Back to Journals » Cancer Management and Research » Volume 7
Clinical management of grade III oligodendroglioma
Authors Simonetti G, Gaviani P, Botturi A, Innocenti A, Lamperti E, Silvani A
Received 28 April 2015
Accepted for publication 14 June 2015
Published 27 July 2015 Volume 2015:7 Pages 213—223
DOI https://doi.org/10.2147/CMAR.S56975
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
G Simonetti, P Gaviani, A Botturi, A Innocenti, E Lamperti, A Silvani
Neurooncology Unit, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
Abstract: Oligodendrogliomas represent the third most common type of glioma, comprising 4%–15% of all gliomas and can be classified by degree of malignancy into grade II and grade III, according to WHO classification. Only 30% of oligodendroglial tumors have anaplastic characteristics. Anaplastic oligodendroglioma (AO) is often localized as a single lesion in the white matter and in the cortex, rarely in brainstem or spinal cord. The management of AO is deeply changed in the recent years. Maximal safe surgical resection followed by radiotherapy (RT) was considered as the standard of care since paramount findings regarding molecular aspects, in particular co-deletion of the short arm of chromosome 1 and the long arm of chromosome 19, revealed that these subsets of AO, benefit in terms of overall survival (OS) and progression-free survival (PFS), from the addition of chemotherapy to RT. Allelic losses of chromosomes 1p and 19q occur in 50%–70% of both low-grade and anaplastic tumors, representing a strong prognostic factor and a powerful predictor of prolonged survival. Several other molecular markers have potential clinical significance as IDH1 mutations, confirming the strong prognostic role for OS. Malignant brain tumors negatively impacts on patients' quality of life. Seizures, visual impairment, headache, and cognitive disorders can be present. Moreover, chemotherapy and RT have important side effects. For these reasons, “health-related quality of life” is becoming a topic of growing interest, investigating on physical, mental, emotional, and social well-being. Understanding the impact of medical treatment on health-related quality of life will probably have a growing effect both on health care strategies and on patients.
Keywords: anaplastic oligodendroglioma, radiotherapy, chemotherapy, molecular markers, treatments toxicity
Epidemiology
Oligodendrogliomas (ODs) represent the third most common type of glioma and comprise 4%–15% of all gliomas. Because of their elevated response to chemotherapy (CHT) and their ability to be divided into prognostic subgroups based on molecular biology, this group of tumors has generated substantial interest over the past decade. Pure oligodendroglial tumors can be classified by the degree of malignancy into grades II and III, according to the WHO classification. The current WHO definition of anaplastic oligodendroglioma (AO) is “an oligodendroglioma tumor characterized by focal or diffuse histological features of malignancy and a less favorable prognosis”. Only 30% of oligodendroglial tumors have anaplastic characteristics (such as high cell density, mitosis, nuclear atypia, microvascular proliferation, and necrosis); therefore, this group of tumors is uncommon. In fact, the prevalence of AO varies from 3.5% of all malignant gliomas to 20%–54% of all oligodendroglial tumors in a reported series.1 Males are more commonly affected, with the male-to-female ratio reaching 2:1. No definite inheritance pattern or known genetic risk factors have been documented; however, occasional familial groupings of OD have been reported.2 AOs primarily occur in adults, with a median age at diagnosis ranging from 45 to 50 years, which is approximately 7 to 8 years older than those with grade II OD; this gap appears to reasonably correspond to the average time of tumor “evolution” from grade II to III WHO. In fact, ODs frequently tend to gradually grow and evolve from a low-grade, well-differentiated glioma into high-grade glioma with anaplastic features. Because these morphologic changes, which are characteristic of high-grade glioma, appear gradually within a glioma, the exact delineation of low- and high-grade OD is unclear. ODs may also present as anaplastic tumors that lack a clinically manifest low-grade precursor lesion. The reported median overall survival (OS) for pure AO is approximately 4.5 years whereas a slightly inferior reported overall survival (OS) for mixed-oligoastrocytoma may be related to a higher astrocytic component and a lower prevalence of the co-deletion of 1p/19q. ODs can be divided into prognostic subgroups based on their clinical and molecular characteristics; however, there is less evidence for AO because of the small number of cases, differences in low-grade or mixed-grade OD, and variations in diagnostic standards and treatment modalities. In the majority of studies, favorable clinical prognostic factors include a younger age, a good performance status, and frontal lobe location, and the favorable molecular prognostic factors include the co-deletion of 1p/19q. In a study by Yang et al,3 the authors analyzed the factors affecting the prognosis of 70 AO patients who were treated surgically. The results showed that CHT, age, primary or secondary tumors, preoperative Karnofsky Performance Scale scores and neurological symptoms, and the presence of epilepsy at initial presentation can correlate with prognosis; in particular, patient age appears to be one of the strongest predictors for survival, with older age associated with more aggressive tumors and a poorer prognosis.3
Molecular characteristics
AO was the first subgroup to be recognized as a discrete subgroup that is uniquely sensitive to CHT (particularly to the combined use of procarbazine, lomustine, and vincristine, also known as the procarbazine-CCNU-vincristine [PCV] schedule). However, approximately one-third of all gliomas that possess characteristics of oligodendroglial lesions do not respond to CHT. It is important to note that histopathological classification based uniquely on morphological criteria makes diagnosis subject to variability and is not perfect in predicting patient outcome. Progress in molecular techniques has allowed for the identification of a number of markers and genetic profiles that can characterize gliomas beyond their histologic criteria; in particular, today, there are a few markers that could be useful for identifying glioma subtypes and that have prognostic and predictive value. In this setting, AOs are also distinguished by a unique constellation of molecular genetic alterations that can distinguish OD from astrocytomas and other glioma subtypes. Allelic losses of chromosomes 1p and 19q represent an important molecular signature of OD and occur in 50%–70% of both low-grade and anaplastic tumors.4 Tumors that contain the combined allelic loss of chromosomes 1p and 19q are associated with a better prognosis and prolonged survival, and they exhibit increased chemosensitivity with a better response to treatment. In 2012, the follow-up results of more than 11 years from two distinct studies (RTOG 9402 and EORTC 26951) showed that the addition of CHT to radiotherapy (RT) could benefit patients with AO tumors with 1p/19q co-deletion in terms of OS.5,6 It is known that the partial deletion of 1p or 19q lacks prognostic value.7
An interesting study by Walker et al8 evaluated the role of molecular alterations in 100 oligodendroglial tumors. The loss of 1p and 19q was noted in 46% of tumors with oligodendroglial component and in up to 70% of AO. As in the other study, the presence of a p53 mutation was inversely associated with 1p and 19q loss. This study confirmed that 1p and 19q loss is a strong prognostic factor and a powerful predictor of prolonged survival. In contrast, the loss of all or part of chromosome 10 was less common in OD than in astrocytomas and has been associated with a poor prognosis.8 Moreover the molecular profiling of AO tumors has identified several other markers with potential clinical significance, such as mutations of IDH and CDKN2A, as well as the candidate tumor suppressor genes targeted by losses of 1p and 19q genes, CIC on 19q13.2 and FUBP1 on 1p31.1, that have only recently been discovered; however, these markers require further validation before they can be implemented as clinical decision-making tools.9 Mutations in Alpha Thalassemia/Mental Retardation Syndrome X-linked (ATRX) and mutations in CIC and FUBP1, which represent potent regulators of cell growth, have been studied in glioma subtypes. ATRX alterations are associated with an alternative lengthening of telomeres. The mutation analysis of ATRX in series of adult gliomas revealed a high incidence of this alteration in diffuse astrocytomas (63%) compared with the lower incidence observed in pure ODs (20%).10 These findings suggest a possible role for ATRX in distinguishing diffuse astrocytic from oligodendroglial tumors. CIC and FUBP1 mutations occurred frequently in ODs (46% and 24%, respectively) but rarely in astrocytomas or oligoastrocytomas (<10%). Conversely, the vast majority of ODs carry a TERT promoter mutation.11 A strong prognostic role was confirmed for IDH 1 mutations in AO tumors with regard to OS without a predictive role for CHT outcome. In particular, in a study by van den Bent et al, a subgroup of 44/91 patients with AO tumors presented with an IDH1 mutation, and this population exhibited a significantly better survival: the 2-year survival rate for the IDH1-intact tumors was 38.3% (95% confidence interval (CI), 24.6–51.8) versus 88.4% (95% CI, 74.3–95.0) for the IDH1-mutated tumors. IDH1 mutations were strongly associated with 1p/19q co-deletion and MGMT promoter methylation. However, the IDH1 mutations showed no relation to the outcome of adjuvant PCV CHT.12
In a multivariate analysis, wild-type IDH is an independent and poor prognostic factor in anaplastic gliomas.13 PTEN deletion and overexpression of p53 protein are typically less frequent in oligodendroglial tumors than in astrocytic tumors.14 EGFRvIII is an EGFR mutant that is expressed in approximately 20%–30% of glioblastoma multiforme (GBM) cases. The EGFRvIII mutant is most common in primary GBM and is rare in secondary GBM, and in patients with anaplastic ODs, the variant is very rare. The prognostic role of this mutation is controversial. Importantly, because EGFRvIII is not expressed in normal tissues, it could be an effective target for immunotherapy. Currently, there are two studies evaluating the therapeutic effect of an EGFRvIII-directed vaccine, rindopepimut, on newly diagnosed or recurrent GBM.15
Diagnosis
The vast majority of all oligodendroglial tumors occur in the supratentorial brain, with the frontal lobe the most common site overall (50%–65%). The temporal lobe (47%) is the second most common location, followed by the parietal lobe (7%–20%) and occipital lobe (1%–4%). Other sites include the cerebellum (3%), brainstem, spinal cord (1%), leptomeninges, cerebellopontine angle, cerebral ventricles, retina, and optic nerve. Multiple lobar involvement is possible. Infratentorial or deep nuclei localization might suggest a more aggressive phenotype.16 The most common symptoms that may occur in a patient affected by AO depend primarily on the tumor location. Seizures are more closely associated with lesions in the frontal, parietal, and temporal lobes; hemiparesis and sensory neglect may also be present in fronto-parietal lesions. Visual impairment and hemianopsia could be present and are generally related to temporal lobe neoplasms. The symptoms of increased intracranial pressure, such as headache, visual disturbances, and papilledema, are generally associated with intraventricular lesions. Although histopathology is essential in the diagnosis and staging of tumor, grade III gliomas are often heterogeneous and have different degrees of malignant degeneration within the same lesion;17 missing the high-grade areas during the biopsy could lead to an underestimation of the actual degree of neoplasm. For this reason, radiological examinations have become an integral part of the diagnostic and therapeutic pathways and ensure a high diagnostic accuracy. Magnetic resonance imaging (MRI)-based imaging is, in fact, a powerful technique that may contribute to assessing the treatment response and improving the clinical management of gliomas.17
On conventional MRI, including axial T1, T2-fast spin echo, T2-fluid attenuated inversion recovery, as well as postcontrast T1-weighted images, which are generally obtained on 1.5–3 T, lesions appear hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging; on postcontrast imaging, lesions could show a “ring-like enhancement” that mimics a glioblastoma. In addition, perfusion imaging can be obtained to assess the vascularization of the lesion, which may correlate with grading and aggressiveness and may show an increased cerebral blood volume.17,18 Spectroscopy can be used to help determine the malignancy grade. As a general rule, as malignancy increases, a decrease in both N-acetylaspartate and creatine can be observed with an increase in colin. Moreover, a peak of lactate and lipids can be found. To improve accuracy, the spectroscopic voxel should be placed over an enhancing region of the tumor, thus avoiding areas of necrosis, hemorrhage, calcification, or cysts.19
Diffusion-weighted imaging shows typically no diffusion restriction in oligodendroglial tumors. In addition, diffusion-weighted imaging has been proposed to distinguish tumor progression from radionecrosis. In the literature, there are contradictory data.20 In fact, the apparent diffusion coefficient values were found to be significantly lower in cases of tumor recurrence compared with radionecrosis in some studies; however, in other series, no significant difference was found between the groups. Moreover, in a study by Sundgren, the mean apparent diffusion coefficient values were found to be significantly higher in the recurrence group.21
Even if MRI is the preferred imaging modality, computed tomography (CT) is often required to confirm calcification and hemorrhages, which are frequently present in AO. CT is useful in oligodendroglial tumors for the detection of calcifications, for example. In fact, oligodendroglial tumors have a frequency of calcifications up to 80%. Standard imaging MRI sequences are considered inferior to CT in detecting and characterizing intracranial calcification, even if there are promising data on susceptibility imaging.22 Moreover, susceptibility-weighted imaging and high-resolution magnetic resonance (MR) spectroscopy have been used to distinguish the genotypic profiles of ODs; however, there is substantial overlap in patients with grade III ODs. It appears that the integration of regional cerebral blood volume and high-resolution MR spectroscopy indices may aid in the detection of ODs with 1p/19q loss of heterozygosity; in particular, in lesions that grow as compact and hypercellular tumors, spectroscopy shows elevated levels of Cho/Cr, and there is a significantly higher susceptibility effect.23
Treatment
Role of surgery
The first approach in AO management is represented by maximal safe surgical resection. The goals of an extended exeresis are manifold: to obtain a precise histopathology diagnosis; to perform molecular analysis; and, often, to improve tumor-related clinical symptoms, such as intracranial pressure or compression. In particular, a resection of >90% of the contrast-enhancing volume appears to improve both progression-free survival (PFS) and OS,16 thereby reducing the incidence of recurrence and the risk of malignant transformation.24–27 In the literature, a prospective study conducted on 560 patients with high-grade gliomas demonstrated that resection is a strong prognostic factor (P<0.0001) compared with biopsy.28
However, most of the available data were derived from small, uncontrolled, retrospective studies, where potential bias, such as tumor location, age, Karnofsky Performance Scale, and patient comorbidities, may alter or confound the true outcome. Moreover, a gross total resection is not always possible due to the infiltrative characteristics of the tumor, which often involves eloquent areas of the brain.29,30
Further randomized trials are needed to establish the association between PFS–OS and the extent of resection in malignant brain tumors.31,32
Hence, as a general rule, it is recommended to perform the largest resection possible to minimize the post-operative sequelae and complications that could reduce performance status. To reach this goal, the surgeon can employ a series of preoperative examinations, such as MR spectroscopy, single-photon emission CT, or positron emission tomography, functional MRI and anatomical studies (Diffuse Tensor Imaging fiber), to display the relationship between the fiber tracts and tumor as well as intraoperative examinations, such as neuronavigation, intraoperative MRI, and brain mapping techniques, to identify and protect functional areas and to maximize the safe resection areas, thereby reducing the risk of damage.30 The resection beyond the area of MR signal abnormalities imaging up to 20 mm is possible with awake surgery: cortical and subcortical electrostimulation mapping allows for a maximal degree of resection while preserving a good quality of life (QoL) and increasing OS by delaying further malignant transformation.30,33 In contrast, biopsy is strongly recommended for cases in which tumor resection is unsafe because the availability of a tissue specimen is useful for assessing the histopathological and molecular structure of the tumor and for planning further treatment.
Role of RT and CHT
If surgery represents the primary therapeutic approach, AOs clearly require a multidisciplinary collaboration that includes surgery, RT, and CHT.
However, the timing and the optimal sequences of treatments have not yet been clarified.
Since the 1990s, radiation therapy has represented an integral part of AOD management.28 The importance and the efficacy of RT were demonstrated in retrospective studies and in recent randomized prospective trials,34,35 thus reinforcing the concept that postoperative irradiation is highly recommended in patients with AO. The effectiveness of RT has not been assessed in randomized trials that specifically examined patients with oligodendroglial tumors. However, external beam RT is usually administered in standard fractions of 1.8–2 Gy and can reach a total dose in the range of 54–60 Gy.27 There are no data suggesting a beneficial effect of doses higher than 60 Gy, although retrospective studies have given conflicting results regarding whether postoperative RT prolongs survival in low-grade oligodendroglial tumors. Nevertheless, postoperative RT has been considered as an integral component of the treatment of patients with AO tumors.
However, whole brain irradiation inevitably affects the surrounding healthy tissue, with well-known negative effects on cognitive functions and neurocognitive sequelae, especially in long-term survivors.36
Stereotactic external beam RT represents an innovative method to provide localized radiation. In particular, stereotactic radiosurgery delivers high doses of radiation with high accuracy in few sessions while saving healthy tissue and minimizing side effects. stereotactic radiosurgery has also recently been used in neuro-oncological fields to treat primary malignant brain tumors as well as brain metastases, spinal cord tumors, and it has presented advantages for patients and for their QoL.37
The role of CHT has been widely discussed, and to date, it remains somewhat controversial.
It is well known that gliomas originating from oligodendrocytes are highly sensitive to nitrosurea-based CHT, such as PCV schedule.38 Originally, chemosensitivity was demonstrated39–44 in patients who relapsed after RT; then, PCV became the subject of study in numerous clinical trials as the chemotherapeutic regimen used as a first-line approach.45,46 As mentioned, recent findings have indicated that AO can be stratified into prognostic groups based on chromosome 1p/19q co-deletion. Growing evidence, in fact, suggests that 1p/19q deletion is a relevant prognostic marker and predictor of chemosensitivity. In light of these findings, less of heterozygosity on chromosome 1p/19q in combination with histological examination is paramount for determining treatment and is essential for optimizing treatment decisions. A retrospective long-term analysis of two large randomized trials, EORTC 26951 and RTOG 9402,5,6 has demonstrated that patients with newly diagnosed AO/anaplastic oligoastrocytoma (AOA), which harbors 1p/19q co-deletion, benefit from the addition of PCV to standard RT. In contrast, in patients without such deletions, OS does not differ significantly.
In particular, in the RTOG 9402 trial, 148 AO/AOA patients were randomized to receive four cycles of PCV followed by RT, and 143 patients received standard RT. The mOS was 4.6 years for the CHT+RT group and 4.7 years for the RT group. After stratifying for patients with 1p/19q co-deletions, the data showed that the mOS was 14.7 years in patients treated with PCV followed by radiation and was 7.3 years in patients treated with only radiation.
In addition, in the EORTC 26951 trial, 368 AO/AOA patients were randomized to receive either RT alone or RT followed by six cycles of PCV. Patients with 1p/19q co-deletions benefited from the addition of PCV in terms of median PFS. The mOS was 112 months with RT alone but had not been reached in the RT and CHT arm for the co-deleted tumors at the time of publication. Conversely, in patients without the 1p/19q co-deletion, the mOS was not significantly different.
Recently, a large retrospective study of >1,000 anaplastic glioma conducted by Lassman et al showed that in patients treated with chemo-RT or CHT alone, temozolomide (TMZ) was used far more often than PCV from 2005 to 2007 (87% vs 2%, respectively), especially in patients with a 1p/19q co-deletion.47
However, many questions regarding the choice between PCV and TMZ remain unanswered.
Although there are no formal direct comparisons demonstrating the superiority of combined CHT (PCV) or single-agent CHT with TMZ, in clinical practice, TMZ is generally preferred; its ease of administration, good tolerability, and minimal myelotoxicity make TMZ a good alternative. If PCV is an old schedule that often induced serious hematological toxicities, then TMZ is an oral alkylating agent that is extremely well tolerated and that has become the standard of care for newly diagnosed glioblastoma. TMZ has also been employed recently in anaplastic gliomas and for the salvage treatment of grade II–IV gliomas.48
There is one other point regarding the association of RT with TMZ or PCV. A study by Speirs et al49 on 111 patients with anaplastic gliomas reported on the clinical outcomes of RT plus TMZ for anaplastic gliomas, stratified by histology and 1p/19q co-deletion. The authors demonstrated that 1p/19q co-deletion status and histology can be used to stratify this heterogeneous population into three risk groups, which suggests that RT vs TMZ may be a promising approach for both co-deleted and non-co-deleted AO/AOA, but not for anaplastic astrocytoma patients.
Furthermore, the issue regarding the optimal order of RT and CHT is also unclear. In the EORTC 26951 trial, PCV was administered after the completion of RT, whereas in the RTOG 9402 trial, four cycles of PCV CHT were administered immediately before RT. According to The National Comprehensive Cancer Network, the recommendation is to administer PCV after RT.
If strong evidence now suggests that 1p/19q co-deleted AO/AOAs should be treated with adjuvant RT+CHT, no standard treatment is yet available for tumors without the 1p/19q co-deletion. However, in the RTOG 9402 and EORTC 26951 trials, no advantages in terms of OS or PFS emerged with the addition of PCV to RT. It is likely that the ongoing Phase III CATNON trial (NCT00626990) for newly diagnosed anaplastic gliomas without 1p/19q co-deletion will provide information regarding this question.
In this type of tumor, disease recurrence is unfortunately the rule, and recurrences are typically local and often of a higher grade (ie, GBM). If possible, the option of second surgery should be always considered. Data on second-line treatment regimens in AO tumors are limited, and postoperative options largely depend on the treatment that has already been used. For patients who were not treated with CHT and who recurred after RT, alkylating CHT is the treatment of choice (TMZ or, alternatively, a nitrosourea-based regimen with no clear evidence of superiority). In patients without a clinical indication of CHT, it would be possible to consider a new hypofractionated RT (with 25–30 Gy in five fractions of 5 or 6 Gy, or with 35 Gy in ten 3.5 Gy fractions) that appears feasible if the recurrences are circumscribed.50 Other chemotherapeutic agents have been previously tested as second-line CHT in patients with anaplastic oligodendroglial tumors, including paclitaxel, irinotecan, carboplatin, and etoposide plus cisplatin, without remarkable results in term of response rates.
Several smaller studies using bevacizumab alone or in combination with other agents in patients with recurrent oligodendroglial tumors have suggested that bevacizumab is also active in these tumors.51,52
However, the exact impact of antiangiogenetic agents on the treatment of oligodendroglial tumors needs to be defined. The TAVAREC trial (NCT01164189), recently closed, on recurrent anaplastic gliomas without 1p/19q co-deletion, may clarify the possible efficacy of bevacizumab in these tumors.28
Another treatment option that was recently reported in the literature includes the use of first-line dose-intense TMZ in patients with AO. Ahluwalia et al enrolled 62 patients in a multicenter Phase II study to investigate the efficacy, in terms of PFS and OS, of 150 mg/m2 TMZ given on days 1–7 and 15–21 every 28 days for eight cycles. Although the sample was relatively small, the data are similar to those obtained following treatment with a standard dose of TMZ and TMZ followed by RT: 75% patients were stable, and the PFS reached 27 months. Stratifying patients for loss of heterozygosity, the median PFS was 62 months. Again, the positive prognostic role of 1p/19q co-deletion was confirmed in this trial.53
Complications and treatment toxicities
Epilepsy
Epileptic seizure is a relatively common disorder in patients with brain tumors, and clinical management plays an important role in seizure control and improving the patient QoL. In recent years, considerable progress has been made in the tumor-related epilepsy field, primarily after the introduction of second- and third-generation antiepileptic drugs (AEDs), which are generally well tolerated and have a low risk of interactions. It is estimated that approximately 60%–100% of patients with a low-grade glioma and 40%–60% of patients with high-grade glioma will experience a seizure during the course of the disease.54 It is well known that approximately half of all patients with low-grade gliomas show the highest incidence of seizures as one of the presenting clinical symptoms of a brain lesion, and it is considered as a good prognostic factor for survival. In contrast, the presence as well as the worsening of epileptic seizures over the course of disease could indicate tumor progression. Seizure onset depends on many factors, including tumor type, location, pathophysiology of peritumoral changes, and genetic factors; seizures can occur as simple or complex seizures with or without secondary generalization.
The main problems related to the medical management of epilepsy in patients with brain tumors relate to the exact time when an AED should be started, the refractoriness of epilepsy, and the interactions between antiepileptic and chemotherapeutic agents.
The decision to start an antiepileptic treatment remains a much-debated question. Based on the current evidence, treatment should be considered after the presence of a single seizure due to the high risk of recurrence. The decision should also be supported by a careful consideration of the presence of any epileptiform abnormalities on electroencephalography, risk factors, and lesion localization.55 In contrast, there is no indication to begin prophylactic AED in patients who have never presented with seizures.56
The choice of initial treatment is not yet standardized and depends on local clinical practice. The most commonly used AEDs are summarized in Table 1.57–61
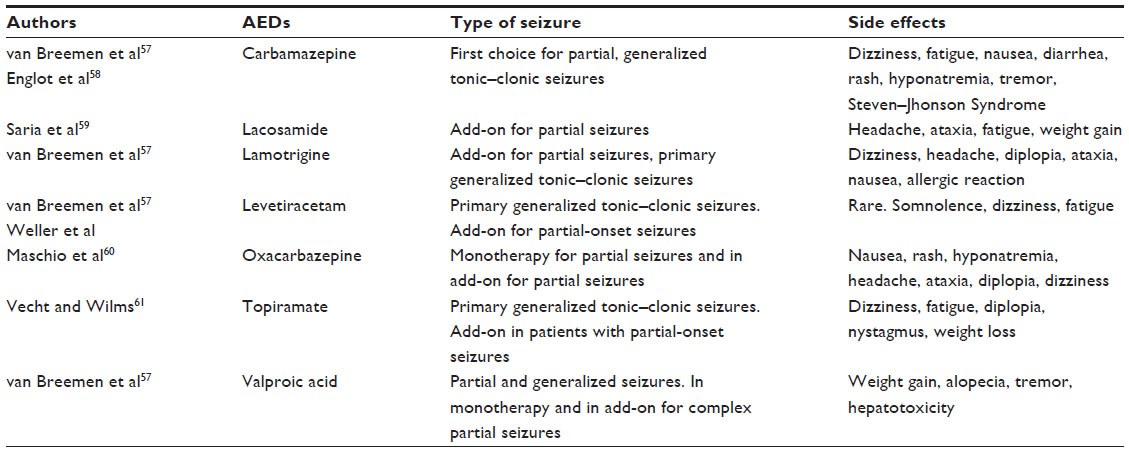 | Table 1 Efficacy and side effects related to most commonly used AEDs |
In patients with tumoral epilepsy, however, the choice of non-enzyme-inducing AED should be the rule because of the low risk of drug interactions between the antiepileptic and CHT. Hence, attention should be paid to phenobarbital, carbamazepine, and phenytoin that belong to the inducing AEDs, privileging valproic acid, lamotrigine, and oxcarbazepine. Recently, a second-generation, non-enzyme-inducing AED that is widely used is levetiracetam. Levetiracetam presents with good tolerability, a favorable safety profile, and a low risk of interaction, making this drug an excellent alternative. If the goal of AED treatment is make the patient seizure-free with a consequent improvement in QoL, then pharmacoresistance does not always allow this objective to be achieved. The International League Against Epilepsy defines pharmacoresistant epilepsy as the “failure of a patient’s seizure to respond to at least two antiepileptic medications that are appropriately chosen and used for an adequate period”. In this particular case, a polytherapy with a combination of up to three AEDs should also be considered, and any side effects should be closely monitored.
Venous thromboembolism
Venous thromboembolism (VTE), including deep vein thrombosis and pulmonary embolism (PE), represents one of the most important causes of morbidity (hospitalization, anticoagulation use, bleeding complications, increased risk of recurrent VTE, and cancer treatment delays) and mortality in cancer patients both during and beyond the perioperative period. VTE is a common complication in patients with cancer, and its frequency is particularly high in malignant glioma, as it occurs in approximately 20%–30% of such patients. The incidence of deep vein thrombosis and/or PE in patients with a brain tumor was found to be 120:100,000 – the second highest rate for any malignancy – according to the Medicare Provider Analysis and Review Record. Both retrospective and prospective studies have suggested a particularly high incidence of VTE in patients with malignant gliomas, ranging from 2% to 60%.62 The pathophysiological mechanism of VTE development is unclear, but the so-called Virchow’s triad (alterations of hemostasis and the alterations of the vessel wall and blood stasis) appears to be the basis for VTE development. Other risk factors include a histologic diagnosis of GBM, a larger tumor size (tumors produce pro-coagulation factors, and larger tumors release them in larger quantities), the presence of leg paresis (the absence of the muscle pump effect favors venous stagnation), older age (pro-coagulant factors increase with age, but anticoagulant proteins remain stable), more-lengthy surgery, CHT (reduces fibrinolytic activity) and steroids.63 A hypercoagulable state, which characterizes all patients with brain tumors, is attributed to the prothrombotic properties of the neoplastic cells through the production of procoagulant. All of these pathophysiological mechanisms, in addition to many other general factors, such as stasis, the presence of inflammation, dysproteinemia, infections, and entrapment, contribute to the hypercoagulable state.
The options for anticoagulation therapy include low- molecular-weight heparin (LMWH) and unfractionated heparin, which are generally first-choice drugs. Unfractionated heparin is reserved for patients with symptomatic PE or a high risk for hemorrhage/bleeding. LMWH is more manageable: it does not require frequent laboratory management, and it has a low potential for interactions and is simply self-administered. The newer oral anticoagulant agents, which are specific inhibitors of activated factor Xa (FXa) (rivaroxaban and apixaban) or thrombin (dabigatran), will likely further improve the current antithrombotic treatment. Anticoagulation therapy should be maintained for 3–6 months and beyond in patients with active malignant disease or with ongoing anticancer therapy. There are also mechanical methods such as intermittent pneumatic compression devices (by providing a wavelike compression on the leg, it is possible to evacuate the leg veins) and graduated compression stockings (with a continuous and graduated pressure on the whole lower extremity, it is possible to improve venous clearance and prevent venous stasis) that could both support treatment and help prevent VTE. Salmaggi et al investigated the thromboprophylaxis treatment in the perioperative period in neurooncological patients in a systematic review of the literature and suggested that the use of mechanical methods, particularly intermittent pneumatic compression, is both safe and protective for neurooncological patients who are undergoing surgery. The addition of pharmacological treatment (LMWH) is even more protective in terms of reduction of VTE, with a modest increase in the frequency of major bleeding: the dose should not exceed 4,000 IU/day, and the administration should begin on the day after surgery.63
Treatment toxicity
CHT-related toxicity
The hematological and non-hematological toxicity linked to CHT for brain tumors is generally less frequent and less debilitating than other neoplasm treatments. Alkylating agents are the most common chemotherapeutic agents that are used in the management of AO. Of these, TMZ and the combined use of procarbazine, lomustine, and vincristine (even if the last belongs to the vinca alkaloids) have similar side effects.
TMZ and PCV are frequently associated with nausea and vomiting (generally within 24 hours of administration) in almost all of patients; thus, a prophylactic anti emetic is strongly recommended prior to the administration of both TMZ and lomustine to prevent these side effects. Other gastrointestinal toxicities are rare but possible and include constipation or diarrhea, stomatitis, and anorexia. Bone marrow toxicity is the most common side effect linked to TMZ and PCV administration; severe myelosuppression can occur, with grade 3 and grade 4 neutropenia and grade 3 and grade 4 thrombocytopenia. For TMZ, as a general rule, nadir is noted between day 21 and day 28, but its incidence varies from patient to patient. For these reasons, an adequate hematologic monitoring before starting treatment is mandatory.
Once patients experience myelosuppression, they may also present with prolonged pancytopenia that can degenerate to aplastic anemia and become fatal in some cases. Moreover, the exposure to concomitant medications associated with aplastic anemia (ie, carbamazepine, phenytoin, and sulfamethoxazole/trimethoprim) can complicate the assessment.
Baseline liver function tests should also be performed; liver enzyme elevation, hyperbilirubinemia, cholestasis, and hepatitis have been reported in patients who were treated with TMZ and PCV, with a major incidence in PCV.
Fatigue is another common side effect, but it is often underestimated and undertreated.
For patients with moderate to severe fatigue, the National Comprehensive Cancer Network guidelines recommend a careful evaluation of contributing factors that can be managed, including pain, medications such as anticonvulsants and opioids, depression or anxiety, sleep disorders, anemia, nutritional deficiencies, and comorbidities.64
TMZ and PCV may have also genotoxic effects. Due to potential effects to the fetus, women should not become pregnant during treatment or in the 6 months after discontinuation. Moreover, it is strongly recommended for male patients to not to father a child up to 6 months after discontinuation and to proceed with the cryopreservation of sperm prior to treatment because of the possibility of irreversible infertility due to anticancer therapies.
RT-related toxicity
Radiation therapy plays an important role in the management of AO, but its side effects can be debilitating in the long term and can damage every tissue within its range of action. Until 1970, it was argued that human brain was resistant to radiation, observing only acute central nervous system syndromes after single doses of 30 Gy and white matter necrosis after fractionated doses of 60 Gy. A decade later, white matter necrosis with vascular abnormalities and demyelination was recognized as a dose-limiting morbidity that was evident in the majority of cases, starting at 6 months from irradiation.65,66 To date, it is well known that impairment of neurocognitive functions is not only the result of the direct effects of the tumor but also the sequelae of therapy. The sensitivity of the central nervous system to radiation therapy is dose limiting, and neurocognitive decline is very common after whole brain radiation therapy, with approximately 90% of patients having memory deficits and attention disorders, which has a clearly negative impact on patient QoL. Radiation damage can be classified as acute effects that occur in the days immediately following RT with swelling of the brain parenchyma (edema) or as late effects (7 months after), including encephalopathy with atrophy, matter changes, and demyelination. Generally, these significant side effects can be noted in patients who received high-dose radiation in a large volume.
Radiation necrosis, defined as coagulation necrosis of the white matter, is a potential long-term (6–36 months) complication of RT. The risk of induced radiation necrosis, in patients who received 60 Gy, is approximately 5%, which causes significant morbidity and neurologic deficits. In clinical practice, this situation can represent a diagnostic dilemma because the distinction between radiation necrosis and recurrent brain neoplasm is quite difficult. On MRI, in fact, the lesion may appear as an enhancing mass that mimics the presence of a neoplasm. Occasionally, PET or single-photon emission CT can help in the differential diagnosis and can show a reduction or increase in glucose metabolism in the case of radionecrosis or recurrence, respectively. It is often necessary to perform a biopsy to guarantee a diagnosis.
Although the modern RT techniques can prevent many cases of acute and late toxicity, many functional deficits, including progressive disorders of memory, attention and executive functions, are still present and have a clearly negative impact on the QoL of long-term survivors.66
Although a small proportion of OD patients are >65 years, it should be emphasized that the elderly population has a high risk of toxicities related to therapies compared with younger patients. In addition, RT could increase the risk of radiation-induced neurotoxicity. To ensure good tolerability in terms of both toxicity and QoL, hypofractionated RT has been proposed as viable treatment alternative. The benefit of hypofractionated radiation therapy is also supported by a prospective trial conducted by Keime-Guibert et al. The authors enrolled 81 GBM patients with a median age of 73 to evaluate OS in patients who were treated with supportive care alone (corticosteroids, anticonvulsant agent) vs supportive care in combination with RT (focal radiation in daily fractions of 1.8 Gy 5 days per week, for a total dose of 50 Gy to minimize the risk of neurotoxicity). The study was discontinued because of the superiority of supportive care plus RT: the mOS after 21 weeks of follow-up was 29.1 weeks in the RT plus supportive care group and 16.9 weeks in those receiving supportive care alone. Moreover, no severe adverse event related to RT was recorded.67
Health-related quality of life
Although considerable progress has been made in the neuro-oncological field, anaplastic glioma remains incurable. The addition of CHT to RT in patients with a favorable genetic profile has certainly improved the mOS, which now ranges between 2.5 and 4.7 years.68 However, it is undeniable that all anticancer therapies are linked to many side effects, such as neurotoxicity and cognitive impairments, with a consequent impact on QoL.
For these reasons, “health-related quality of life” (HRQoL) is becoming a topic of growing interest. HRQoL is a relatively new, broad, and multi-dimensional concept that includes physical, mental, emotional, and social well-being. It is usually assessed by using specific and validated questionnaires. Obtaining information regarding long-term cognitive functioning and HRQoL in patients who underwent radio-CHT could influence the choice of postoperative treatments. Although several recent studies have started to investigate this aspect and because the baseline HRQoL scores are considered as a strong independent prognostic factor for survival in cancer patients,69–71 data regarding patients with brain tumor are still lacking.
Between September 2006 and March 2009, Yavas et al enrolled 118 patients with high-grade glioma. Their HRQoL scores were assessed throughout the study using a specific validate psychological scale (European Organization for Research and Treatment of Cancer Quality of Life Questionnaire 30 EORTC-C30; Brain Cancer Module-20 BN-20; Minimental State Examination; Hospital Anxiety and Depression Scale [HADS]) at baseline, at the end of RT and during follow-up. The data showed that for the global EORTC-C30 and BN-20 scores, the physical, emotional function, insomnia, and appetite loss scores were significantly related to disease progression and to seizure and leg weakness, drowsiness and bladder control, motor dysfunction, future uncertainty, visual disorder, and communication deficit symptoms. Minimental State Examination recorded an evident worsening of the orientation, attention, calculation, and language scores. For the anxiety and depression scores, the authors did not find significant differences between baseline and after 18 months of follow-up. The authors concluded that many changes afflicted patients with high-grade glioma over the course of the disease, most of which are related to disease progression.72 In addition, Habets et al68 evaluated the cognitive functioning and HRQoL in a cohort of long-term survivors who were affected by AO by using EORTC QLQ-C30 EORTC QLQ-BN20 neuropsychological tests. The results were compared with healthy controls and with the results obtained at baseline 2.5 years prior. The data collected showed a worse HRQoL compared with the control, which was similar to the data obtained at baseline. As the authors stated, the data are highly variable; however, the patients without progression had a relatively stable HRQoL, but a substantial proportion of the survivors suffered from cognitive impairment. Due to the small sample size, no indication regarding the effect of the addition of PCV on cognition or HRQoL can be offered.
Future studies are needed to explore the associations between treatment and HRQoL/cognitive functioning in the long term. Understanding the impact of medical treatment on HRQoL will likely have increasing effects both on health care strategies and on patients. This useful information could play a key role in the neurooncological field and could help health care professionals to identify patients who require psychosocial intervention programs to maintain or improve their QoL.
Disclosure
The authors report no conflicts of interest in this work.
References
Koeller KK, Rushing EJ. From the archives of the AFIP: oligodendroglioma and its variants: radiologic-pathologic correlation. Radiographics. 2005;25(6):1669–1688. | |
Kros JM, Lie ST, Stefanko SZ. Familial occurrence of polymorphous oligodendroglioma. Neurosurgery. 1994;34(4):732–736. | |
Yang LS, Huang FP, Zheng K, et al. Factors affecting prognosis of patients with intracranial anaplastic oligodendrogliomas: a single institutional review of 70 patients. J Neurooncol. 2010;100(1):113–120. | |
Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473–1479. | |
van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31(3):344–350. | |
Cairncross G, Wang M, Shaw E, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31(3):337–343. | |
Hofer S, Rushing E, Preusser M, et al. Molecular biology of high grade gliomas: what should the clinician know? Chin J Cancer. 2014; 33(1):4–7. | |
Walker C, du Plessis DG, Joyce KA, et al. Molecular pathology and clinical characteristics of oligodendroglial neoplasms. Ann Neurol. 2005;57:855–865. | |
Sahebjam S, McNamara MG, Mason WP. Emerging biomarkers in anaplastic oligodendroglioma: implications for clinical investigation and patient management. CNS Oncol. 2013;2(4):351–358. | |
Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3(7):709–722. | |
David E Reuss, Felix Sahm, Daniel Schrimpf, et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015;129:133–146. | |
van den Bent MJ, Dubbink HJ, Marie Y, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res. 2010;16(5):1597–1604. | |
Shibahara I, Sonoda Y, Shoji T, et al. Malignant clinical features of anaplastic gliomas without IDH mutation. Neuro Oncol. 2015;17(1):136–144. | |
Appin CL, Gao J, Chisolm C, et al. Glioblastoma with oligodendroglioma component (GBM-O): molecular genetics and clinical characteristics. Brain Pathol. 2013;23(4):454–461. | |
Siegal T. Clinical impact of molecular biomarkers in gliomas. J Clin Neurosci. 2015;22(3):437–444. | |
Polivka J Jr, Polivka J, Rohan V, et al. New treatment paradigm for patients with anaplastic oligodendroglial tumors. Anticancer Res. 2014; 34(4):1587–1594. | |
Laws ER, Parney IF, Huang W, et al. Survival following surgery and prognostic factors for recently diagnosed malignant glioma: data from the Glioma Outcomes Project. J Neurosurg. 2003;99(3):467–473. | |
Mangla R, Ginat DT, Kamalian S, et al. Correlation between progression free survival and dynamic susceptibility contrast MRI perfusion in WHO grade III glioma subtypes. J Neurooncol. 2014;116(2):325–331. | |
John R. Hesselink, Fundamentals of MR Spectroscopy. Available from: http://spinwarp.ucsd.edu/NeuroWeb/Text/mrs-TXT.htm. Accessed July 21, 2015. | |
Raimbault A, Cazals X, Lauvin MA, et al. Radionecrosis of malignant glioma and cerebral metastasis: a diagnostic challenge in MRI. Diagn Interv Imaging. 2014;95(10):985–1000. | |
Sundgren PC. MR spectroscopy in radiation injury. Am J Neuroradiol. 2009;30(8):1469–1476. | |
Zulfiqar M, Dumrongpisutikul N, Intrapiromkul J, et al. Detection of intratumoral calcification in oligodendrogliomas by susceptibility-weighted MR imaging. AJNR Am J Neuroradiol. 2012;33(5):858–864. | |
Chawla S, Krejza J, Vossough A, et al. Differentiation between oligodendroglioma genotypes using dynamic susceptibility contrast perfusion-weighted imaging and proton MR spectroscopy. AJNR Am J Neuroradiol. 2013;34(8):1542–1549. | |
Sanai N, Berger MS. Glioma extent of resection and its impact on patient outcome. Neurosurgery. 2008;62(4):753–764; discussion 264–266. | |
Smith JS, Chang EF, Lamborn KR, et al. Role of extent of resection in the long-term outcome of low-grade hemispheric gliomas. J Clin Oncol. 2008;26(8):1338–1345. | |
Brandes A. Linee Guida Neoplasie Cerebrali. Milano: Associazione Italiana di Oncologia Medica (AIOM); 2012. | |
Brandes A. Linee Guida Neoplasie Cerebrali. Milano: Associazione Italiana di Oncologia Medica (AIOM); 2014. | |
Roth P, Wick W, Weller M. Anaplastic oligodendroglioma: a new treatment paradigm and current controversies. Curr Treat Opt Oncol. 2013;14(4):505–513. | |
Anderson MD, Gilbert MR. Treatment recommendations for anaplastic oligodendrogliomas that are codeleted. Oncology. 2013;27(4):315–320, 322. | |
Maris D, Nica D, Mohan D. Multidisciplinary management of adult low grade gliomas. Chirurgia. 2014;109(5):590–599. | |
Tsitlakidis A, Foroglou N, Venetis CA, et al. Biopsy versus resection in the management of malignant gliomas: a systematic review and meta-analysis. J Neurosurg. 2010;112(5):1020–1032. | |
Veeravagu A, Jiang B, Ludwig C, et al. Biopsy versus resection for the management of low- grade gliomas. Cochrane Database Syst Rev. 2013;4. | |
Duffau H. The challenge to remove diffuse low-grade gliomas while preserving brain functions. Acta Neurochir. 2012;154(4):569–574. | |
van den Bent MJ, Carpentier AF, Brandes AA, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006;24:2715–2722. | |
Cairncross G, Berkey B, Shaw E, et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group trial 9402. J Clin Oncol. 2006;24:2707–2714. | |
Mulhern RK, Merchant TE, Gajjar A, et al. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004;5(7):399–408. | |
Combs SE, Widmer V, Thilmann C, et al. Stereotactic radiosurgery (SRS): treatment option for recurrent glioblastoma multiforme (GBM). Cancer. 2005;104(10):2168–2173. | |
Cairncross JG, Macdonald DR, Ramsay DA. Aggressive oligodendroglioma: a chemosensitive tumor. Neurosurgery. 1992;31(1):78–82. | |
Brandes AA, Tosoni A, Vastola F, et al. Efficacy and feasibility of standard procarbazine, lomustine, and vincristine chemotherapy in anaplastic oligodendroglioma and oligoastrocytoma recurrent after radiotherapy. A Phase II study. Cancer. 2004;101:2079. | |
Cairncross G, Macdonald D, Ludwin S, et al. Chemotherapy for anaplastic oligodendroglioma. National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 1994;12:2013. | |
van den Bent MJ, Kros JM, Heimans JJ, et al. Response rate and prognostic factors of recurrent oligodendroglioma treated with procarbazine, CCNU, and vincristine chemotherapy. Dutch Neuro-oncology Group. Neurology. 1998;51:1140. | |
Soffietti R, Rudà R, Bradac GB, Schiffer D. PCV chemotherapy for recurrent oligodendrogliomas and oligoastrocytomas. Neurosurgery. 1998;43:1066. | |
Mason WP, Krol GS, DeAngelis LM. Low-grade oligodendroglioma responds to chemotherapy. Neurology. 1996;46:203. | |
Buckner JC, Gesme D Jr, O’Fallon JR, et al. Phase II trial of procarbazine, lomustine, and vincristine as initial therapy for patients with low-grade oligodendroglioma or oligoastrocytoma: efficacy and associations with chromosomal abnormalities. J Clin Oncol. 2003;21:251. | |
Brandes AA, Tosoni A, Cavallo G, et al. Correlations between O6-methylguanine DNA methyltransferase promoter methylation status, 1p and 19q deletions, and response to temozolomide in anaplastic and recurrent oligodendroglioma: a prospective GICNO study. J Clin Oncol. 2006;24:4746. | |
van den Bent MJ, Taphoorn MJ, Brandes AA, et al. Phase II study of first-line chemotherapy with temozolomide in recurrent oligodendroglial tumors: the European Organization for Research and Treatment of Cancer Brain Tumor Group Study 26971. J Clin Oncol 2003;21:2525. | |
Lassman AB, Iwamoto FM, Cloughesy TF, et al. International retrospective study of over 1000 adults with anaplastic oligodendroglial tumors. Neuro Oncol. 2011;13(6):649–659. | |
Wick W, Wiestler B, Platten M. Treatment of anaplastic glioma. Cancer Treat Res. 2015;163:89–101. | |
Speirs CK, Simpson JR, Robinson CG, et al. Impact of 1p/19q codeletion and histology on outcomes of anaplastic gliomas treated with radiation therapy and temozolomide. Int J Radiat Oncol Biol Phys. 2015;91(2):268–276. | |
Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15(9):e395–e403. | |
Taillibert S, Vincent LA, Granger B, et al. Bevacizumab and irinotecan for recurrent oligodendroglial tumors. Neurology. 2009;72:1601–1606. | |
Chamberlain MC, Johnston S. Bevacizumab for recurrent alkylator- refractory anaplastic oligodendroglioma. Cancer. 2009;115:1734–1743. | |
Ahluwalia MS, Xie H, Dahiya S, et al. Efficacy and patient-reported outcomes with dose- intense temozolomide in patients with newly diagnosed pure and mixed anaplastic oligodendroglioma: a phase II multicenter study. J Neurooncol. 2015;122(1):111–119. | |
Kahlenberg CA, Fadul CE, Roberts DW, et al. Seizure prognosis of patients with low-grade tumors. Seizure. 2012;21(7):540–545. | |
Perucca E. Optimizing antiepileptic drug treatment in tumoral epilepsy. Epilepsia. 2013;54(Suppl 9):97–104. | |
Glantz MJ, Cole BF, Forsyth PA, et al. Practice parameter: anticonvulsant prophylaxis in patients with newly diagnosed brain tumors. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;54(10):1886–1893. | |
van Breemen MS, Rijsman RM, Taphoorn MJ, et al. Efficacy of anti-epileptic drugs in patients with gliomas and seizures. J Neurol. 2009;256(9):1519–1526. | |
Englot DJ, Berger MS, Chang EF, et al. Characteristics and treatment of seizures in patients with high-grade glioma: a review. Neurosurg Clin N Am. 2012;23(2):227–235. | |
Saria MG, Corle C, Hu J, et al. Retrospective analysis of the tolerability and activity of lacosamide in patients with brain tumors: clinical article. Neurosurgery. 2013;118(6):1183–1187. | |
Maschio M, Dinapoli L, Sperati F, et al. Oxcarbazepine monotherapy in patients with brain tumor-related epilepsy: open-label pilot study for assessing the efficacy, tolerability and impact on quality of life. J Neurooncol. 2012;106(3):651–656. | |
Vecht CJ, Wilms EB. Seizures in low- and high-grade gliomas: current management and future outlook. Expert Rev Anticancer Ther. 2010; 10(5):663–669. | |
Simonetti G, Trevisan E, Silvani A, et al. Safety of bevacizumab in patients with malignant gliomas: a systematic review. Neurol Sci. 2014;35(1):83–89. | |
Salmaggi A, Simonetti G, Trevisan E, et al. Perioperative thromboprophylaxis in patients with craniotomy for brain tumours: a systematic review. J Neurooncol. 2013;113(2):293–303. | |
Schiff D, Lee EQ, Nayak L, et al. Medical management of brain tumors and the sequelae of treatment. Neuro Oncol. 2015;17(4):488–504. | |
Schultheiss TE, Stephens LC. Permanent radiation myelopathy. Br J Radiol. 1992;65:737–753. | |
Greene-Schloesser D, Robbins ME. Radiation-induced cognitive impairment – from bench to bedside. Neuro Oncol. 2012;14(Suppl 4):37–44. | |
Keime-Guibert F, Chinot O, Taillandier L, et al. Radiotherapy for glioblastoma in the elderly. N Engl J Med. 2007;356(15):1527–1535. | |
Habets EJ, Taphoorn MJ, Nederend S. Health-related quality of life and cognitive functioning in long-term anaplastic oligodendroglioma and oligoastrocytoma survivors. J Neurooncol. 2014;116(1):161–168. | |
Osoba D, Brada M, Prados MD, et al. Effect of disease burden on health-related quality of life in patients with malignant gliomas. Neuro Oncol. 2000;2(4):221–228. | |
Montazeri A, Milroy R, Hole D, et al. Quality of life in lung cancer patients: as an important prognostic factor. Lung Cancer. 2001; 31(2–3):233–240. | |
Kaasa S, Mastekaasa A, Lund E. Prognostic factors for patients with inoperable non-small cell lung cancer, limited disease. The importance of patients’ subjective experience of disease and psychosocial well-being. Radiother Oncol. 1989;15(3):235–242. | |
Yavas C, Zorlu F, Ozyigit G, et al. Health-related quality of life in high-grade glioma patients: a prospective single-center study. Support Care Cancer. 2012;20(10):2315–2325. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.