Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Clinical features of limbic encephalitis with LGI1 antibody
Authors Wang ML, Cao XY, Liu QX, Ma W, Guo XQ, Liu XW
Received 11 March 2017
Accepted for publication 28 April 2017
Published 16 June 2017 Volume 2017:13 Pages 1589—1596
DOI https://doi.org/10.2147/NDT.S136723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Meiling Wang,1,2,* Xiaoyu Cao,2,* Qingxin Liu,2 Wenbin Ma,1,2 Xiaoqian Guo,1,3 Xuewu Liu1
1Department of Neurology, Qilu Hospital of Shandong University, Jinan, 2Department of Neurology, Binzhou Medical University Hospital, Binzhou, 3Department of Neurology, Jining First People’s Hospital, Jining, Shandong, People’s Republic of China
*These authors contributed equally to this work
Objective: The objective of this study was to analyze the clinical manifestation, course, evolution, image manifestation, and treatments of LGI1 limbic encephalitis (LE).
Patients and methods: Studies confirmed that LE with the complex antibody of voltage-gated potassium channels is LGI1 LE. Since then, LE cases have been reported. In this study, 10 typical LE cases were searched in PubMed. These cases and one additional case, which we reported herein, were retrospectively analyzed.
Results: All the patients suffered from recent memory deterioration. The following cases were observed: eight with faciobrachial dystonic seizures (FBDS), six with different kinds of epileptic seizures (four complex partial seizures, one myoclonus seizure, and one generalized tonic–clonic seizure), four with FBDS and different kinds of epileptic seizures at the same time, five with mental disorders (one visual hallucination, one paranoia, one depression, one anxiety, and one dysphoria), five with hyponatremia, and two with sleep disorder. The brain MRI of nine patients revealed abnormalities in the mediotemporal lobe and the hippocampus. The LGI1 antibodies in the blood and/or cerebrospinal fluid (CSF) were positive. The content of the CSF protein of two patients increased slightly. The tumor marker of all the patients was normal, but capitate myxoma was detected in the combined pancreas duct of one patient. Gamma globulin and hormone treatments were administered to nine patients. Of these patients, six received a combination of antiepileptic drugs. The clinical symptoms of all the patients improved.
Conclusion: LGI1 LE is an autoimmune encephalitis whose clinical manifestations are memory deterioration, FBDS, epileptic seizure, mental disorders, and hyponatremia. Brain MRI shows that this autoimmune disease mainly involves the mediotemporal lobe and the hippocampus. This condition can also be manifested with other autoimmune encephalitis cases but can be rarely associated with tumors. After patients with LGI1 LE receive gamma globulin and hormone treatments, their clinical prognosis is good.
Keywords: positive LGI1, limbic encephalitis, epilepsy, FBDS, immunotherapy
Introduction
Limbic encephalitis (LE) was first used by English neuropathologists and neurologists in the 1960s.1–3 In 2004, the PNSEURONET organization (http://www.pnseuronet.org),4 composed of neurologists, established the diagnostic criteria of related encephalitis. Clinical symptoms among adults manifest during the disease course of not >5 years, which is a prerequisite for diagnosis. Patients should exhibit at least one of the following clinical symptoms: episodic dysmnesia, epileptic seizure, emotional instability, lack of emotion suppression, and involvement of the central or peripheral nervous system. Patients should also suffer from the following: tumors, related LE antibody, high fluid-attenuated inversion recovery (FLAIR)/T2 signal, and pathologically examined brain tissues, especially chronic lymphocyte–microglial cells. LE is regarded as a disease associated with epilepsy, memory deterioration, behavioral disorders, and sleep disorders; LE is also related to the paraneoplastic disease of antibodies in cerebellar cells.5 LE, implicating voltage-gated potassium channels (VGKCs), is a potentially reversible disease affecting the central nervous system and is quickly responsive to immunotherapy.6 Most of the involved antibodies are LGI1, and they present a good relationship with antibody titers and clinical manifestations. Therefore, they exhibit pathogenicity. Immunopathology studies have shown neuronal loss and lymphocyte infiltration around the blood vessels of the hippocampus and amygdala.7 Some studies and case reports have found that LGI1 LE is mainly non-paraneoplastic, tumor markers and radiological examinations are negative, and these observations are different from those of typical LE. However, further studies should be performed to clarify the pathogenesis of LE. Traditional primary treatments for LE include hormones and antiepileptic drugs. With research on LE pathogenesis, clinical treatments mainly involve a combination of hormones and immunosuppressants, which can alleviate clinical symptoms and improve patient prognosis. There is short of case studies’ research about LGI1 LE currently, and its treatments are also only empirical and symptomatic. There is no literature that has conducted evidence-based medicine research of the pathogenesis, clinical manifestations, diagnosis, and treatment of LGI1 LE until now. This study discussed all of above problems of LE through a retrospective analysis of 11 patients diagnosed with LE.
Patients and methods
In addition to the case report of one patient diagnosed in our hospital, 10 LE patients were searched in PubMed by using the following key words: LGI1 antibody, LE, epilepsy, faciobrachial dystonic seizures (FBDS), and immunotherapy. The 10 patients were diagnosed with LE (Table 1).
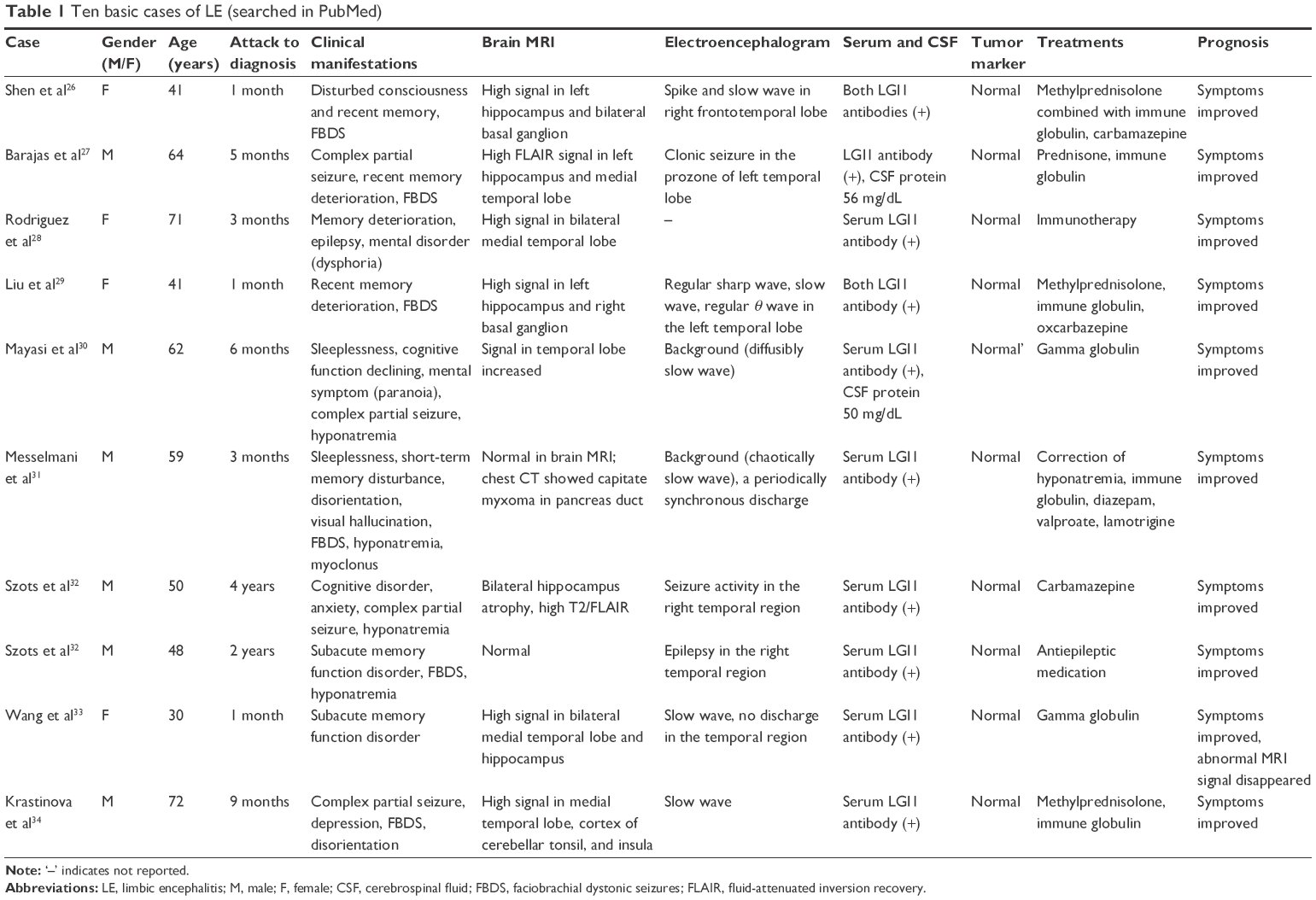 | Table 1 Ten basic cases of LE (searched in PubMed) |
Case report
The patient was a 69-year-old male teacher who manifested an untreated hypothyroidism upon physical examination in May 2013. In July 2013, he exhibited a paroxysmal involuntary twitch of the upper limbs and the right face. This condition was relieved within several seconds but was observed at a frequency of 10 times/day. However, the patient remained untreated. In the beginning of October, he experienced memory loss and failed to identify his home in some instances. On October 20, he lost consciousness and suffered from involuntary eye rolling, trismus, foaming at the mouth, and limb stretching without gatism. These symptoms lasted 5 min until they subsided. No generalized tonic–clonic seizure was noted after treatment with oral levetiracetam and phenobarbital; however, the patient had recurring symptoms of wild eyes, deviated mouth, getting terrified after sitting up forcedly and then relieved in a few seconds, and significant memory loss. As such, he was sent to a hospital. No apparent positive signs were found after he completed a nervous system examination. Brain MRI (Figures 1 and 2) showed an abnormal signal (no enhancement) in the bilateral hippocampus region. Electroencephalogram revealed spiked and slow waves in the right temporal lobe. Thyroid function tests indicated 3.61 pmol/L T3, 15.08 pmol/L T4, 7.750 mIU/L thyroid-stimulating hormone (TSH), 20.33 IU/mL anti-thyroid peroxidase (TPO), 763.10 IU/mL anti-thyroglobulin (TG), 130.4 mmol/L serum sodium, and 94.1 mmol/L serum chloride. The levels of the cerebrospinal fluid (CSF), protein, immunoglobulin, antinuclear antibody, rheumatoid factors, and tumor markers were in normal ranges. The patient was treated with Euthyrox (levothyroxine), levetiracetam, and dexamethasone. As a result, his memory improved significantly after 14 days, epileptic seizure was relieved, and blood sodium returned to normal levels upon reexamination.
 | Figure 1 Brain MRI FLAIR showed abnormal signal in bilateral hippocampus. |
 | Figure 2 Brain MRI showed enhancement in bilateral hippocampus. |
In April 2016, the patient suffered from 39°C fever and diarrhea and could not recognize his family. The condition improved after the fever was alleviated. After 7 days, his memory, particularly his recent memory, declined gradually and exacerbated progressively. He was sent to the hospital again in August because of memory deterioration and epileptic seizure. His score of Montreal Cognitive Assessment (MoCA) is 14, Mini Mental State Examination (MMSE) is 20. Brain MRI (Figure 3) showed bilateral hippocampus atrophy. Thyroid function test results were as follows: 3.68 mmol/L T3, 9.88 pmol/L T4, 4.660 mL U/L TSH, 123.4 mmol/L serum sodium, and 92.7 mol/L serum chloride. Blood antinuclear antibody, anticardiolipin antibody, tuberculosis antibody, herpes simplex virus antibody, rheumatoid factors, anti-O, IgA, IgM, and IgG were normal. NMDA, AMPA1, AMPA2, Casper2, GABA2, Hu, Yo, Ri, and Ma2 in the serum and CSF were negative. The antibodies of the serum and CSF LGI1 were positive. Considering the possibility of LGI LE, we treated the patient with immunoglobulin (0.4 g/kg/d, continuing for 5 days), dexamethasone (10 mg/d, continuing for 2 weeks), and prednisone (dosage was tapered gradually, and its usage was 50 mg/d, reducing 10 mg/month for 3 months. Since then, 5 mg per month was subtracted until the prednisone withdrawal. The total course was 9 months levothyroxine, oxcarbazepine, and levetiracetam). After 2 months, MoCA: 20, MMSE: 25, and memory deterioration were alleviated, and epileptic seizure was relieved. The patient has provided written informed consent for the case details including images to be published for this study.
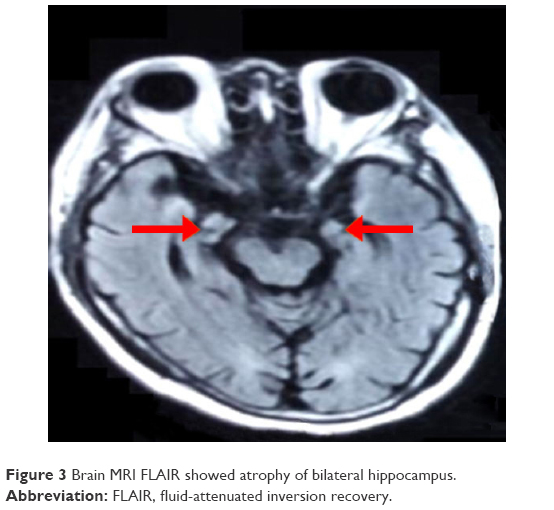 | Figure 3 Brain MRI FLAIR showed atrophy of bilateral hippocampus. |
Results
Among the 11 patients, 7 were males, aged 48–72 years (average age 60 years) and 4 were females, aged 30–71 years (average age 45 years). All the patients had recent memory deterioration; eight patients, comprising 72.7%, had FBDS; six patients had different kinds of epileptic seizures (four complex partial seizure, one myoclonus, and one generalized tonic–clonic seizure); four patients had FBDS and different kinds of epileptic seizures (FBDS before epileptic seizures); five patients had dysphrenia (one visual hallucination, one paranoid, one depression, one anxiety, and one dysphoria); five patients had hyponatremia; and two patients had sleep disorder. Brain MRI results of nine cases were abnormal, and the focus involved mediotemporal lobe and hippocampus. Examination of CSF yielded the following results: protein levels of two patients increased and routine CSF, glucose, and chloride levels were all normal. LGI1 antibodies of blood and CSF were positive. Tumor marker of all patients was normal, except one case in which capitate myxoma was found in the combined pancreas duct. The thyroid function of one patient decreased. Nine patients, of whom six were given antiepileptic drugs, were treated with gamma globulin and combined hormone, and all the patients’ clinical symptoms improved.
Discussion
Diseases affecting antibody-mediated central nervous system are usually caused by a combination of antibodies and expressed proteins on the neuronal surface. LE, which is a kind of autoimmune disease with the main manifestations of acute and subacute hypomnesia, cognitive function disorder, and epilepsy, is rare clinically.
VGKCs are the potassium channels adjusting the neuronal excitability of central and peripheral nervous systems. VGKC antibody was first found in patients with Isaacs’ syndrome. These kinds of patients may have muscular fibrillation and skin perspiration caused by increasing excitability of peripheral nerve. LE is usually regarded as an immune system disease caused by cancer, such as small cell lung, thymoma, and testicular cancers, but it is also found in some patients who had no paraneoplastic syndrome or neuromuscular overexcitation.8 VGKCs mainly include LGI1 antibody (maximum) and Casper-2 antibody. LGI1 is the glycoprotein released by presynaptic membrane, and it can interact with presynaptic membrane ADAM metallopeptidase domain 22 and presynaptic membrane ADAM metallopeptidase domain 23 to affect the signal transduction between the synapses by the VGKCs and postsynaptic membrane α−amino-3-hydroxy-5-methyl-4-isoxazole propionate receptors.1 This combination is necessary for inhibition of signal conduction. LGI1 antibody reduces the interaction of LGI1–ADAM and aggregation of AMPAR (reversibly). Protein gene disruption of LGI1 causes temporal lobe epilepsy. The mouse without LGI1 gene may die of fatal epilepsy, and this phenomenon explains the importance of LG11 in information transduction.9 After the attack of disease, the patient had significant mood and sleep disorders, which are mainly related to the damage of hippocampus and amygdaloid nucleus.10 In the case reports, 60% of autoimmune encephalitis related to LGI1 antibody had intractable hyponatremia. Some scholars think that this hyponatremia may be caused by improper secretion syndrome of antidiuretic hormone, and it might be related to the simultaneous LGI1 expressions of the hypothalamus and kidney.1
In this study, the patients’ disease symptoms were similar, usually as acute or subacute disease, and the main manifestations are as follows: FBDS, cognitive disorder (mainly recent memory deterioration), epilepsy, mental disorder, and hyponatremia, together with disorientation and sleep disorder. The specific explanations are as follows:
- Epilepsy: Epilepsy commonly comprises partial epileptic seizure, myoclonus, and generalized tonic–clonic seizure. Among the patients with positive LGI antibody, ~20%–40%11 have FBDS, which is short, frequent, and unconscious, before LE, together with dystonia, simple upper limbs spasm and contraction, and ipsilateral face twitch (not long, <3 s, several times a day). Minor improvements were noted after the regular use of antiepileptic drugs, but the FBDS remarkably reduced and disappeared after treatment with oral steroid.7 The patients with FBDS have basal ganglia lesion.9 FBDS usually arises before the cognitive decline, and LE can be prevented through the timely treatment with immunotherapy.12 Among the 11 patients in this study, six had different types of epileptic seizures (four complex partial seizure, one myoclonus, and one generalized tonic–clonic seizure). Four patients had FBDS and different types of epileptic seizures (FBDS before seizure, without FBDS after the seizures). The results of the electroencephalogram examination of all patients were as follows: 10 cases of slow wave, spike wave, and spike and slow waves and one normal case. In this study, 72.7% of patients had FBDS. Therefore, an increasing number of LE patients were diagnosed with FBDS. Moreover, treatments with immunotherapy, which can reduce the epileptic seizures and prevent long-term complications, including hippocampal atrophy and persistent memory damage, should be administered.13 In a prospective study of 20 months,14 the authors mentioned that three patients had paroxysmal epileptic discharge and 10 patients had positive VGKC antibody, of which nine had particularly positive LGI1 antibody; this indicated that epileptic seizures easily occurred in patients who had positive LGI1 antibody. Lai et al1 thought that LGI1 mutation would lead to autosomal dominant inheritance, that is, temporal lobe epilepsy of autosomal dominant inheritance has hallmark syndromes of paroxysmal hearing and visual hallucination. In the third case in Table 1, the patient had FBDS with normal electroencephalogram (no epileptic wave); therefore, the FBDS might be dystonia. However, Irani et al15 thought that FBDS was a kind of epileptic seizure. Studies have also yet to determine whether FBDS is an epileptic seizure or dystonia.
- Cognitive disorder: The manifestations are memory disturbance and disorientation of acute or subacute disease. Memory disorder mainly affects the recent memory.9 The main cognitive disorder of LGI1 patient is memory deterioration, which is not significant in the early stage of disease. Majoie et al8 found that 89% of LE patients with 8-month disease course had memory disorder. All the patient cases in this study had different degrees of memory disorder, of which three cases had combined disorientation. Malter et al11 confirmed that the decrease in cognitive function was related to the disease course before immunotherapy.
- Hyponatremia: As was reported,4 60% of patients had hyponatremia after the early treatment of disease. Some scholars thought that hyponatremia was caused by abnormal secretion of antidiuretic hormone and it might be related to the simultaneous LGI1 expression of the hypothalamus and kidney. Among the 11 patients in this study, 5 (45.5%) had hyponatremia, and the serum sodium level increased as the condition improved. Therefore, we thought that, to some extent, serum sodium level reflects the disease severity, and it can be one of the markers to test LGI1 antibody.
- Dysphrenia: Main manifestations are personality and behavior disorders, irascibility, anxiety, impulsive behavior, mental hallucination, paranoia, and stupor; some patients mainly have emotionally mental disorder and visual hallucination.1,13 The onset of patients’ symptoms was delayed because they were treated in psychiatric hospitals. Among the 11 patients in this study, 5 patients (45.5%) had mental symptoms (one paranoia, one depression, one dysphoria, one anxiety, and one visual hallucination), coinciding with previous report.15 Mental symptoms disappeared after immunotherapy. Therefore, mental symptoms are possibly the main clinical manifestations of LGI LE. To some degree, these symptoms can reflect disease severity.
- Brain MRI: The imaging scans of LE patients mainly show high T2 signal and FLAIR in bilateral temporal lobe, and a few patients had abnormal signal in one side of the hippocampus region. Moreover, the temporal lobe and basal ganglia region are often involved.16 Among the 11 patients in this study, MRIs of two patients were normal; meanwhile, lesion involvements were seen in the hippocampus of six patients, in the medial temporal lobe of five patients, in the basal ganglia region of two patients, in the insular lobe of one patient, and in the cerebellar tonsil cortex of one patient. The disease course of the seven patients and the patient reported in this study were 4 years and 3 years, respectively. They had hippocampus atrophy in the later period with bilateral/unilateral disease involvements, and these symptoms were similar with previous reports. Irani et al14 found that the LGI LE, without any immunotherapy, might gradually develop into hippocampus atrophy with related nervous system symptoms. In this report, brain MRI reexaminations of two patients showed bilateral hippocampus atrophy, which agreed with clinical tests.
- Laboratory examination: No specificity in the routine examination of blood serum and CSF was noted; a few patients had a slight increase of CSF. Two patients in this study had increased CSF protein, whereas others had normal levels. All blood LGI1 antibodies were positive, whereas five LGI1 antibodies of CSF were positive. This result indicated that blood was more sensitive than CSF, which might be related to antibody titer and disease course. The possibility of LE patients with positive VGKC antibody, particularly positive LGI1 antibody, having tumors is minimal.2,17 Tumor marker examination of the 11 patients was normal, but one of them had capitate myxoma in the pancreas duct. Therefore, we think that LGI1 LE is seldom a tumor comorbidity. In this study, combined thyroid function of patients decreased, indicating that LE can be a comorbidity of other autoimmune disease.
Different diagnosis
Viral encephalitis
Viral encephalitis entails previous infection and includes symptoms such as fever, headache, and mental and behavioral disorders. This disease is the most common viral meningitis of herpes simplex. Brain MRI results show abnormal signals in medial temporal lobe and temporal lobe. Antiviral treatment is effective. If the CSF examination is normal and the patient has no significant headache or fever, the disease can be ruled out.
Hashimoto’s encephalopathy
This is a kind of encephalopathy related to autoimmune thyroid disease. The patients may have symptoms of consciousness disturbance, epileptic seizure, edema, and increased antithyroid antibodies. The thyroid function can be divided into normal, hyperfunction, and hypofunction. The disease course is as follows: relapse, remission, and progression. A significant improvement can be noted after hormone therapy. Hashimoto’s encephalopathy has no clear pathogenesis, and pathologic changes mainly are lymphocyte infiltration, myelin sheath damage, and axon damage around the blood capillary (in brain parenchyma), arteries, veins, and meninx (especially around the veins).18
Creutzfeldt–Jakob Disease (CJD)
This is the most common prion disease and mainly involves the cortex, basal nuclei, and spinal cord. CJD is also called corticostriatospinal degeneration. The main manifestations are progressive dementia, myoclonus twitches, and injury of pyramidal tract and extrapyramidal system. The main different diagnosis point of CJD and LE is the negative LGI1 antibody of CSF, 14-3-3 positive protein.19,20
Anti-NMDA receptor encephalitis
This is related to NMDA receptor. Anti-NMDA receptor encephalitis has good reaction to treatment. It may be a comorbidity of paraneoplastic LE encephalitis and mainly affects young women with benign teratoma. The NMDA receptor antibody can be found in blood and CSF examination. The disease has severe clinical manifestations and is potentially fatal. Most patients can recover after tumor removal and immunotherapy.21
Treatment
Although we have no official test, we generally think that clinical symptoms of LGI1 LE patient can be relieved after hormone therapy and immunotherapy, including methylprednisolone injection, immunoglobulin injection, plasma exchange, and immune support. After the immunotherapy, the patients no longer have epileptic seizure and have improved memory. Epilepsy, particularly FBDS, disappears immediately, but the cognitive function improves slowly.9 Irani et al22 found that cognitive function damage only affects patients who had no immunotherapy, indicating that early treatment has good prognosis and can prevent disease progression. After immunotherapy, cognitive function and epilepsy can be improved significantly. Symptom remission is usually related to antibody titer level; therefore, reducing LGI1 antibody titer is important. In the early stage, the patients are given the treatments of intervention, particularly immunoglobulin, and blood exchange combined with hormone therapy is better than simplex glucocorticoid.23,24 Shin et al23 found that large doses of glucocorticoid combined with blood exchange/intravenous infusion may protect the structure and function of the mediotemporal lobe and can avoid the possibility of refractory epilepsy in the future. Some studies suggest that effective and long-term immunotherapy, which not only reduces the epileptic seizure but also prevents long-term complications, including hippocampus atrophy and persistent memory damage, should be given.11 This study reports that after hormone therapy, the symptoms improved and then reappeared. Meanwhile, the symptoms improved after treatment with a combination of hormone therapy and immunoglobulin. A total of six of the 11 patients in this study took the antiepileptic drugs. Therefore, we think that antiepileptic drugs should be given to patients with epileptic seizures, except FBDS.
Prognosis
In this study, all patient symptoms improved, but the symptoms relapsed after infection and improved again after immunotherapy. The recurrence rate of LE patients (positive LGI1) is 0%–20%. Moreover, relapse 7 years after the first treatment is possible.1,2,8
Conclusion
LE is rare clinically and can be easily missed or misdiagnosed. Therefore, we should enhance our understanding of this disease. Of course in our research, there are some deficiencies such as less sample size, short follow-up times, absence of antibodies titer by quantitative,25 and so on. We need some large samples to conducting evidence-based medicine research for improving diagnosis and treatment. The main symptoms of LE (positive LGI1 antibody) are cognitive disorders (mainly recent memory decrease), epileptic seizure, FBDs, hyponatremia, dysphrenia, and sleep disorders. Brain MRI scans show abnormal signals in the mediotemporal lobe and hippocampus. LGI1 antibody can be detected in the blood/CSF. This condition can be associated with an autoimmune disease and can be rarely manifested with tumors. Once diagnosed, patients should be subjected to immunotherapy and long-term maintenance treatment to alleviate their symptoms, improve prognosis, and avoid hippocampal atrophy and intractable epilepsy.
Acknowledgment
Dr Meiling Wang and Dr Xiaoyu Cao are the joint first authors of this paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGl1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9(8):776–785. | ||
Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain. 2010;133(9):2734–2748. | ||
Brierley JB, Corsellis JA, Hierons R, et al. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain. 1960;83:357–368. | ||
Bien CG, Elger CE. Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav. 2007;10(4):529–538. | ||
Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10(8):759–772. | ||
Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001;50(1):73–78. | ||
Gastaldi M, Thouin A, Vincent A, et al. Antibody-mediated autoimmune encephalopathies and immunotherapies. Neurotherapeutics. 2016;13(1):147–162. | ||
Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res. 2006;71(2–3):135–141. | ||
van Sonderen A, Schreurs MW, Wirtz PW, Sillevis Smitt PA, Titulaer MJ. From VGKC to LGI1 and Caspr2 encephalitis: the evolution of a disease entity over time. Autoimmun Rev. 2016;15(10):970–974. | ||
Peter-Derex L, Device P, Rogemond V, et al. Full recovery of agrypnia associated with anti-Lgil antibodies encephalitis under immunomodulatory treatment: a case report with sequential polysomnographic assessment. Sleep Med. 2012;13(5):554–556. | ||
Malter MP, Frisch C, Schoene-Bake JC, et al. Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol. 2014;261(9):1695–1705. | ||
Vincent A. Developments in autoimmune channelopathies. Autoimmun Rev. 2013;12(6):678–681. | ||
Lalic T, Pettingill P, Vincent A, Capogna M. Human limbic encephalitis serum enhances hippocampal mossy fiber-CA3 pyramidal cell synaptic transmission. Epilepsia. 2010;52(1):121–131. | ||
Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain. 2013;136(10):3151–3162. | ||
Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede LGI1 antibody limbic encephalitis. Ann Neurol. 2011;69(5):892–900. | ||
Fauser S, Talazko J, Wagner K, et al. FDG-PET and MRI in potassium channel antibody-associated non-paraneoplastic limbic encephalitis: correlation with clinical course and neuropsychology. Acta Neurol Scand. 2005;111(5):338–343. | ||
Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in cases. Ann Neurol. 2012;72(2):241–255. | ||
Maki Y, Takashima H. Clinical features and treatment of Hashimoto encephalopathy. Brain Nerve. 2016;68(9):1025–1033. | ||
Sun L, Cao J, Liu C, Lv Y. Creutzfeldt-Jakob disease versus anti-LGI1 limbic encephalitis in a patient with progressive cognitive dysfunction, psychiatric symptoms, involuntary facio-brachio-crural movement, and an abnormal electroencephalogram: a case report. Neuropsychiatr Dis Treat. 2015;11(11):1427–1430. | ||
Takada LT, Kim MO, Cleveland RW, et al. Genetic prion disease: experience of a rapidly progressive dementia center in the United States and a review of the literature. Am J Med Genet B Neuropsychiatr Genet. 2017;174(1):36–69. | ||
Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167–177. | ||
Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain. 2010;133(6):1655–1667. | ||
Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGl1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol. 2013;265(1–2):75–81. | ||
Quek AM, Britton JW, McKeon A, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol. 2012;69(5):582–593. | ||
Hart IK, Maddison P, Newsom-Davis J, Vincent A, Mills KR. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain. 2002;125(8):1887–1895. | ||
Shen N, Ren H, Wu J, Feng J, Cui L, Fang S. A rare case of limbic encephalitis with anti leucine-rich gliomainactivated-1(LGI1) antibodies. Neuro Endocrinol Lett. 2014;35(2):95–97. | ||
Barajas RF, Collins DE, Cha S, Geschwind MD. Adult-onset drug-refractory seizure disorder associated with anti-voltage-gated potassium-channel antibody. Epilepsia. 2010;51(3):473–477. | ||
Rodríguez Cruz PM, Pérez Sánchez JR, Alarcón Morcillo C, et al. Alternating faciobrachial dystonic seizures in LGI1-antibody limbic encephalitis. Pract Neurol. 2016;16(5):387–388. | ||
Liu J, Li M, Li G, Zhou C, Zhang R. Anti-leucine-rich glioma-inactivated 1 limbic encephalitis: a case report and literature review. Exp Ther Med. 2016;11(1):315–317. | ||
Mayasi Y, Takhtani D, Garg N, et al. Leucine-rich glioma-inactivated protein 1 antibody encephalitis, a case report. Neurol Neuroimmunol Neuroinflamm. 2014;1(4):1–3. | ||
Messelmani M, Fekih-Mrissa N, Zaouali J, et al. Limbic encephalitis associated with leucine-rich glioma-inactivated 1 antibodies. Ann Saudi Med. 2015;35(1):76–79. | ||
Szots M, Marton A, Kover F, et al. Natural course of LGI1 encephalitis: 3–5 years of follow-up without immunotherapy. J Neurol Sci. 2014;343(1–2):198–202. | ||
Wang SJ, Zhao YY, Wang QZ, et al. Limbic encephalitis associated with positive anti-LGI1 and antithyroid antibodies. Neurology. 2016;86(2):16–18. | ||
Krastinova E, Vigneron M, Le Bras P, Gasnault J, Goujard C. Treatment of limbic encephalitis with anti-glioma-inactivated 1 (LGI1) antibodies. J Clin Neurosci. 2012;19(11):1580–1582. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.