Back to Archived Journals » Journal of Neurorestoratology » Volume 3
Clinical neurorestorative progress in Alzheimer’s disease
Authors Qiao L, Huang H, Muresanu D
Received 30 September 2014
Accepted for publication 31 October 2014
Published 15 December 2014 Volume 2015:3 Pages 1—9
DOI https://doi.org/10.2147/JN.S74143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Hari Shanker Sharma
Liyan Qiao,1 Hongyun Huang,2,3 Dafin F Muresanu4,5
1Department of Neurology, Tsinghua University Yuquan Hospital, 2Beijing Rehabilitation Hospital of Capital Medical University, 3Beijing Hongtianji Neuroscience Academy, Beijing, People's Republic of China; 4Department of Clinical Neurosciences, Iuliu Hatieganu University of Medicine and Pharmacy, 5RoNeuro Institute for Neurological Research and Diagnosis, Cluj-Napoca, Romania
Abstract: Alzheimer’s disease (AD) is the most prevalent type of dementia, and its neuropathology is characterized by the deposition of insoluble β-amyloid peptides and intracellular neurofibrillary tangles and the loss of diverse neurons. Although much is known about the neurobiology of AD, few treatments are available to arrest or slow the illness. There is an urgent need for novel therapeutic approaches for AD. We reviewed the recent improvements in the neurorastorlogy strategies for AD, including medicine, bioengineering and neuromodulation and clinical cell therapy. We emphasized that cell therapy may be an promising treatment for AD.
Keywords: Alzheimer's disease, neurorastorlogy, cell therapy, medicine
Introduction
Alzheimer’s disease (AD) is the most prevalent type of dementia, and its neuropathology is characterized by deposition of insoluble β-amyloid (Aβ) peptides, intracellular neurofibrillary tangles, amyloid angiopathy, age-related brain atrophy, synaptic pathology, white matter rarefaction, granulovacuolar degeneration, neuron loss, and neuroinflammation.1 Currently, it is estimated that 5.2 million Americans of all ages have AD. Among them, 5 million people are aged 65 years of age.2
With the exception of the rare cases of AD caused by known genetic mutations, AD, similar to other common chronic diseases, develops as a result of multiple factors rather than a single cause. The known risk factors for AD include age, family history, presence of the apolipoprotein (Apo)E ε4 gene, mild cognitive impairment, traumatic brain injury, cardiovascular disease risk factors, lack of social and cognitive engagement, and lower education level.1,2
Abnormal accumulation of Aβ peptide is widely believed to be the underlying mechanism of pathologic and clinical changes seen in AD.1,3 Aβ appears earliest in the cerebral neocortex, prior to onset of symptoms, and reaches the cerebellar cortex in advanced clinical disease.4 Although Aβ plaques may play a key role in AD pathogenesis, the severity of cognitive impairment correlates best with the burden of neocortical neurofibrillary tangles.5 Cholinergic dysfunction in the basal and rostral forebrain is associated with even early cognitive impairments observed in AD.6 In addition to abnormal accumulation of the Aβ peptide, the formation of neurofibrillary tangles, and the degeneration of the cholinergic pathway, AD has also been associated with environmental and genetic factors, mitochondrial dysfunction, vascular factors, immune system dysfunction, and infectious agents. Neurovascular dysfunction plays an essential role in this multifactorial pattern. The neurovascular unit encompasses neurons, interneurons, astrocytes, smooth muscle cells, pericytes, endothelial cells, and the extracellular matrix.7 A broad spectrum of insults to the neurovascular unit, such as blood–brain barrier break down, cerebral amyloid angiopathy, loss or abnormal cholinergic innervation of blood vessels, arterial hypercontractility, atherosclerosis, vascular anatomical defects, mitochondrial abnormalities of the endothelial cells, and degeneration of pericytes, has been implicated.8,9
The treatment of AD mainly includes nonpharmacologic and pharmacologic therapies. None of the treatments available for AD, except for neurorestorative strategies, have been shown to slow or stop the malfunction and death of neurons in the brain that cause AD symptoms. Here, we review the recent clinical progress of neurorestorative strategies.
Clinical progress of neurorestorative strategies
Medicine and factors
Currently, no single molecule has been approved for the treatment of AD since the advent of cholinesterase inhibitors and memantine. However, potential neurorestorative treatments that have been tested in AD patients are emerging.
γ-secretase inhibitors (GSIs) and modulators
γ-secretase cleaves amyloid precursor proteins (APPs) to release Aβ peptides, which play a causative role in the pathogenesis of AD. GSIs have been developed and used for clinical studies. Studies in animal models of AD have indicated that GSIs administered by the oral route are able to lower Aβ concentrations. However, GSIs cause nondiscriminatory inhibition of Notch (a transmembrane receptor involved in regulating cell-fate decisions) signaling, overall APP processing, and other substrate cleavages. These findings have called for the development of disease-modifying agents called γ-secretase modulators, which target γ-secretase activity and lower Aβ42 production without blocking the overall processing of γ-secretase substrates. Animal studies of this generation of γ-secretase modulators have shown encouraging preclinical profiles.10,11
Two large Phase III clinical trials of semagacestat (LY-450139) in patients with mild to moderate AD were prematurely interrupted due to the observation of detrimental effects on cognition and functionality in patients receiving the drug compared with those receiving placebo.12 Two large Phase III studies of tarenflurbil (r-flurbiprofen), a putative γ-secretase modulator, in patients with mild AD were also completely negative. The failures were ascribed to the inhibition of the cleavage of Notch, and low potency and brain penetration.13,14
Immunology
Active and passive immunotherapy targeting either Aβ or tau has been successful in repeated AD model animal trials.15,16 In the last decade, an increased number of clinical trials for AD immunotherapy have started, failed, and are continuing to be pursued.17 AN-1792 was the first drug to actively immunize humans with Aβ peptide. Approximately 5% of treated patients presented with meningoencephalitis, which halted its Phase II trial prematurely. It is likely that the severe inflammatory symptoms from AN-1792 related significantly to the immunological adjuvant.18 Bapineuzumab (AAB-001) is a humanized mouse monoclonal antibody to the N-terminal portion of Aβ. Its Phase II trial has shown increased side effects, including vasogenic edema of the brain, in ApoEε4 carriers, but evidence suggestive of a benefit has also been found, particularly in noncarriers of the ApoE ε4 allele. A different humanized mouse monoclonal antibody to the midportion of Aβ, solanezumab (LY2062430), has been shown to clear soluble Aβ, accompanied by biomarker changes in cerebrospinal fluid (CSF) in a Phase II trial.
Pilot studies of human intravenous immunoglobulin (IVIG) resulted in the stabilization of cognitive functioning in patients with mild to moderate AD. The putative mechanism for IVIG involves low levels of polyclonal anti-Aβ antibody that possibly modify the immune response. However, the subsequent Phase III trial found no evidence for slowing of AD progression. These disappointing results have reduced enthusiasm for IVIG as a possible treatment for AD.19
Anti-inflammatory drugs
Long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs) reduces the risk of developing AD and delays its onset, as shown by epidemiological studies. In addition, a subset of NSAIDs, such as ibuprofen, indomethacin, and flurbiprofen, may have direct Aβ-lowering properties in cell cultures as well as in transgenic models of AD-like amyloidosis.20 However, treatment for 1 to 3 years with naproxen or celecoxib did not protect against cognitive decline in older adults with a family history of AD.21
Neurotrophic factors (NTFs)
Based on current scientific knowledge, treatment with NTFs would be a desirable intervention for brain diseases with a complex pathology, such as dementia. Unlike the classical single-target molecules used for current therapies, NTFs allow for designing treatment strategies based on multimodal activity (Figure 1).
 | Figure 1 Mode of action of the pleiotropic multimodal drug Cerebrolysin® in AD. |
Cerebrolysin® is a parenterally administered neuropeptide preparation that contains active fragments of some important NTFs and has displayed activity closely matching that of endogenous NTFs in preclinical research models of neurological disorders; it exerts neuromodulatory therapeutic effects, including pleiotropic neuroprotective effects (such as antiapoptotic, anti-inflammatory, and antioxidative activity and the reduction of toxic protein aggregates) and neurorestorative effects (including the stimulation of neurogenesis, neuronal sprouting and synaptogenesis, oligodendrogenesis, and neurite remodeling). Cerebrolysin reduces toxic protein aggregation as well as activation of proapoptotic genes by modulating key regulatory elements of the neurotrophic signaling pathway, namely glycogen synthase kinase-3β (GSK3β) and cyclin-dependent kinase-5 (CDK5).22 Cerebrolysin has been shown to protect brain tissue against amyloid plaque formation by modifying APP maturation, a process regulated by these phosphorylating enzymes. By modulating the activity of these enzymes, Cerebrolysin reduces the cellular transport of APP to synaptic sites, thus lowering the titer of Aβ peptides released into the extracellular matrix, which results in reduced formation of amyloid plaques. Treatment with Cerebrolysin also results in a significant decrease in the levels of tau tangles, by modulating activity of the same enzymes.23
Several clinical trials have been performed to investigate the safety and efficacy of Cerebrolysin in patients suffering from mild to moderately severe AD. Clinical data have strongly shown that Cerebrolysin is an effective therapeutic option for patients diagnosed with AD.24–29 Most of the randomized, double-blind, placebo-controlled trials have shown significant improvement in cognitive, behavioral, functional, and global domains after treatment with Cerebrolysin. These effects were largely maintained over several months, indicating a long-term beneficial influence of Cerebrolysin on the disease. Due to the drug’s multimodal activity and pleiotropic neuroprotective effects, which target distinct molecular pathways in the pathologic cascade, treatment with Cerebrolysin goes far beyond pure symptomatic improvement, suggesting a potential delay in disease progression.30–32
It is well known that there are close interactions between AD and vascular dementia; in the majority of cases, it is difficult to determine the difference between these two types of dementia.33,34 Coexistence of degenerative and vascular pathology has both additive and synergistic effects on dementia progress. Cerebrolysin also has beneficial effects on cognitive and global function in elderly patients with vascular dementia of mild to moderate severity, as shown in a recent Cochrane review.35
Bioengineering
Genetically modified neural stem/progenitor cells (NSCs) and mesenchymal stromal cells (MSCs) that stably express and secrete Aβ-degrading enzyme, human choline acetyltransferase, and nerve growth factor (NGF) gene have been tested in transgenic mouse models of AD. These approaches was shown to enhance synaptic plasticity, elevate acetylcholine level, and improve the cognitive function in AD animal models.36–43 Tuszynski et al44 performed a Phase I trial by implanting autologous fibroblasts genetically modified to express human NGF into the forebrain of eight individuals with mild AD. No long-term adverse effects of NGF occurred. A cognitive evaluation suggested improvement, and robust growth responses to NGF were observed in one brain autopsy. Injections of DNA coding for brain-derived neurotrophic factor (BDNF) has been the subject of a Phase I experiment.45As NGF or BDNF are not the sole candidates as a possible contributor to brain homeostasis among 50 different trophic factors in the brain, their neuroprotective effect are still being considered.
Clinical cell therapy
Cortical cholinergic loss is strongly associated with the reduction of progenitors.46 However, viable precursor cells do remain in AD and aged normal control brain specimens and can be induced to differentiate.47 Lower stem cell factor plasma levels were found in AD patients.48 These results raise the possibility of stimulation of inherent precursor cells of aged individuals or AD patients to replace neurons lost in aging and/or neurodegeneration.49
Sanchez-Ramos et al34 investigated the effects of a 5-day schedule of granulocyte colony stimulating factor (G-CSF) administration in eight patients with mild to moderate stage AD and found it was tolerated and safe, and it improved cognition. A South Korea group conducted a Phase I trial in nine patients with mild to moderate AD, using umbilical cord stromal cells. The cells were injected bilaterally in the hippocampus and in the right precuneus. There were no serious adverse events. The Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-cog) suggested possible improvements in two patients; seven patients showed cognitive decline at 1 month and 3 months. Pittsburgh compound B (PiB) positron emission tomography (PET) scans showed a decreased PiB retention ratio in four patients, but five patients showed an increased PiB retention ratio at 1 month and 3 months. Among 12 Neuropsychiatric Inventory (NPI) items, the scores in ten items decreased at 3 months and the scores in two items increased at 3 months compared with baseline.50
Neuromodulation
Deep brain stimulation (DBS)
By targeting specific structures within known memory circuits, including the entorhinal, and hippocampal areas, and activating the brain’s default mode network, DBS can have effects in AD patients and is therefore a potentially promising avenue for novel dementia treatments. It has been shown that no complications occurred and that the stimulation was perfectly tolerated, after DBS treatment in AD patients. After 1 year of stimulation, the memory scores were either stabilized or possible improved compared with baseline, in pilot studies.51,52
Repetitive transcranial magnetic stimulation (rTMS)
rTMS is a noninvasive tool for modulating cortical activity and a potential therapy choice for AD. However, no recommendation for rTMS was suggested in AD patients.53 Nevertheless, high-frequency rTMS seems to have better effects on improving cognitive function in AD patients compared with low-frequency rTMS, especially in combination with cognitive therapy.54,55
Discussion
The rationality for cell therapy in AD
The rationale for cell therapy in AD is based on the following findings:
Impaired neurogenesis in the subventricular zone has been found in early AD.56,57 Aβ impairs the proliferation and neuronal differentiation of neuronal precursor cells and contributes to the depletion of neurons and cognitive impairment in AD.58 APP, especially APP possessing the protease inhibitor domain, regulates the growth of neuronal precursor cells during development of the nervous system.59
Either a wild-type or a familial AD (FAD) mutant PS-1 transgene can reduce the number of neural progenitors in the dentate gyrus. However, the wild-type, but not the FAD mutant, PS-1 promoted the survival and differentiation of progenitors, leading to the generation of more immature granule cell neurons in PS-1 wild-type-expressing animals.60
As well, some evidence shows compensatory increase in neurogenesis after PS-1 mutation, which represents FAD pathogenesis.61
Finally, in separate work, the deafferented hippocampus provided proper microenvironment for the survival and neuronal differentiation of neural progenitors.62
The mechanism of cell therapy in AD
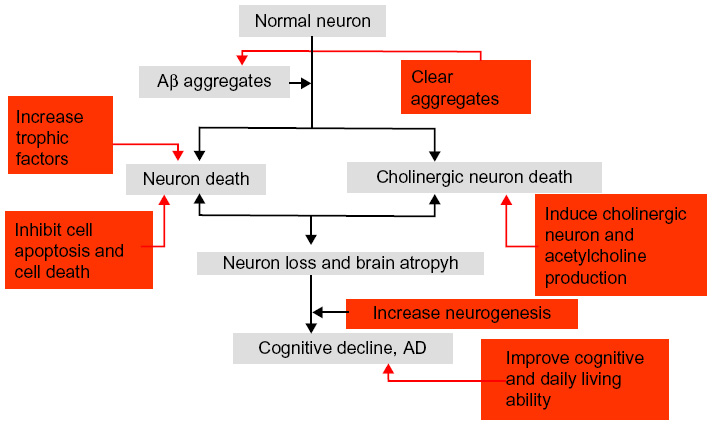
Cell types that have been used in AD include NSCs, MSCs, embryonic stem cells (ESCs), and induced pluripotent stem cells. These cell types have been shown to be beneficial for increasing neurogenesis and neuronal differentiation, enhancing Aβ clearance, inhibiting cell death, recovering acetylcholine levels, and improving clinical symptoms, including memory and visuospatial impairment (Figure 2).
| Figure 2 The potential mechanism of cell therapy in AD. |
Increased neurogenesis
MSCs increase hippocampal neurogenesis, neuronal differentiation, and the formation of neurites. These outcomes may be achieved by enhancing the Wnt signaling pathway in AD models.63,64
Inhibition of cell apoptosis and cell death
MSCs can inhibit cell apoptosis and cell death caused by either Aβ or tau,65,66 which are accompanied by memory improvement.67
Clearance protein aggregates
MSCs have capacity to clear Aβ but not tau. The possible mechanism for clearing Aβ is by enhancing autophagy function.51,68,69
Increased trophic factors
Human adipose tissue-derived MSCs could enhance the concentrations of BDNF and NGF, which would be accompanied by an improvement in cognitive function.70 Another group showed that MSCs, via intravenous or intracerebral transplantation, significantly rescued memory deficit and neuropathology, by upregulating cytokine interleukin (IL)-10 and vascular endothelial growth factor (VEGF).71
Induction of acetylcholine production in AD
Increasing the acetylcholine level is crucial for current therapy methods for AD. In neurorestorative treatment, two methods can be used to achieve the goal of increasing the acetylcholine level: bioengineering and cell therapy. As mentioned above, the acetylcholine level could be increased by gene expression via a vector. For the latter, several cell types have been shown to have the potential to differentiate into cholinergic cell phenotypes. Transplanted NSCs were shown to differentiate into cholinergic neurons in a rat AD model.72 Transplantation of mouse ESC-derived neuronal precursor cells, following commitment to a cholinergic cell phenotype, were able to promote behavioral recovery in a rodent model of AD. No tumors were formatted, which indicated that ESCs may be safe for transplantation.73
Neuronal precursors with cholinergic neuron phenotype derived from human induced pluripotent stem cells survived in APP mouse hippocampus and improved their spatial memory loss.74 Induced pluripotent stem cells derived from dermal fibroblasts of AD patients and differentiated into cholinergic neurons might be a promising novel tool for disease modeling and drug discovery for the sporadic form of AD75 and FAD.76 In one study, human adipose tissue-derived MSCs were intravenously or intracerebroventricularly transplanted into the brains of 18-month-old mice. Transplantation improved both locomotor activity and cognitive function in the aged animals, in parallel with recovery of acetylcholine levels in brain tissues.
Inducing endogenous cell in AD therapy
AD subjects have significantly fewer viable precursor cells in the hippocampus compared with age-matched normal control subjects. However, viable precursor cells remaining in AD and aged normal control brain specimens can be induced to differentiate.77 These endogenous precursor cells can be induced to show neuroprotective effects by chemical compounds and stimulating factors, such as allopregnanolone (Apα), fluoxetine, G-CSF, AMD3100, stromal cell-derived factor-1a (SDF-1α), and others.78
Recent studies have proposed that chronic treatment with antidepressants increases neurogenesis in the adult hippocampus. Chang et al79 showed the dose-dependent effects of fluoxetine, a common antidepressant, on the proliferation and neural differentiation of fetal derived NSCs. Fluoxetine, even at nanomolar concentrations, stimulated proliferation and differentiation of NSCs. In addition, fluoxetine has protective effects against cell death induced by Aβ.42 Chadwick et al80 showed amitriptyline, another common antidepressant, mediated cognitive enhancement in aged AD mice that was associated with neurogenesis and neurotrophic activity.
Even though the distribution of G-CSF and G-CSF receptor has not been shown to substantially differ between AD and control brains, even in the hippocampus,81 there are studies suggesting that G-CSF can be used in AD treatment. G-CSF application was shown to improve memory and neurobehavior in an Aβ-induced experimental model of AD.82,83 G-CSF has also been shown to decrease the brain amyloid burden.84
SDF-1α is an effective adjuvant in inducing migration of endogenous bone morrow-derived hematopoietic progenitor cells, which are mobilized by G-CSF, into the brain, suggesting that these two molecules can act synergistically to produce a therapeutic effect in AD.85
GLP-1 is a growth factor that has neuroprotective properties. GLP1 receptors are present on neuronal progenitor cells, and the GLP1 analogue liraglutide was shown to increase cell proliferation and differentiation into neurons in an AD mouse model.86
The therapy time for AD
MSCs can promote the reduction of Aβ and the improvement of synaptic transmission, both in AD mice and in predementia AD mice, which indicates that MSCs may exert their effects in different stages of AD.
The aging effect
AD is common in the elderly, and it is not known whether transplanted cells can work in the aging environment. Human adipose tissue-derived MSCs were intravenously or intracerebroventricularly transplanted into the brains of 18-month-old mice. Transplantation improved both locomotor activity and cognitive function in the aged animals, in parallel with recovery of acetylcholine levels in brain tissues through enhanced concentrations of BDNF and NGF.
Conclusion
Although much is known about the neurobiology of dementia and AD, very few conventional therapies are available to arrest or slow the illness. There is an urgent need for novel therapeutic approaches for AD. Translating neurorestorative strategies with positive preclinical results for AD into clinical studies has allowed patients to receive clinical neurorestoration to a certain extent, via medicine/factors, cell therapy, neuromodulation, neurorehabilitation and combined therapies. Cell-based neurorestorative strategies are promising for AD. To gain better neurorestorative effect, earlier interventional neurorestorative strategies for AD should be recommended because dementia and other clinical presentations occur well after the pathological findings in AD. In the future, there should be greater encouragement to translate effective preclinical neurorestorative strategies to clinic practice as quickly as possible.
Disclosure
The authors report no conflicts of interest in this work.
References
Victor M, Ropper AH. Degenerative diseases of the nervous system. In: Ropper AH, Samuels MA. Adams and Victor’s Principles of Neurology. 1106–1174. | |
Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10(2):e47–e92. | |
Drachman DA. The amyloid hypothesis, time to move on: Amyloid is the downstream result, not cause, of Alzheimer’s disease. Alzheimers Dement. 2014;10(3):372–380. | |
Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. | |
Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–381. | |
Parsons CG, Danysz W, Dekundy A, Pulte I. Memantine and cholinesterase inhibitors: complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox Res. 2013;24(3):358–369. | |
Muoio V, Persson PB, Sendeski MM. The neurovascular unit – concept review. Acta Physiol (Oxf). 2014;210(4):790–798. | |
Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009; 118(1):103–113. | |
Baloyannis SJ, Baloyannis IS. The vascular factor in Alzheimer’s disease: a study in Golgi technique and electron microscopy. J Neurol Sci. 2012;322(1–2):117–121. | |
Panza F, Frisardi V, Solfrizzi V, et al. Interacting with γ-secretase for treating Alzheimer’s disease: from inhibition to modulation. Curr Med Chem. 2011;18(35):5430–5447. | |
Crump CJ, Johnson DS, Li YM. Development and mechanism of γ-secretase modulators for Alzheimer’s disease. Biochemistry. 2013;52(19):3197–3216. | |
Doody RS, Raman R, Farlow M, et al; Alzheimer’s Disease Cooperative Study Steering Committee; Semagacestat Study Group. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341–350. | |
Imbimbo BP, Giardina GA. γ-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: disappointments and hopes. Curr Top Med Chem. 2011;11(12):1555–1570. | |
Green RC, Schneider LS, Amato DA, et al; Tarenflurbil Phase 3 Study Group. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302(23):2557–2564. | |
Wisniewski T, Goñi F. Immunotherapy for Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):499–507. | |
Lambracht-Washington D, Rosenberg RN. Anti-amyloid beta to tau – based immunization: Developments in immunotherapy for Alzheimer disease. Immunotargets Ther. 2013;2013(2):105–114. | |
Lambracht-Washington D, Rosenberg RN. Advances in the development of vaccines for Alzheimer’s disease. Discov Med. 2013;15(84):319–326. | |
Honig LS. Translational research in neurology: dementia. Arch Neurol. 2012;69(8):969–977. | |
Loeffler DA. Intravenous immunoglobulin and Alzheimer’s disease: what now? J Neuroinflammation. 2013;10(1):70. | |
Townsend KP, Praticò D. Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19(12):1592–1601. | |
ADAPT-FS Research Group. Follow-up evaluation of cognitive function in the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial and its Follow-up Study. Alzheimers Dement. Epub July 8, 2014. | |
Rockenstein E, Torrance M, Mante M, et al. Cerebrolysin decreases amyloid-beta production by regulating amyloid protein precursor maturation in a transgenic model of Alzheimer’s disease. J Neurosci Res. 2006;83(7):1252–1261. | |
Ubhi K, Rockenstein E, Doppler E, et al. Neurofibrillary and neurodegenerative pathology in APP-transgenic mice injected with AAV2-mutant TAU: neuroprotective effects of Cerebrolysin. Acta Neuropathol. 2009;117(6):699–712. | |
Ruether E, Husmann R, Kinzler E, et al. A 28-week, double-blind, placebo-controlled study with Cerebrolysin in patients with mild to moderate Alzheimer’s disease. Int Clin Psychopharmacol. 2001;16(5):253–263. | |
Panisset M, Gauthier S, Moessler H, Windisch M; Cerebrolysin Study Group. Cerebrolysin in Alzheimer’s disease: a randomized, double-blind, placebo-controlled trial with a neurotrophic agent. J Neural Transm. 2002;109(7–8):1089–1104. | |
Muresanu DF, Rainer M, Moessler H. Improved global function and activities of daily living in patients with AD: a placebo-controlled clinical study with the neurotrophic agent Cerebrolysin. J Neural Transm Suppl. 2002;(62):277–285. | |
Alvarez XA, Cacabelos R, Laredo M, et al. A 24-week, double-blind, placebo-controlled study of three dosages of Cerebrolysin in patients with mild to moderate Alzheimer’s disease. Eur J Neurol. 2006;13(1):43–54. | |
Alvarez XA, Sampedro C, Cacabelos R, et al. Reduced TNF-α and increased IGF-I levels in the serum of Alzheimer’s disease patients treated with the neurotrophic agent cerebrolysin. Int J Neuropsychopharmacol. 2009;12(7):867–872. | |
Álvarez XA, Fuentes P. Cerebrolysin in Alzheimer’s Disease. Drugs Today (Barc). 2011;47(7):487–513. | |
Plosker GL, Gauthier S. Cerebrolysin: a review of its use in dementia. Drugs Aging. 2009;26(11):893–915. | |
Álvarez XA, Fuentes P. Cerebrolysin in Alzheimer’s disease. Drugs Today (Barc). 2011;47(7):487–513. | |
Allegri RF, Guekht A. Cerebrolysin improves symptoms and delays progression in patients with Alzheimer’s disease and vascular dementia. Drugs Today (Barc). 2012;48 Suppl A:S25–S41. | |
Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120(3):287–296. | |
Muresanu DF, Popa-Wagner A, Stan A, Buga AM, Popescu BO. The vascular component of Alzheimer’s disease. Curr Neurovasc Res. 2014;11(2):168–176. | |
Chen N, Yang M, Guo J, Zhou M, Zhu C, He L. Cerebrolysin for vascular dementia. Cochrane Database Syst Rev. 2013;1:CD008900. | |
Blurton-Jones M, Spencer B, Michael S, et al. Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem Cell Res Ther. 2014;5(2):46. | |
Park D, Lee HJ, Joo SS, et al. Human neural stem cells over-expressing choline acetyltransferase restore cognition in rat model of cognitive dysfunction. Exp Neurol. 2012;234(2):521–526. | |
Lee HJ, Lim IJ, Park SW, Kim YB, Ko Y, Kim SU. Human neural stem cells genetically modified to express human nerve growth factor (NGF) gene restore cognition in the mouse with ibotenic acid-induced cognitive dysfunction. Cell Transplant. 2012;21(11):2487–2496. | |
Wu L, Sluiter AA, Guo HF, et al. Neural stem cells improve neuronal survival in cultured postmortem brain tissue from aged and Alzheimer patients. J Cell Mol Med. 2008;12(5A):1611–1621. | |
Cenciarelli C, Budoni M, Mercanti D, et al. In vitro analysis of mouse neural stem cells genetically modified to stably express human NGF by a novel multigenic viral expression system. Neurol Res. 2006;28(5):505–512. | |
Marei HE, Farag A, Althani A, et al. Human olfactory bulb neural stem cells expressing hNGF restore cognitive deficit in Alzheimer’s disease rat model. J Cell Physiol. 2015;230(1):116–130. | |
Li LY, Li JT, Wu QY, et al. Transplantation of NGF-gene-modified bone marrow stromal cells into a rat model of Alzheimer’ disease. J Mol Neurosci. 2008;34(2):157–163. | |
Liang J, Wu S, Zhao H, et al. Human umbilical cord mesenchymal stem cells derived from Wharton’s jelly differentiate into cholinergic-like neurons in vitro. Neurosci Lett. 2013;532:59–63. | |
Tuszynski MH, Thal L, Pay M, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11(5):551–555. | |
Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011; 10(3):209–219. | |
Ziabreva I, Perry E, Perry R, et al. Altered neurogenesis in Alzheimer’s disease. J Psychosom Res. 2006;61(3):311–316. | |
Sanchez-Ramos J, Cimino C, Avila R, et al. Pilot study of granulocyte-colony stimulating factor for treatment of Alzheimer’s disease. J Alzheimers Dis. 2012;31(4):843–855. | |
Laske C, Stellos K, Stransky E, et al. Decreased plasma and cerebrospinal fluid levels of stem cell factor in patients with early Alzheimer’s disease. J Alzheimers Dis. 2008;15(3):451–460. | |
Lovell MA, Geiger H, Van Zant GE, Lynn BC, Markesbery WR. Isolation of neural precursor cells from Alzheimer’s disease and aged control postmortem brain. Neurobiol Aging. 2006;27(7):909–917. | |
Seo SW, Lee JII, Kim CH, et al. A phase I trial of parenchymal injection of umbilical cord stem cells in Alzheimer’s disease. Alzheimers Dement. 2013;9(Suppl 4):S291. | |
Laxton AW, Tang-Wai DF, McAndrews MP, et al. A phase I trial of deep brain stimulation of memory circuits in Alzheimer’s disease. Ann Neurol. 2010;68(4):521–534. | |
Fontaine D, Deudon A, Lemaire JJ, et al. Symptomatic treatment of memory decline in Alzheimer’s disease by deep brain stimulation: a feasibility study. J Alzheimers Dis. 2013;34(1):315–323. | |
Lefaucheur JP, André-Obadia N, Antal A, et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS). Clin Neurophysiol. Epub June 5, 2014. | |
Rabey JM, Dobronevsky E, Aichenbaum S, Gonen O, Marton RG, Khaigrekht M. Repetitive transcranial magnetic stimulation combined with cognitive training is a safe and effective modality for the treatment of Alzheimer’s disease: a randomized, double-blind study. J Neural Transm. 2013;120(5):813–819. | |
Eliasova I, Anderkova L, Marecek R, Rektorova I. Non-invasive brain stimulation of the right inferior frontal gyrus may improve attention in early Alzheimer’s disease: A pilot study. J Neurol Sci. Epub August 29, 2014. | |
Demars M, Hu YS, Gadadhar A, Lazarov O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J Neurosci Res. 2010;88(10):2103–2117. | |
Rodríguez JJ, Jones VC, Verkhratsky A. Impaired cell proliferation in the subventricular zone in an Alzheimer’s disease model. Neuroreport. 2009;20(10):907–912. | |
Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J Neurochem. 2002;83(6):1509–1524. | |
Hayashi Y, Kashiwagi K, Ohta J, Nakajima M, Kawashima T, Yoshikawa K. Alzheimer amyloid protein precursor enhances proliferation of neural stem cells from fetal rat brain. Biochem Biophys Res Commun. 1994;205(1):936–943. | |
Wen PH, Shao X, Shao Z, et al. Overexpression of wild type but not an FAD mutant presenilin-1 promotes neurogenesis in the hippocampus of adult mice. Neurobiol Dis. 2002;10(1):8–19. | |
Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol. 2005;167(1):151–159. | |
Zhang X, Jin G, Tian M, Qin J, Huang Z. The denervated hippocampus provides proper microenvironment for the survival and differentiation of neural progenitors. Neurosci Lett. 2007;414(2):115–120. | |
Oh SH, Kim HN, Park HJ, Shin JY, Lee PH. Mesenchymal stem cells increase hippocampal neurogenesis and neuronal differentiation by enhancing the Wnt signaling pathway in Alzheimer’s disease model. Cell Transplant. March 7, 2014. | |
Zilka N, Zilkova M, Kazmerova Z, Sarissky M, Cigankova V, Novak M. Mesenchymal stem cells rescue the Alzheimer’s disease cell model from cell death induced by misfolded truncated tau. Neuroscience. 2011;193:330–337. | |
Shin JY, Park HJ, Kim HN, et al. Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models. Autophagy. 2014;10(1):32–44. | |
Zilka N, Zilkova M, Kazmerova Z, Sarissky M, Cigankova V, Novak M. Mesenchymal stem cells rescue the Alzheimer’s disease cell model from cell death induced by misfolded truncated tau. Neuroscience. 2011;193:330–337. | |
Lee JK, Jin HK, Bae JS. Bone marrow-derived mesenchymal stem cells attenuate amyloid β-induced memory impairment and apoptosis by inhibiting neuronal cell death. Curr Alzheimer Res. 2010;7(6):540–548. | |
Lee JK, Jin HK, Bae JS. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-beta deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci Lett. 2009;450(2):136–141. | |
Bae JS, Jin HK, Lee JK, Richardson JC, Carter JE. Bone marrow-derived mesenchymal stem cells contribute to the reduction of amyloid-β deposits and the improvement of synaptic transmission in a mouse model of pre-dementia Alzheimer’s disease. Curr Alzheimer Res. 2013;10(5):524–531. | |
Park D, Yang G, Bae DK, et al. Human adipose tissue-derived mesenchymal stem cells improve cognitive function and physical activity in ageing mice. J Neurosci Res. 2013;91(5):660–670. | |
Kim S, Chang KA, Kim Ja, et al. The preventive and therapeutic effects of intravenous human adipose-derived stem cells in Alzheimer’s disease mice. PLoS One. 2012;7(9):e45757. | |
Xuan AG, Luo M, Ji WD, Long DH. Effects of engrafted neural stem cells in Alzheimer’s disease rats. Neurosci Lett. 2009;450(2):167–171. | |
Moghadam FH, Alaie H, Karbalaie K, Tanhaei S, Nasr Esfahani MH, Baharvand H. Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differentiation. 2009;78(2–3):59–68. | |
Fujiwara N, Shimizu J, Takai K, et al. Restoration of spatial memory dysfunction of human APP transgenic mice by transplantation of neuronal precursors derived from human iPS cells. Neurosci Lett. 2013;557 Pt B:129–134. | |
Ooi L, Sidhu K, Poljak A, et al. Induced pluripotent stem cells as tools for disease modelling and drug discovery in Alzheimer’s disease. J Neural Transm. 2013;120(1):103–111. | |
Yagi T, Ito D, Okada Y, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. | |
Lovell MA, Geiger H, Van Zant GE, Lynn BC, Markesbery WR. Isolation of neural precursor cells from Alzheimer’s disease and aged control postmortem brain. Neurobiol Aging. 2006;27(7):909–917. | |
Choi SS, Lee SR, Kim SU, Lee HJ. Alzheimer’s disease and stem cell therapy. Exp Neurobiol. 2014;23(1):45–52. | |
Chang KA, Kim JA, Kim S, Jet al. Therapeutic potentials of neural stem cells treated with fluoxetine in Alzheimer’s disease. Neurochem Int. 2012;61(6):885–891. | |
Chadwick W, Mitchell N, Caroll J, et al. Amitriptyline-mediated cognitive enhancement in aged 3× Tg Alzheimer’s disease mice is associated with neurogenesis and neurotrophic activity. PLoS One. 2011;6(6):e21660. | |
Ridwan S, Bauer H, Frauenknecht K, Hefti K, von Pein H, Sommer CJ. Distribution of the hematopoietic growth factor G-CSF and its receptor in the adult human brain with specific reference to Alzheimer’s disease. J Anat. 2014;224(4):377–391. | |
Prakash A, Medhi B, Chopra K. Granulocyte colony stimulating factor (GCSF) improves memory and neurobehavior in an amyloid-β induced experimental model of Alzheimer’s disease. Pharmacol Biochem Behav. 2013;110:46–57. | |
Tsai KJ, Tsai YC, Shen CK. G-CSF rescues the memory impairment of animal models of Alzheimer’s disease. J Exp Med. 2007;204(6):1273–1280. | |
Sanchez-Ramos J, Song S, Sava V, et al. Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer’s mice. Neuroscience. 2009;163(1):55–72. | |
Shin JW, Lee JK, Lee JE, et al. Combined effects of hematopoietic progenitor cell mobilization from bone marrow by granulocyte colony stimulating factor and AMD3100 and chemotaxis into the brain using stromal cell-derived factor-1α in an Alzheimer’s disease mouse model. Stem Cells. 2011;29(7):1075–1089. | |
Parthsarathy V, Hölscher C. Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS One. 2013;8(3):e58784. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.