Back to Journals » Drug Design, Development and Therapy » Volume 10
Clinical implications for substandard, nonproprietary medicines in multiple sclerosis: focus on fingolimod
Authors Correale J ![]() , Chiquete E, Boyko A, Beran R, Barahona Strauch J, Milojevic S, Frider N
, Chiquete E, Boyko A, Beran R, Barahona Strauch J, Milojevic S, Frider N
Received 20 February 2016
Accepted for publication 4 May 2016
Published 30 June 2016 Volume 2016:10 Pages 2109—2117
DOI https://doi.org/10.2147/DDDT.S106802
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Jorge Correale,1 Erwin Chiquete,2 Alexey Boyko,3 Roy G Beran,4–6 Jorge Barahona Strauch,7,8 Snezana Milojevic,9 Nadina Frider10
1Department of Neurology, Raúl Carrea Institute for Neurological Research, Foundation for the Fight against Infant Neurological Illnesses (FLENI), Buenos Aires, Argentina; 2Department of Neurology and Psychiatry, Salvador Zubirán National Institute of Medical Sciences and Nutrition, Mexico City, Mexico; 3Clinical and Research Center “MS and Other Demyelinating Diseases” at the Neuroclinical Hospital, Department of Neurology, Neurosurgery and Medical Genetics of the Pirogov Russian National Research Medical University, Moscow, Russia; 4South Western Clinical School, University of New South Wales, Liverpool, 5Department of Neurology, Liverpool Hospital, Sydney, NSW, 6School of Medicine, Griffith University, Southport, QLD, Australia; 7Department of Neurology, Clínica Alemana de Santiago, 8School of Medicine, Universidad del Desarrollo, Santiago, Chile; 9Novartis Pharma AG, Basel, Switzerland; 10Novartis Latin America and Canada Region, Buenos Aires, Argentina
Abstract: Both proprietary and nonproprietary medicines are expected to undergo rigorous preapproval testing and both should meet stringent health authority regulatory requirements related to quality to obtain approval. Nonproprietary (also known as copy, or generic) medicines, which base their authorization and use on the proprietary documentation and label, are often viewed as a means to help lower the cost and, thus, increase patient access. If these medicines fail to meet quality standards, such as good manufacturing practice and bioequivalence (in humans), they are then defined as substandard copies and can pose serious risks to patients in terms of safety and efficacy. Potentially noncontrolled or different manufacturing process and excipients in nonproprietary medicines may result in poor batch-to-batch reproducibility (accurate and consistent quantity of each ingredient in each capsule/tablet) and lower quality. Substandard, nonproprietary copies of medicines that are immunomodulatory or immunosuppressive are of concern to patients due to their possible untoward safety and lack of efficacy events. This article reviews the potential risks associated with nonproprietary medicines that do not meet the regulatory requirements of the United States Food and Drug Administration, the European Medicines Agency, or the World Health Organization. The clinical implications for patients are described. This article focuses on nonproprietary medicines for multiple sclerosis, particularly fingolimod, that are not identical to proprietary versions and could thus fail to meet efficacy expectations or have different impact on the safety of patients with multiple sclerosis.
Keywords: bioequivalence, fingolimod, multiple sclerosis, proprietary, substandard copies, toxicity
Introduction
Safe, high-quality medicines are essential to ensure optimal clinical impact for patients. Use of ineffective, poor-quality, harmful medicines can cause therapeutic failure, disease exacerbation, resistance to medicines, and sometimes death.1,2 It undermines confidence in health systems, health professionals, pharmaceutical manufacturers, and distributors.1 Proprietary medicines follow new drug application review requirements (Figure 1),3 undergo preapproval testing in animal studies and clinical trials, must meet stringent health authority regulatory requirements for quality, and are submitted to long-term safety monitoring (Figure 2).2,4–6
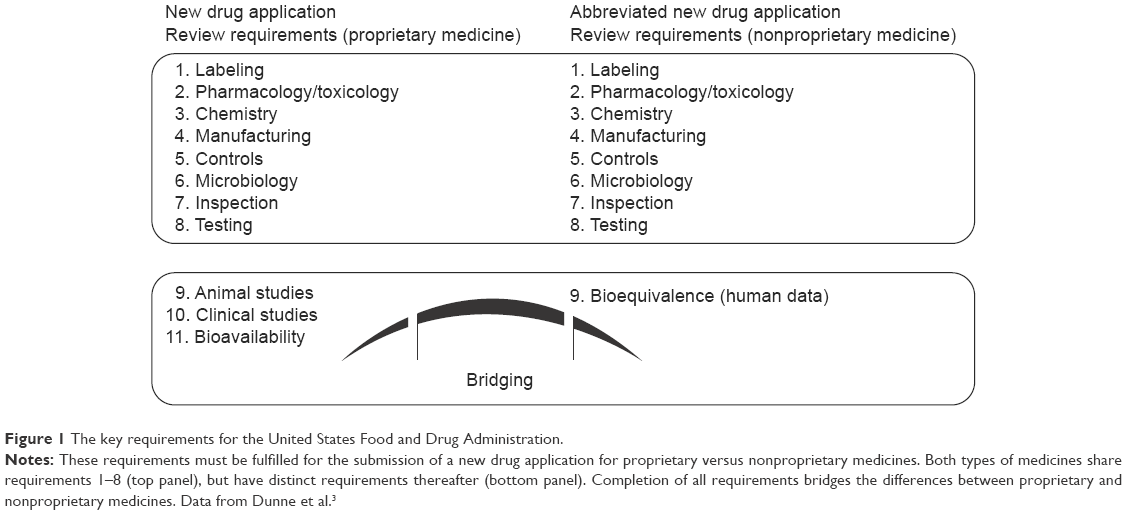 | Figure 1 The key requirements for the United States Food and Drug Administration. |
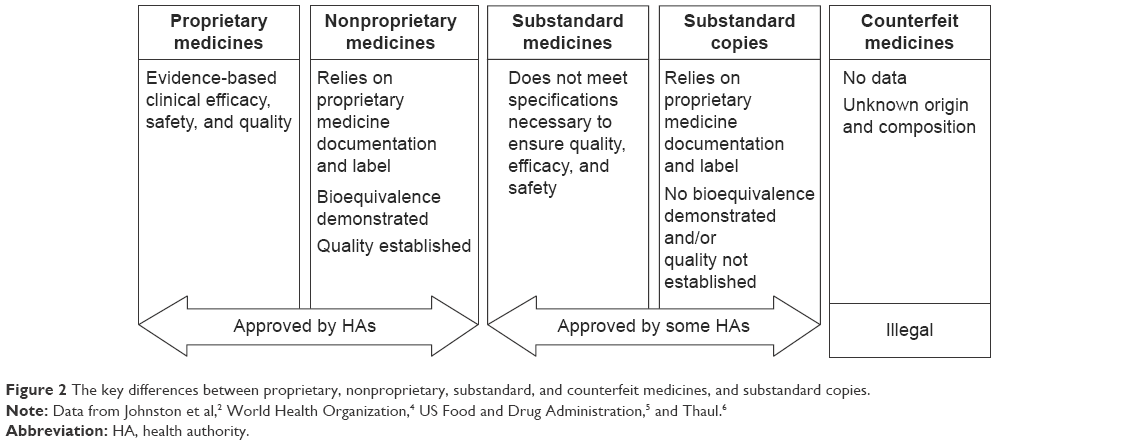 | Figure 2 The key differences between proprietary, nonproprietary, substandard, and counterfeit medicines, and substandard copies. |
Nonproprietary (also known as copy, or generic) medicines must fulfill the requirements on quality and undergo good manufacturing practice before approval. In contrast to biologic copies, most synthetic copies must be additionally tested in bioequivalence studies.7,8 Substandard medicines are genuine medicines produced by manufacturers authorized by the national medicines regulatory authorities that do not meet the quality specifications set for them by national standards (Figure 2).2,4–6 Such medicines are prevalent in regions where health authorities do not enforce registration regulations that are as stringent as those enforced by the United States Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the World Health Organization (WHO), and include defined requirements for quality standards. Substandard medicines can be approved by local health authorities.2 Globally, ~15% of medicines produced are counterfeit and substandard;9 rates in Asia and Africa reach 40%–50%.9,10 The proportion in highly regulated markets (Australia, Canada, the European Union [EU], and the US) is ~1%. This is likely to be an underestimation as demonstrated by a recent increase in reports of substandard medicine recalls and investigations into these medicines.9,11,12 India and China reportedly supply 50%–70% of active pharmaceutical ingredients (APIs) used in the EU.13 Medicines imported into the EU should comply with EU good manufacturing practice for active substances as addressed in Directive 2011/62/EU.14
The contribution of nonproprietary substandard copies resulting in cases of toxicity, disability, death, failure to slow or prevent disease progression, and waste of public financial resources has been reviewed.1,2,7 Quality issues include: cleavage, deamination, and oxidation not present in the proprietary product;15 failed thin-layer chromatography and disintegration tests to determine the levels of specified and unspecified impurities;16,17 determination of genotoxic impurities;16 and absence of, or variations in, the APIs18 (Table 1). Copy manufacturers have also experienced problems leading to product recalls and withdrawals.19
 | Table 1 Examples of a link between quality parameters and their potential clinical impact |
Substitution with nonproprietary carbamazepine (Tegretol®; Novartis Pharmaceuticals, East Hanover, NJ, USA), a common antiepileptic medication, resulted in case reports of breakthrough seizures and higher incidence of neurological side effects and skin rash.7 Carbamazepine has a narrow therapeutic index and demands careful patient monitoring, in addition to its low water solubility and nonlinear pharmacokinetics. Clinical failure, higher side effect incidences (dizziness and ataxia), and changes in dissolution characteristics have been reported with carbamazepine copies.20 A US publication reported that 53 lots (70 million tablets) of carbamazepine 200 mg were recalled by the manufacturer because of concerns over reports of clinical failures.21
Proprietary medicine manufacturers may encounter similar problems, as exemplified by a case involving proprietary lamotrigine (Lamictal®; GlaxoSmithKline, Research Triangle Park, NC, USA), an antiepileptic medication.22 Over a defined period, reports of safety issues (fatigue, ataxia, impaired cognition, and unacceptably high levels of lamotrigine) were documented in patients with epilepsy using this medicine. It emerged that the manufacturer had changed the manufacturing site, which may have resulted in a change in compound excipients’ source. The finished product was not subjected to the same rigorous testing as the product manufactured at the initial site.
There are regional inconsistencies regarding the nomenclature for copies,23,24 which pose extreme difficulties for comparative cross-national studies on policies.25 In Latin America, many policymakers, patients, and prescribing physicians interchange the terms “similar” and “generic”. There are differences between countries in promoting the use of copies: some health authorities promote their use if the proprietary medicines are off patent. In Mexico, locally produced copies have legally explicit advantages for production, marketing, and public tendering for access to the health care system. These measures promote economic growth and technological progress in developing countries.
This article reviews the clinical implications of substandard copy (nonproprietary) multiple sclerosis (MS) medicines that do not meet the FDA, EMA, and WHO regulatory requirements. In MS, which is treated using immunomodulatory medicines, substandard copies can expose patients to unnecessary risks, as demonstrated by reports of efficacy and tolerability problems with β-interferon biosimilars.26,27
Importance of bioequivalence studies
The concepts of bioequivalence and interchangeability of proprietary and nonproprietary medicines have important implications for manufacturers seeking regulatory approval. Statistical testing of bioequivalence is a key regulatory requirement mandated by the FDA, EMA, and WHO.28–30 The guidelines contain safety precautions comparable with the risk evaluation and mitigation strategy for the proprietary medicine.31 Health authorities that do not require bioequivalence to be demonstrated for registered copies are at increased risk of substandard medicines reaching the market.
A bioequivalence study typically compares the copy with the proprietary medicine in single-dose studies involving small numbers of healthy volunteers. Bioequivalence frequently relies on pharmacokinetic endpoints such as peak plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC), which reflect the rate and extent of absorption, respectively.29 The FDA criteria consider two products bioequivalent if the 90% confidence intervals of the relative mean Cmax and AUC of the test are 80%–125% of those of the reference product in the fasting state.29 For products with a narrow therapeutic index, the EMA recommends tightening the acceptance intervals for AUC to 90%–111%.28 Where Cmax is of particular importance for safety, efficacy, or drug level monitoring, this acceptance interval should also be applied for this parameter.28 Copies at either end of the spectrum of 80%–125% are considered bioequivalent to the proprietary medicine, despite a potentially substantial range in bioavailability. This is important for medicines with a narrow therapeutic index, where slight dose changes can have a large impact on clinical outcomes.32 In practice, some copies may have a bioavailability of ~70%–140% of that of the reference product.7 In bioequivalence studies, it is essential to compare one copy with its proprietary medicine, rather than comparing multiple copies in one study where bioequivalence variations can be masked.
Bioequivalence studies often have small sample sizes. According to the EMA, the number of evaluable volunteers in a bioequivalence study should be 12 or more.28 In contrast, proprietary medicines undergo testing in multiple clinical trials for all phases with large sample sizes. Random variation among small samples may be masked, giving a false impression of the medicine’s safety or efficacy. In a study of 103 transplant patients who were treated with tacrolimus (Prograf®; Astellas Pharma US, Inc., Northbrook, IL, USA) or its copy, copy substitution resulted in a mean reduction of 15.9% and 11.9% in the concentration/dose ratio in liver and kidney transplant patients, respectively.33 Large variations were observed between individual patients; there were increases and decreases in concentration/dose ratios reaching ~50% following substitution.33,34
Populations in bioequivalence studies usually consist of healthy volunteers, which may not reflect medicine efficacy in patients, causing further concern. Patients with schizophrenia can tolerate doses of dopamine-blocking antipsychotics that cause severe adverse events in healthy volunteers.35 The FDA recommends that patients with stable disease, rather than healthy controls, should be enrolled in bioequivalence studies.29
Switching from proprietary to nonproprietary medicines: lessons from diverse therapy areas
Switching to nonproprietary medicines is common in many therapy areas to reduce costs and burden on public health care systems. Switching to a substandard alternative may increase long-term health care costs and exposes patients to ineffective treatment, suggesting that decisions about prioritizing cost over quality should be made in an informed manner. Reports of toxicity, or reduced efficacy, in patients with epilepsy who have switched to copies7,36 have led to increased scrutiny by professional bodies to prevent or reduce the use of antiepileptic copy medicines.7 A study of renal transplant patients in the US revealed higher total health care costs associated with generic cyclosporin A. The cause of cost differences was an unintended higher rate of exposure to immunosuppressants in patients receiving generic cyclosporin A, in addition to suboptimal efficacy.37,38
The patient’s requirement for proprietary forms of medicine must still be considered. As reported by a treating physician,39 a patient with well-controlled, primarily generalized epilepsy received a prescription for proprietary lamotrigine and was prohibited from using a generic copy because of the narrow therapeutic window of the proprietary medicine. Her local pharmacist ignored the instructions and further directed the patient to use a nonproprietary copy.39 This would expose the patient to a product with a bioequivalence value potentially outside the narrow therapeutic window. The patient refused to accept anything other than the prescribed brand. For patients with epilepsy, adherence to medication over time may change if the patients receive a copy with altered efficacy.40 Safety concerns must outweigh cost savings, and several health authorities advise that the decision to treat a patient with a copy should be made only after consultation with a qualified health care professional.
Clinical implications of substandard copies for patients: examples from MS
Disease-modifying therapies for MS are expensive, and the large numbers of patients with this condition present a challenge to health care systems. Copies may reduce treatment costs, but the assumption that a drastic price reduction for disease-modifying therapies will occur is probably unrealistic.3
The β-interferons are routinely used in MS treatment. CinnoVex™ (CinnaGen, Tehran, Iran) and Genfaxon® (Genfa, Geneva, Switzerland), two biosimilars of the proprietary β-interferons Avonex® (Biogen Idec Inc., Cambridge, MA, USA) and Rebif® (EMD Serono, Inc., Rockland, MA, USA) are available commercially in Russia and Ukraine.26,27 In clinical trials, CinnoVex was shown to be less effective than Avonex, while Genfaxon displayed an unfavorable tolerability profile compared with Rebif.26,27 Adverse events included the development of flu-like symptoms, local skin reactions, and mood changes and were reported more frequently than for Avonex or Rebif. Nonproprietary biosimilars undergo a three-stage assessment of pharmaceutical quality, laboratory testing, and clinical data to ensure similar safety and efficacy to the proprietary medicine.41 The greatest safety concern for biosimilars is immunogenicity42 because biosimilars are derived from biological molecules and can induce an immune response in the patient.41,42 Glycosylation, impurities, changes in the three-dimensional structure, and factors related to the patient’s human leukocyte antigen type can elicit an immune response.42,43 Small alterations in the product’s manufacturing process can lead to functional changes that alter safety and efficacy,44 stressing the necessity for careful monitoring through appropriately designed clinical trials and, potentially, premarketing testing, as is done for proprietary medicines.
Some MS medicines with immunomodulatory properties have been associated with progressive multifocal leukoencephalopathy45 and need careful patient monitoring. Increased safety controls should be implemented for such medicines, regardless of their proprietary or copy status.
For medicines with the potential to penetrate the central nervous system, health authorities require the manufacturer to complete specific, additional clinical investigations.46,47 Fingolimod (Gilenya®; Novartis Pharma AG, Basel, Switzerland) is a nonselective sphingosine-1-phosphate receptor modulator that crosses the blood–brain barrier.48 Substandard fingolimod copies that do not undergo such investigations may cause side effect problems for patients. Fingolimod sequesters lymphocytes to lymphoid tissues, thus keeping pathogenic lymphocytes out of the central nervous system,49 and also acts directly within the central nervous system.49 The initial phase of the manufacturing process incorporates a chemical that has genotoxic properties.16 As part of the quality assessment of proprietary fingolimod, genotoxic levels should be below the threshold of toxicological concern (1.5 μg/day), as set in the guidelines of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.50 Copies of fingolimod that become commercially available should follow proper legal approval and conform to good manufacturing practice and other quality evaluations.
The quality parameters of a product’s APIs and excipients are important for patient outcomes and should also be controlled.2 Some excipients alter API bioavailability or modify the product’s shelf life.51 In cases where the excipient nature and source differ from those in the proprietary medicine, this has adversely affected medicine efficacy and patient safety.22 In one study, the API of proprietary fingolimod was assessed across a series of quality parameters and compared with the APIs of eleven nonproprietary products.16 The parameters included microparticle size distributions, heavy metal content, and inorganic impurities. The APIs of all eleven copies failed to meet international or proprietary specifications for more than one parameter. Proprietary fingolimod exhibits Biopharmaceuticals Classification System Class 2 behavior (low solubility, high permeability);52 consequently, according to FDA and EMA requirements, any nonproprietary product of this group should demonstrate bioequivalence in humans.16
Fingolimod product: a case study
Proprietary fingolimod, like other proprietary medicines, uses a tightly designed manufacturing process that ensures batch-to-batch reproducibility (to ensure an accurate and consistent quantity of each ingredient in each capsule) and quality. Manufacturers of nonproprietary fingolimod must comply fully with all aspects of quality evaluation and manufacturing. Additional risks related to use of the medicine are determined using a long-term risk management plan and first-dose monitoring. These procedures control for safety issues in the patient, and apply regardless of whether the medicine is proprietary or a copy. These risks are unrelated to safety issues that arise as a result of poor, uncontrolled, and substandard quality evaluation.
The risk management plan identifies the product safety profile, prevents or minimizes the risks associated with fingolimod, and documents post-authorization obligations.53 First-dose monitoring is an integral part of the risk evaluation and mitigation strategy.54 Key considerations for fingolimod first-dose monitoring relate to its potential cardiovascular effects shortly after initial administration, which are a consequence of the known mechanism of action.55,56
Nonproprietary fingolimod product, not meeting specifications, can fail to meet efficacy or have different impact on safety2 when substandard quality evaluation is employed.
A commercially available fingolimod copy was sourced and tested for a number of quality parameters (Novartis laboratories). The selected parameters were compared in proprietary and nonproprietary fingolimod using high-performance liquid chromatography (Table 2).
 | Table 2 Comparison of selected parameters for proprietary versus nonproprietary fingolimod |
The nonproprietary product was tested 19 months before the stated expiration date. It exhibited fingolimod assay, total degradants, individual unspecified impurity, and content uniformity that were close to or out of proprietary and/or internationally acceptable specifications. Specified degradation products (not shown in table) for the nonproprietary and proprietary product were within proprietor’s specifications.
The assay value in the nonproprietary product was 93.1%, which was at the low end of the proprietary or internationally acceptable specifications of 90.0%. Total degradation products reached 9.4%, almost three times more than proprietor’s total degradation products’ specification (not >3.5%).
One unspecified impurity was measured at 7.6%, which was 14-fold higher than the proprietor’s specification (not >0.5%). In addition, this is seven to eight times over the internationally acceptable specification.57 According to the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines,57 for a product with a daily dose of under 1.0 mg, any impurity over 1.0% must be investigated, identified, and qualified in toxicology studies. The proprietor has implemented a rigorous process to continuously assess potential genotoxic impurities: 1) risk assessment: in silico testing for genotoxic alert structures in starting materials, process intermediates, reagents, and related impurities and 2) risk mitigation: levels of genotoxins are monitored and controlled to ensure that the levels remain below the threshold of toxicological concern (1.5 μg/day).50 This highlights that tight control of potential genotoxins is an essential element of the quality management of fingolimod.
Fingolimod is a potent, active substance and, based on data from clinical trials, is used at very low doses (0.5 mg once daily). The critical parameters to ensure batch-to-batch consistency and, therefore, quality and performance of the product are blend and content uniformity. The uniformity of the dosage units in a batch, with regard to the active compound, is often critical to the safety and efficacy of the dosage units.58 In proprietary fingolimod, this has been achieved by careful selection of excipients, drug particle size, and a robust blending and encapsulation process. In the nonproprietary product addressed here, content uniformity had an acceptance value of 14.4%. Although this narrowly met the content uniformity acceptance value of ≤15.0%,59 it indicates high capsule-to-capsule variability. Individual assay ranged from 86.4% to 97.0% (not shown in the table).
In summary, any manufacturer of nonproprietary fingolimod would have to comply fully with all aspects of quality evaluation and manufacturing to ensure the expected, essential elements of efficacy and safety. Potentially noncontrolled or different manufacturing process may result in: 1) poor batch-to-batch reproducibility (accurate and consistent quantity of each ingredient in each capsule) and 2) lower quality. Such a nonproprietary product would not be identical to proprietary fingolimod and could fail to meet efficacy or have different impact on the safety of patients with MS. Given that a disparity in bioavailability may exist between the medicines deemed bioequivalent under the current guidelines, first-dose monitoring of copies, currently not a requirement for their approval, would provide greater reassurance of their safety.
These data reinforce the necessity for health authorities to implement the highest standards of quality and bioequivalence requirements before copy versions of fingolimod are made available to patients with MS.
Summary and conclusion
To reduce costs and increase access to health care, nearly every country has adopted laws, policies, and/or regulations that encourage substitution of proprietary medicines with copies. Pharmacovigilance programs for copies strengthen safety monitoring by promoting early detection of adverse events and assessing, minimizing, and communicating the risks for patients. Legislation should assure consumers that bioequivalent medicines are safe and effective for their intended use and that all labeling and packaging is truthful and informative.
There is a lack of enforcement of regulatory requirements governing the quality of copies in some markets. In certain developing countries, the standards are less stringent when compared with the FDA, EMA, or WHO. Even for copies approved in highly regulated markets, there are several strong arguments for incorporating greater robustness in the testing procedures that are used to grant approval. Pharmacovigilance is essential to meet the challenges posed by the ever-increasing range and potency of medicines. When adverse events and toxicity emerge, it is vital that they are reported, analyzed, and communicated effectively to relevant audiences. If this does not occur, patients living in countries with lax controls, and those who import substandard copies may be at risk.
There can be serious consequences when using inadequately tested, substandard copies, particularly in patients who require well-controlled treatments.32 There can be a lack of therapeutic equivalence for copies that may be bioequivalent but still fall outside the recommended range of bioavailability. The bioequivalence studies that use small numbers of volunteers may not reflect the efficacy and safety of the product in a larger population. Cost savings envisaged by using copies over proprietary medicines may be outweighed by the cost of adverse consequences. Potential medical and legal concerns may arise for patients who did not give informed consent, based on clear and unequivocal discussion of unwanted effects, before copy substitution.
This review of the literature on copies illustrates the risks in using products that are not adequately tested. For a medicine like fingolimod, to ensure patient safety, it is paramount to control impurities (owing to the genotoxic nature of the starting material for synthesis of the API and the low dose of the API), implement an risk management plan, and demonstrate bioequivalence. It is the responsibility of all stakeholders, including regulators, manufacturers of medicines, physicians, nongovernment organizations, patients, and their caregivers, to continue to take action against low-quality medicines to ensure that patients receive the medicine they need.
Acknowledgments
The authors would like to thank Shangdong Zhan and Alessandro Allodoli for their valuable contributions. Editorial assistance was provided by Robert M Gillies, PhD and Noëlle L O’Regan, PhD from PharmaGenesis London, London, UK, and was funded by Novartis Pharma AG.
Disclosure
Jorge Correale is a board member of Merck Serono Argentina, Novartis Argentina, Genzyme Latin America, Genzyme Global, Biogen Idec Latin America, and Merck Serono Latin America; and has received reimbursement for developing educational presentations from Merck Serono Argentina, Merck Serono Latin America, Biogen Idec Argentina, Genzyme Argentina, Novartis Argentina, Novartis Latin America, Novartis Global, and Teva Argentina as well as professional travel and accommodation stipends. Erwin Chiquete has received research grants from Sanofi and Ferrer Grupo; has served as a research advisor for Sanofi, Novartis, Teva, and Genzyme; and has received speaker honoraria from Novartis Mexico, Genzyme, and Ferrer Grupo. Alexey Boyko is a member of advisory boards and a speaker and participant in clinical trials for Novartis, Merck Serono, Teva, Genzyme, Biogen Idec, and Bayer Schering. Roy G Beran has received travel grants and sponsorship from Bayer, Biogen Idec, Merck Serono, Genzyme, Novartis, Teva, and UCB. Jorge Barahona Strauch has received speaker honoraria from Biogen Idec Chile, Bayer Chile, Genzyme Chile, Novartis Chile, Lundbeck Chile, and Teva (Laboratorios Chile). Snezana Milojevic and Nadina Frider are employees and stockholders of Novartis Pharmaceuticals Corporation. The authors report no other conflicts of interest in this work.
References
World Health Organization. Regional Office for the Eastern Mediterranean. Pharmaceutical Products. Geneva: World Health Organization; 2009. Available from: http://www.emro.who.int/health-topics/pharmaceutical-products/index.html. Accessed October 23, 2015. | ||
Johnston A, Holt DW. Substandard drugs: a potential crisis for public health. Br J Clin Pharmacol. 2014;78(2):218–243. | ||
Dunne S, Shannon B, Dunne C, Cullen W. A review of the differences and similarities between generic drugs and their originator counterparts, including economic benefits associated with usage of generic medicines, using Ireland as a case study. BMC Pharmacol Toxicol. 2013;14:1. | ||
World Health Organization. What are Substandard and Counterfeit Medicines? Geneva: World Health Organization; 2009. Available from: http://www.who.int/medicines/services/counterfeit/faqs/QACounterfeit-October2009.pdf. Accessed October 21, 2015. | ||
US Food and Drug Administration. FDA Ensures Equivalence of Generic Drugs. Silver Spring, MD: US Food and Drug Administration; 2002. Available from: http://www.fda.gov/Drugs/EmergencyPreparedness/BioterrorismandDrugPreparedness/ucm134444.htm. Accessed July 6, 2015. | ||
Thaul S. How FDA approves drugs and regulates their safety and effectiveness. Congressional Research Service; 2012. https://fas.org/sgp/crs/misc/R41983.pdf. Accessed July 10, 2015. | ||
Crawford P, Feely M, Guberman A, Kramer G. Are there potential problems with generic substitution of antiepileptic drugs? A review of issues. Seizure. 2006;15(3):165–176. | ||
Sankar R, Glauser TA. Understanding therapeutic equivalence in epilepsy. CNS Spectr. 2010;15(2):112–123. | ||
Cockburn R, Newton PN, Agyarko EK, Akunyili D, White NJ. The global threat of counterfeit drugs: why industry and governments must communicate the dangers. PLoS Med. 2005;2(4):e100. | ||
Shakoor O, Taylor RB, Behrens RH. Assessment of the incidence of substandard drugs in developing countries. Trop Med Int Health. 1997;2(9):839–845. | ||
Almuzaini T, Choonara I, Sammons H. Substandard and counterfeit medicines: a systematic review of the literature. BMJ Open. 2013;3(8):e002923. | ||
Almuzaini T, Sammons H, Choonara I. Quality of medicines in Canada: a retrospective review of risk communication documents (2005–2013). BMJ Open. 2014;4(10):e006088. | ||
Bennet S. The falsified medicines directive: impacts on API imports. Specialty Chemicals Magazine; 2013. Available from: http://www.specchemonline.com/articles/view/the-falsified-medicines-directive#.VaeqePlVhHw. Accessed July 16, 2015. | ||
Directive 2011/62/EU of the European Parliament and of the Council of 8 June 2011. Available from: http://ec.europa.eu/health/files/eudralex/vol-1/dir_2011_62/dir_2011_62_en.pdf. Accessed October 23, 2015. | ||
Jiang H, Wu SL, Karger BL, Hancock WS. Mass spectrometric analysis of innovator, counterfeit, and follow-on recombinant human growth hormone. Biotechnol Prog. 2009;25(1):207–218. | ||
Correale J, Chiquete E, Milojevic S, Frider N, Bajusz I. Assessing the potential impact of non-proprietary drug copies on quality of medicine and treatment in patients with relapsing multiple sclerosis: the experience with fingolimod. Drug Des Devel Ther. 2014;8:859–867. | ||
Lon CT, Tsuyuoka R, Phanouvong S, et al. Counterfeit and substandard antimalarial drugs in Cambodia. Trans R Soc Trop Med Hyg. 2006;100(11):1019–1024. | ||
World Health Organization. Medicines: Spurious/Falsely-labelled/Falsified/Counterfeit (SFFC) Medicines. Geneva: World Health Organization; 2012. Available from: http://www.who.int/mediacentre/factsheets/fs275/en/. Accessed March 1, 2015. | ||
Pharmafile. Dr Reddy’s recalls blood pressure drug; 2014. Available from: http://www.pharmafile.com/news/188481/dr-reddy-s-recalls-blood-pressure-drug. Accessed July 31, 2015. | ||
Neuvonen PJ. Bioavailability and central side effects of different carbamazepine tablets. Int J Clin Pharmacol Ther Toxicol. 1985;23(4):226–232. | ||
Meyer MC, Straughn AB, Jarvi EJ, Wood GC, Pelsor FR, Shah VP. The bioinequivalence of carbamazepine tablets with a history of clinical failures. Pharm Res. 1992;9(12):1612–1616. | ||
Patel V, Cordato DJ, Dias M, Beran RG. Changed constitution without change in brand name – the risk of generics in epilepsy. Epilepsy Res. 2012;98(2–3):269–272. | ||
Álvarez AA, Mysler E, Ruiz de Castilla EM, Flores-Murrieta FJ, Hughes J, Feijó Azevedo V. Recommendations for the regulation of biosimilars and their implementation in Latin America. GaBI. 2014;3(3):143–148. | ||
Homedes N, Ugalde A. Multisource drug policies in Latin America: survey of 10 countries. Bull World Health Organ. 2005;83(1):64–70. | ||
Alfonso-Cristancho R, Andia T, Barbosa T, Watanabe JH. Definition and classification of generic drugs across the world. Appl Health Econ Health Policy. 2015;13(Suppl 1):S5–S11. | ||
Borets OG, Davydovskaia MV, Demina TL, et al. [Experience of using interferon beta-1a biosimilars (Cinnovex and Genfaxon-44) in the Moscow Multiple Sclerosis Center]. Zh Nevrol Psikhiatr Im S S Korsakova. 2015;115(2 Vypusk 2 Rasseiannyi skleroz):51–56. | ||
Kasatkin DS, Spirin NN, Boiko AN, Vlasov IV. [Unified assessment of adverse events of multiple sclerosis disease-modifying drugs.]. Zh Nevrol Psikhiatr Im S S Korsakova. 2014;114(2 Vypusk 2 Rasseiannyi skleroz):78–82. | ||
European Medicines Agency. Guideline on the Investigation of Bioequivalence. London: European Medicines Agency; 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf. Accessed July 1, 2015. | ||
US Food and Drug Administration. Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted under an ANDA. Silver Spring, MD: US Food and Drug Administration; 2013. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm377465.pdf. Accessed August 13, 2015. | ||
World Health Organization. Technical Report Series, No. 937. Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability. Geneva: World Health Organization; 2006. http://apps.who.int/prequal/info_general/documents/TRS937/WHO_TRS_937__annex7_eng.pdf. Accessed September 21, 2015. | ||
US Food and Drug Administration. Guidance for Industry: ANDA Submissions – Content and Format of Abbreviated New Drug Applications. Silver Spring, MD: US Food and Drug Administration; 2014. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm400630.pdf. Accessed August 13, 2015. | ||
Yu LX, Jiang W, Zhang X, et al. Novel bioequivalence approach for narrow therapeutic index drugs. Clin Pharmacol Ther. 2015;97(3):286–291. | ||
Momper JD, Ridenour TA, Schonder KS, Shapiro R, Humar A, Venkataramanan R. The impact of conversion from Prograf to generic tacrolimus in liver and kidney transplant recipients with stable graft function. Am J Transplant. 2011;11(9):1861–1867. | ||
Klintmalm GB. Immunosuppression, generic drugs and the FDA. Am J Transplant. 2011;11(9):1765–1766. | ||
Cutler NR. Pharmacokinetic studies of antipsychotics in healthy volunteers versus patients. J Clin Psychiatry. 2001;62 Suppl 5:10–13; discussion 23–24. | ||
Shaw SJ, Hartman AL. The controversy over generic antiepileptic drugs. J Pediatr Pharmacol Ther. 2010;15(2):81–93. | ||
Helderman JH, Kang N, Legorreta AP, Chen JY. Healthcare costs in renal transplant recipients using branded versus generic ciclosporin. Appl Health Econ Health Policy. 2010;8(1):61–68. | ||
Gothe H, Schall I, Saverno K, et al. The impact of generic substitution on health and economic outcomes: a systematic review. Appl Health Econ Health Policy. 2015;13 (Suppl 1):S21–S33. | ||
Beran RG. Generic substitution in contravention of doctor’s prescriptions. Intern Med J. 2015;45(10):1093. | ||
Cramer JA, Glassman M, Rienzi V. The relationship between poor medication compliance and seizures. Epilepsy Behav. 2002;3(4):338–342. | ||
Warren JB. Generics, chemisimilars and biosimilars: is clinical testing fit for purpose? Br J Clin Pharmacol. 2013;75(1):7–14. | ||
Zuniga L, Calvo B. Biosimilars: pharmacovigilance and risk management. Pharmacoepidemiol Drug Saf. 2010;19(7):661–669. | ||
Calvo B, Zuniga L. EU’s new pharmacovigilance legislation: considerations for biosimilars. Drug Saf. 2014;37(1):9–18. | ||
Declerck PJ. Biosimilar monoclonal antibodies: a science-based regulatory challenge. Expert Opin Biol Ther. 2013;13(2):153–156. | ||
Faulkner M. Risk of progressive multifocal leukoencephalopathy in patients with multiple sclerosis. Expert Opin Drug Saf. 2015;14(11):1737–1748. | ||
US Food and Drug Administration. Guidance for Industry: Suicidal Ideation and Behavior: Prospective Assessment of Occurrence in Clinical Trials. Silver Spring, MD: US Food and Drug Administration; 2012. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM225130.pdf. Accessed November 16, 2015. | ||
US Food and Drug Administration. Guidance for Industry: Assessment of Abuse Potential of Drugs. Silver Spring, MD: US Food and Drug Administration; 2010. Available from: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm198650.pdf. Accessed November 16, 2015. | ||
Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101. | ||
Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci. 2013;328(1–2):9–18. | ||
European Medicines Agency. ICH Guideline M7 on Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. London: European Medicines Agency; 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/02/WC500139217.pdf. Accessed October 21, 2015. | ||
Garcia-Arieta A. Interactions between active pharmaceutical ingredients and excipients affecting bioavailability: impact on bioequivalence. Eur J Pharm Sci. 2014;65:89–97. | ||
US Food and Drug Administration. Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. Silver Spring, MD: US Food and Drug Administration; 2000. Available from: http://www.gmp-compliance.org/guidemgr/files/3618FNL.PDF. Accessed October 21, 2015. | ||
European Medicines Agency. Guideline on Good Pharmacovigilance Practices (GVP): Module V – Risk Management Systems. London: European Medicines Agency; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129134.pdf. Accessed July 1, 2015. | ||
Novartis Pharmaceuticals Corporation. NDA 22-527. Gilenya (fingolimod) 0.5mg capsules: risk evaluation and mitigation strategy (REMS); 2015. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/rems/Gilenya_2015-05-14_Full.pdf. Accessed October 22, 2015. | ||
US Food and Drug Administration. Drug Safety Communication: Revised Recommendations for Cardiovascular Monitoring and Use of Multiple Sclerosis Drug Gilenya (Fingolimod). Silver Spring, MD: US Food and Drug Administration; 2015. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm303192.htm. Accessed July 3, 2015. | ||
Kuperman G. Cardiac safety profile during first-dose monitoring period of fingolimod (Gilenya) treatment in patients with relapsing remitting MS: data from the Argentinean registry (REAL). Interim results. Neurology. 2015;84(14):Supplement P3.250. | ||
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use harmonised tripartite guideline. Impurities in new drug products Q3B(R2). Current Step 4 version. 2006. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3B_R2/Step4/Q3B_R2__Guideline.pdf. Accessed September 21, 2015. | ||
US Food and Drug Administration. Quality by Design for ANDAs: An Example for Immediate-release Dosage Forms. Pharmaceutical Development Report. Example QbD for IR Generic Drugs. Silver Spring, MD: US Food and Drug Administration; 2012. Available from: http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM304305.pdf. Accessed October 21, 2015. | ||
The United States Pharmacopeial Convention. Stage 6 harmonization. Uniformity of dosage units. 2011. Available from: http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/2011-02-25905UNIFORMITYOFDOSAGEUNITS.pdf. Accessed October 21, 2015. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.