oral eliglustat tartrate (Genz-112638) treatment: 2-year results of a phase 2 study. Blood. 2010;116(20):4095–4098. | |
Wenstrup RJ, Kacena KA, Kaplan P, et al. Effect of enzyme replacement therapy with imiglucerase on BMD in type 1 Gaucher disease. J Bone Miner Res. 2007;22(1):119–126. | |
Dasouki M, Lukina E, Ben Dridi M-F, et al. Effects of Oral Eliglustat on Bone Disease in Gaucher Disease Type 1: Results from the Randomized, Placebo-Controlled ENGAGE Trial. Presented at 12th International Congress of Inborn Errors of Metabolism, Barcelona, Spain, 3-6 September 2013. | |
Ben Turkia H, Lukina E, Amato D, et al. ENGAGE: A Phase III, Randomized, Double-Blind, Placebo-controlled, Multicenter Study To Investigate The Efficacy and Safety of Eliglustat in Adults with Gaucher Disease Type 1: 9 Month Results. Presented at 12th International Congress of Inborn Errors of Metabolism, Barcelona, Spain, 3-6 September 2013. | |
Cox TM, Drelichman G, Cravo R, et al. ENCORE: A Multi-national, Randomized, Controlled, Open-Label Non-inferiority Study Comparing Eliglustat with Imiglucerase in Gaucher Disease Type 1 Patients on Enzyme Replacement Therapy Who Have Reached Therapeutic Goals. Presented at 12th International Congress of Inborn Errors of Metabolism, Barcelona 3-6 September 2013. | |
Mankoski R, Peterschmitt JM, Puga AC, Cox, et al. Eliglustat Safety Profile: A Pooled Analysis from Four Trials in Gaucher Disease Type 1. American Society of Human Genetics Annual Meeting Boston, USA, Oct 2013 Poster. | |
Chérin P, Rose C, de Roux-Serratrice C, et al. The neurological manifestations of Gaucher disease type 1: the French Observatoire on Gaucher disease (FROG). J Inherit Metab Dis. 2010;33(4):331–338. | |
Larsen SD, Wilson MW, Abe A, et al. Property-based design of a glucosylceramide synthase inhibitor that reduces glucosylceramide in the brain. J Lipid Res. 2012;53(2):282–291. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.