")
Back to Journals » Patient Preference and Adherence » Volume 16
Clinical Evaluation of Siponimod for the Treatment of Secondary Progressive Multiple Sclerosis: Pathophysiology, Efficacy, Safety, Patient Acceptability and Adherence
Authors Sabsabi S, Mikhael E, Jalkh G, Macaron G , Rensel M
Received 22 February 2022
Accepted for publication 5 May 2022
Published 24 May 2022 Volume 2022:16 Pages 1307—1319
DOI https://doi.org/10.2147/PPA.S221882
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Johnny Chen
Sajida Sabsabi,1 Elio Mikhael,2 Georges Jalkh,1 Gabrielle Macaron,1,3 Mary Rensel3
1Department of Neurology, Hotel Dieu de France Hospital, Saint Joseph University, Beirut, Lebanon; 2Department of Internal Medicine, Hotel Dieu de France Hospital, Saint Joseph University, Beirut, Lebanon; 3Mellen Center for Multiple Sclerosis Treatment and Research, Cleveland Clinic, Cleveland, OH, USA
Correspondence: Mary Rensel, Email [email protected]
Introduction: A number of disease-modifying therapies have been approved for use in relapsing-remitting multiple sclerosis (MS) in the past two decades. However, only few treatment options are available for patients with secondary progressive multiple sclerosis (SPMS). Siponimod has recently been approved for use in patients with active forms of SPMS (who experience clinical relapses or new lesions on MRI superimposed on secondary progression independent of relapse activity).
Objective: The aim of this article is to provide a comprehensive review on the mechanism of action, efficacy, safety, cost effectiveness and patient adherence with siponimod.
Methods: We performed a PubMed search using the search terms: “siponimod”, “secondary progressive multiple sclerosis”, “sphingosine 1-phosphate modulators”. Titles and abstract were screened and selected for relevance to the key section of this article.
Findings: Siponimod is an oral sphingosine-1-phosphate receptor (S1PR) modulator with selectivity to S1PR-1 and 5. Modulation of this receptor on lymphocytes causes its internalization and degradation, preventing their egress from lymphoid tissues to the blood. In the pivotal Phase 3 randomized controlled trial EXPAND, siponimod was superior to placebo in reducing the risk of disability progression confirmed at 3 and 6 months, as well as the development of new MRI lesions and the rate of brain volume loss. Secondary analysis also showed a benefit on measures of cognitive functioning. The risk of lymphopenia and first-dose bradycardia appears to be lower with siponimod compared to non-selective S1P1R modulators. Different CYP2C9 genotypes affect the metabolism of siponimod; hence, genetic testing is required to adapt the titration and final dose accordingly.
Conclusion: Long-term extension and real-world studies will allow further evaluation of efficacy and safety in this population. Future research should focus on better defining SPMS, and identifying biomarkers of progression and outcome measures of treatment response in this category of patients.
Keywords: secondary progressive multiple sclerosis, siponimod, efficacy, safety
Introduction
Multiple sclerosis (MS) is a chronic inflammatory, autoimmune disease of the central nervous system (CNS) and a common cause of non-traumatic disability in young adults.1 The most common disease course is characterized by the development of sub-acute clinical demyelinating events (relapses) with complete or incomplete recovery, referred as relapsing remitting MS (RRMS). Inflammatory lesion formation and secondary axonal transection within the lesions is frequent in RRMS, especially early in the disease course.2 Data from natural history cohorts suggest that about 85% of patients will eventually evolve into a secondary progressive course (SPMS), whereas data from recent cohorts in the treatment era have shown a much lower rate of conversion (15–30)%3,4 with a median time of 19 years from first MS symptom and 12 years from MS diagnosis.5,6 SPMS is a retrospective clinical diagnosis and there is no established criteria or biomarker to determine the exact time of conversion from RRMS to SPMS. Active lesions are a less prominent feature of progressive MS, which is mostly driven by more diffuse pathological mechanisms such as diffuse axonal injury, microglial activation in normal appearing grey and white matter, whole brain atrophy, smoldering lesions, and subpial lesions. Patients with SPMS can develop superimposed relapses (clinical activity) or new lesion formation (radiological activity).7 The distinction between active and inactive progressive MS has important therapeutic implication as most approved disease-modifying therapies (DMTs) are effective on the inflammatory component causing clinical relapses and new lesion formation in the brain and spinal cord.
Recent studies suggest that the early use of high-efficacy DMTs in RRMS is associated with a lower risk of conversion to SPMS.8–11 However, the escalation approach (starting with lower efficacy DMTs and switching in case of breakthrough disease activity) remains widely used.9 In the past decade, several newer DMTs have been approved for use in MS. Injectable DMTs such as interferons β and glatiramer acetate as well as intravenous mitoxantrone were the only approved therapies in 2000 followed by natalizumab in 2006.3 Injectable DMTs, with some exceptions such as newer sub-cutaneous anti-CD-20 therapies are limited by their route of administration, lower efficacy, and poor tolerability. Mitoxantrone is limited by its toxicity and its use has been abandoned in MS. Oral DMTs are now available with a more practical route of administration, higher efficacy, and better tolerability. Sphingosine-1-phosphate receptor modulators (S1PRM) represent an important DMT category in MS therapeutics. Fingolimod (Gilenya®, Novartis), a non-selective S1PRM, was FDA-approved as the first oral drug for RRMS in 2010. Since then, other more selective S1PRM have been developed, including siponimod (Mayzent®, Sanofi Genzyme), which received its FDA approval in March 2019 for use in RRMS and active SPMS.
In this review article, we will discuss the characteristics of SPMS, diagnostic and treatment challenges, and provide a detailed review of the efficacy, safety, patient acceptance and adherence of siponimod.
Secondary Progressive Multiple Sclerosis (SPMS)
Classifications and terminologies in progressive MS have gone through several modifications. In the latest disease course definition by Lublin et al in 2014,7 the importance of distinguishing active vs inactive progressive MS was highlighted (based on the presence or absence of relapses or new MRI lesions during the follow-up time), as well as the recognition of progressive disability worsening (based on the presence or absence of progression independent of relapses during the previous year).7 SPMS, as opposed to primary progressive MS (PPMS), requires an initial course of RRMS which progresses into SPMS. A recent study revealed that a quarter of RRMS patients progress to SPMS within the first 10 years, then half of them by 20 years, and more than 75% by 30 years.12 The estimated prevalence of SPMS across studies and countries is variable, and this may be attributable to differences in SPMS definition, study design, and genetic differences between populations.
Nevertheless, the early detection of SPMS remains a challenge for clinicians. In fact, there are no clear clinical, biological, or imaging biomarkers for the diagnosis of SPMS.7,13 Consequently, the diagnosis of SPMS is frequently retrospective with an estimated diagnosis delay of 3 years.14 Irreversible axonal damage and disability accumulation in MS might not initially be present as clinically significant disabilities due to compensatory mechanisms, but can manifest later in the disease course when these mechanisms are surpassed.15–17 Therefore, the term “silent progression” has been recently used to describe this phase of the disease.18
Detecting the transition from RRMS to SPMS also has implications when it comes to treatment options. Focus on reducing relapse occurrence and new lesion formation is the mainstay of RRMS treatment target, whereas delaying progression of disability remains the primary goal in progressive MS.3,19 While the range of pharmaceutical options have been rapidly expanding in MS during the last ten years, it has more so been the case for RRMS than for progressive forms of MS. One of the reasons behind the less pronounced advances in SPMS compared to RRMS are linked to trial methodology.20–23 Phase 3 trials often last 2 years or less, which might not be enough to detect an effect of therapy on the slope of disability worsening in progressive MS.24 Moreover, patients included in trials are not representative of all progressive MS patients, as they are often younger and have evidence of disease activity.20–23 Finally, efficacy outcomes in SPMS trials are not clearly defined and often reflect inflammatory processes rather than neurodegeneration.20–23 Efforts are significantly increasing to enhance research in progressive MS.
DMTs approved for use in SPMS include siponimod and cladribine, both approved by the FDA and EMA25 (European Medicines Agency). National guidelines such as the European Committee of Treatment and Research in Multiple Sclerosis (ECTRIMS)/European Academy of Neurology (EAN) and the American Academy of Neurology (AAN) clinical practice guidelines, both published in 2018 (prior to approval of Siponimod) are aligned on general treatment concepts.25,26 However, for active SPMS, the ECTRIMS/EAN guidelines recommend initiation of interferons, mitoxantrone, cladribine, or ocrelizumab, whereas the AAN guidelines make no specific recommendation.27 Since many decisions that arise in the real-world settings are not supported by clear practice guidelines, incorporating evidence from observational studies into recommendations will be increasingly necessary.27
Sphingosine 1 phosphate Receptor Modulators
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid which binds to different G-protein-coupled receptors called (S1PR).28 These receptors are present in different cells such as lymphocytes, cardiomyocytes, endothelial cells, fibroblasts, neurons, astrocytes, macular cells, microglia and oligodendrocytes.29 Table 1 details S1PR distribution and relation to active S1PRM. Specifically, S1PR1-3 are ubiquitous, S1PR-4 is mostly expressed in lymphoid tissue, and S1PR-5 in the spleen and oligodendrocytes.28 S1P is involved in lymphocytes egress from thymus and lymph nodes to the blood, cardiac function, inflammation, cell migration and vasogenesis among other several functions.30 S1PRM indirectly antagonize the receptor’s function by binding to one or several receptor subtypes (S1PR-1 to 5) causing their internalization and degradation.
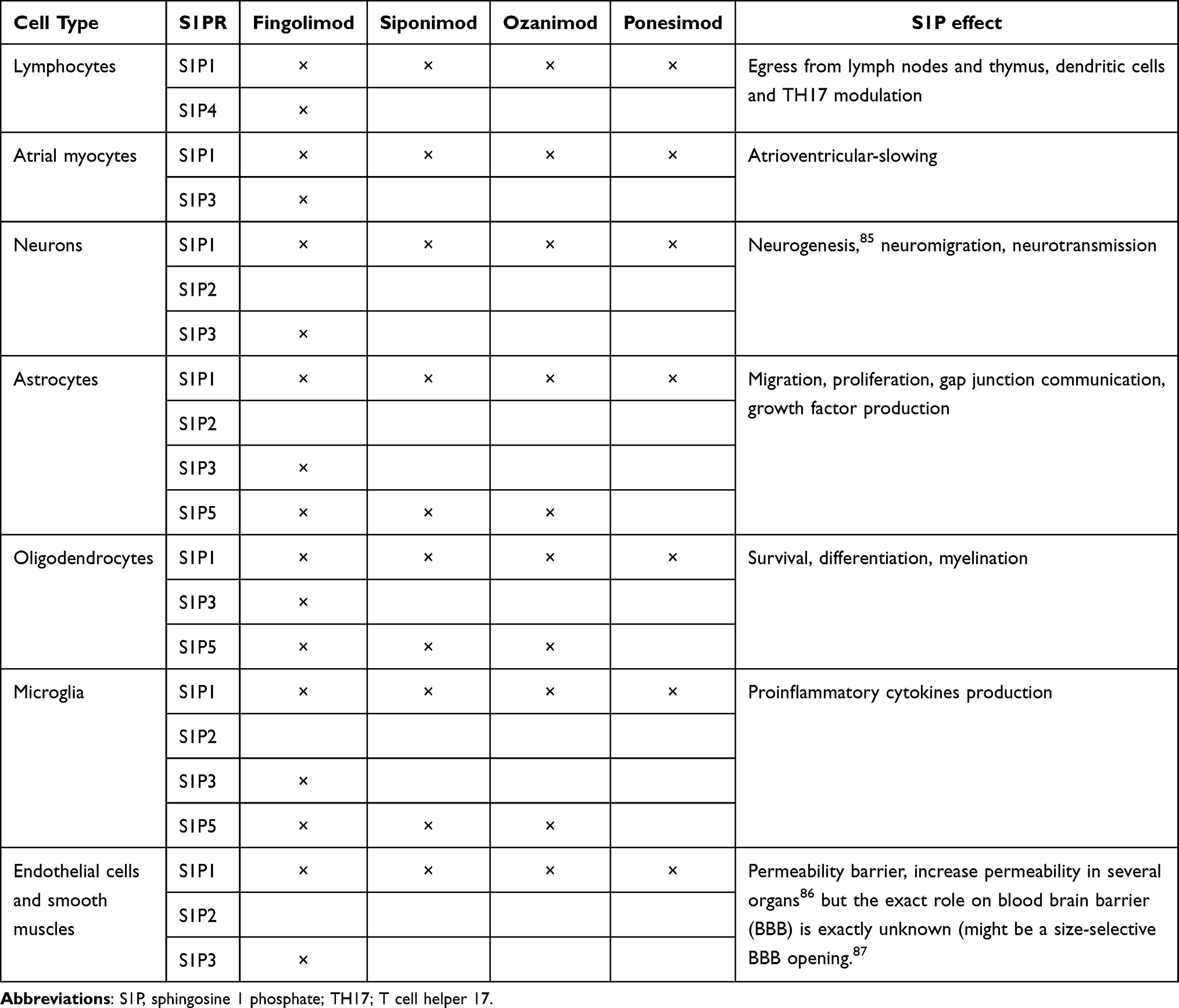 |
Table 1 Distribution of Sphingosine 1 Phosphate Receptors by Cell Type, Effect and Corresponding S1P Modulators |
Fingolimod (FTY720, Gilenya®, Novartis Pharmaceuticals AG, Basel, Switzerland) was the first developed oral DMT and S1PRM for MS.31 Fingolimod undergoes phosphorylation by sphingosine kinase to become active and then binds to S1PR-1, 3, 4, and 5. S1PR-1 antagonism prevents lymphocyte egress from lymphoid tissues. S1PR-3 modulation induces atrioventricular slowing in rodent models,32 however, this adverse effect in humans is mainly secondary to S1PR-1 modulation.28 In the pivotal trials, first-degree atrioventricular block occurred in 4.7%, symptomatic bradycardia in 0.5%, and second-degree atrioventricular block occurred in 0.2% of participants after the first dose, which resulted in the institution of a strict mandatory monitoring protocol for 6 hours after the first dose.33 Other side effects as macular edema, bronchoconstriction and vasoconstriction were also noted. Fingolimod has a long half-life (>30 hours).30 Despite its efficacy in RRMS, fingolimod failed to improve the course of PPMS (INFORMS trial).34
In light of the unique mechanism of action and efficacy of fingolimod, newer, more selective small-molecule S1PRM with shorter half-lives and potentially less adverse effects were developed. Siponimod (BAF312, Mayzent®, Novartis Pharmaceuticals AG, Basel, Switzerland) selectively binds to S1PR-1 and 5. It is the first FDA-approved drug for the treatment of active SPMS.34 Other selective S1PR modulators demonstrated their efficacy and safety in RRMS such as ozanimod (selective for S1PR-1 and 5) and ponesimod (selective for S1PR-1).35 Their similarity to siponimod regarding the selectivity pattern makes these drugs potential candidates for SPMS treatment.
Unlike fingolimod, siponimod binds directly to S1PR-1 and 5 without undergoing phosphorylation.29 For a better understanding of the mechanism of action of S1PRM, it is important to know the interaction between CNS cells in the active progressive phase of MS. How and why the first immune attack to CNS occurs is unclear but inflammation and demyelination within the CNS, leads to more tissue damage and antigen release to the periphery and subsequent recruitment of more lymphocytes to invade the CNS again.3 Moreover, pro-inflammatory reactions are mediated by microglia within the CNS in response to autoimmune process. In addition, astrocytes are considered the most important regulators of immunocompetent T lymphocytes and they seem to be implicated in astrocyte-induced neurodegeneration. It is believed that immune-mediated demyelination, axonal injury, astrocytic gliosis and microglial activation play an important role in the activity and the progressive course of MS.3
Siponimod inhibits the chemotaxis of immune cells and the migration of lymphocytes to blood and CNS, but also acts directly on the CNS. This drug promotes oligodendrocytes remyelination by acting on S1PR5, limits the pro-inflammatory role of microglia and modulates astrocytic function. The latter role was confirmed in in vitro experiments, showing that siponimod activates glial Nrf2, inducing in vivo anti-oxidant, anti-inflammatory and neuroprotective responses and inhibiting astrocytic NFkB primarily involved in pro-inflammatory reactions, scar formation and neurodegeneration.36 The direct effect of siponimod on CNS cells was also observed after continuous direct intracerebroventricular infusion of the molecule to the brain of mice, which reduced in autoimmune encephalomyelitis (EAE) model severity of EAE without even affecting the number of peripheral lymphocytes.31
Siponimod
Efficacy
EXPAND Trial results
The pivotal trial comparing the efficacy of siponimod to placebo in SPMS, EXPAND,37 was a Phase III, double-blind, randomized controlled trial, enrolling 1651 patients with SPMS from 292 centers in 31 countries, assigned to receive oral siponimod 2 mg per day or placebo (2:1) and followed for up to 3 years. Included patients were between 18 and 60 years old, with moderate-to-advanced disability (Expanded Disability Status Scale (EDSS) score of 3·0–6·5 at screening), and no evidence of relapses in the 3 months prior to randomization. Patients with immunological, cardiac, or pulmonary conditions, ongoing macular edema, uncontrolled diabetes, CYP2C9*3/*3 genotype, and varicella zoster virus antibody negative status were excluded. The primary end-point was 3-month confirmed disability progression (CDP) defined as a 1-point increase in EDSS if the baseline score was 3·0–5·0, or a 0·5-point increase if the baseline score was 5·5–6·5. Secondary end-points included the time to 3-month confirmed worsening of the timed 25-foot walk test (T25FW) of at least 20% and baseline change in T2 lesion volume on brain MRI, 6-month CDP, annualized relapse rate (ARR), time to first relapse, proportion of relapse-free patients, and percentage change in brain volume from baseline among others.
Mean age of participants at baseline was 48 years, mean disease duration was 16.8 years, and mean time to conversion to SPMS was 3.8 years. Importantly, 64% of patients did not experience a clinical relapse in the past 2 years and 56% needed assistance for walking. In other words, this study included a typical SPMS cohort, an under-represented population in classic trials. Siponimod was superior to placebo in reducing the risk of 3-month and 6-month CDP. Siponimod was not superior to placebo on reducing worsening on the T25FW. Siponimod was also superior to placebo on other secondary outcome measures (Figure 1). Importantly, the cumulative number of gadolinium-enhancing lesions and mean number of new or enlarging T2 lesions on all post-baseline MRIs were lower with siponimod (adjusted mean (standard deviation) 0.08 (0.07–0.10) and 0.70 (0.58–0.84) respectively) compared to placebo (adjusted mean (standard deviation) 0.60 (0.47-0.76); relative risk 0.14 (0.10–0.19) and 3.60 (3.03–4.29); relative risk 0.19 (0.16–0.24), respectively, p < 0.0001 for both). Moreover, there was an in-between group difference in percent brain volume change from baseline of 0.15% (0.07–0.23; p-value 0.0002) in favor of siponimod.
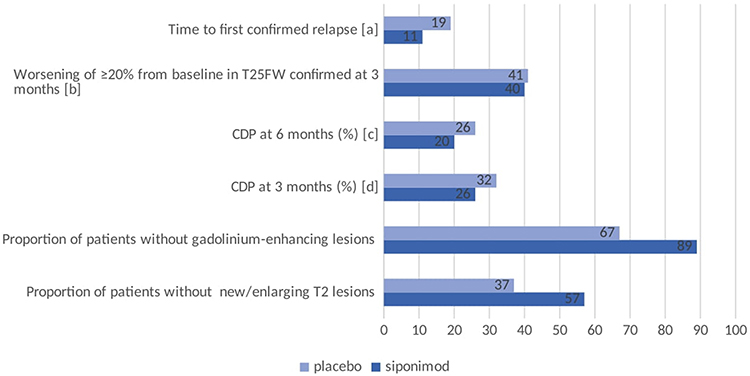 |
Figure 1 Efficacy of siponimod vs placebo on different outcome measures in the EXPAND trial37 add all outcome measures. [a] Hazard ratio [HR] 0·54, 95% CI 0·41–0·70; risk reduction 46%; p < 0·0001. [b] HR 0·94, 95% CI 0·80–1·10; risk reduction 6%; p = 0·44. [c] HR 0·74, 95% CI 0·60–0·92; p = 0·0058. [d] HR 0·79, 95% CI 0·65–0·95; risk reduction 21%; p = 0·013. Data from Kappos et al.37 Abbreviations: T25FW, timed 25-foot walk; CDP, confirmed disability progression. |
Long-Term Efficacy
Extension studies are essential in assessing long-term efficacy of therapies and are currently not available for siponimod. The AMASIA (impact of Mayzent [siponimod] on secondary progressive multiple sclerosis patients in a long-term non-interventional study in Germany) trial, an open label study across 250 medical centers in Germany was initiated in February 2020 and is expected to terminate in 2025.38
Siponimod and Cognition
Benedict et al assessed the impact of siponimod on cognitive function as a secondary analysis of the EXPAND trial. Cognitive tests were applied at baseline, every 6 months, and at the end of trial. Treatment with siponimod was associated with a lower risk of sustained clinically significant decrease in the Symbol Digit Modalities Test (SDMT, a measure of processing speed) score (hazard ratio [HR] 0.79 [0.65–0.96]; p = 0.0157), and even higher chances of a sustained increase in the SDMT score (HR 1.28 [1.05–1.55]; p = 0.0131). The effect size was larger after 12 months of siponimod use.39 However, no effect on other cognitive tests was observed.
Fingolimod vs Siponimod
The efficacy of fingolimod was evaluated in patients with PPMS in a phase III, double blind, randomized, placebo-controlled trial (INFORMS), and showed that fingolimod was not superior to placebo in slowing disability progression in this cohort.40 An indirect treatment comparison between siponimod in SPMS and fingolimod in PPMS was performed in a recent study. This comparison was feasible because trial design, patient’s baseline characteristics and their inclusion/exclusion criteria and outcome definitions were similar between the two trials. For 3 and 6-months CDP, siponimod and fingolimod were not significantly different (HR 0.80, 95% CI 0.52–1.22; p = 0.3, and HR 0.76, 95% CI 0.48–1.20; p = 0.24 respectively).40,41
Safety of Siponimod
Safety in EXPAND
The safety of selective S1PRM was the key feature as compared to fingolimod. Atrio-ventricular slowing was thought to be secondary to S1PR-3 modulation, as demonstrated in mice model. However, cardiac side effects also occur in S1PR-3-sparing agents. Studies concluded that in humans, cardiac AEs were also attributable to S1PR-1 modulation, via activation of G protein-inwardly rectifying potassium channels (GIRK) expressed on atrial myocytes. The finding outlined species-specific differences in S1PR specificity.42,43
In EXPAND, AEs occurred in 89% in the siponimod vs 82% in the placebo group. Eight percent in the siponimod arm discontinued treatment because of AEs vs placebo (Figure 2).
 |
Figure 2 Frequency of side effects in the siponimod arm compared to the placebo arm in the EXPAND tria. Data from Kappos et al.37 Abbreviation: *LFT, liver function tests. |
On day 7 post treatment with siponimod, the mean decrease of HR was 3.1 beats per minute. Telemetry for up to 6 days after the first siponimod dose did not register any second degree AV block (Mobitz II) or third degree AV block.37 While treatment with fingolimod was associated with an increased risk of basal cell carcinoma and malignant melanoma, this risk did not differ between siponimod and placebo patients.37 Long-term data are however needed to confirm this finding. Infectious AEs were similar between placebo and siponimod group except for the risk of varicella zoster reactivation and herpes infection which was higher with siponimod. The risk of developing cardiac AEs is mitigated by a dose titration protocol (Table 2).44
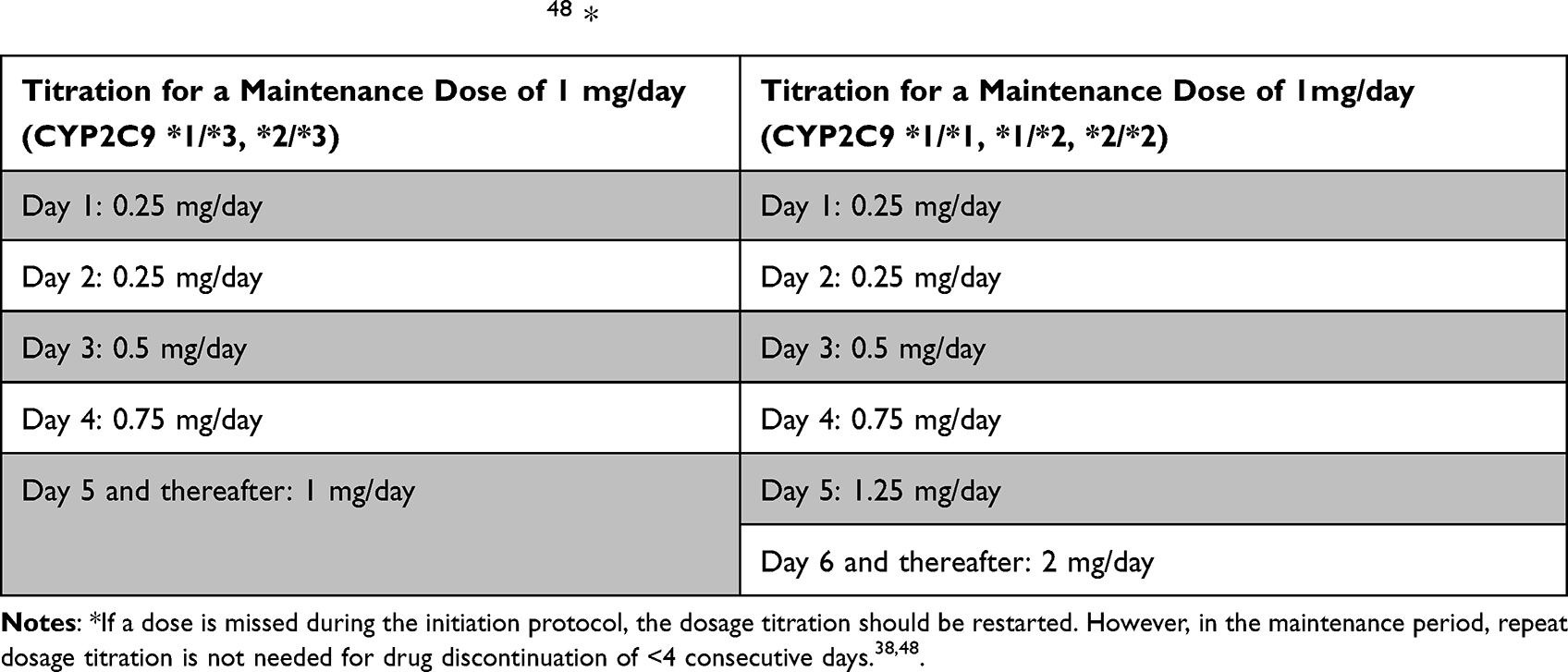 |
Table 2 Siponimod Titration protocol48 * |
Lymphopenia is a potential risk expected with all S1P1R modulators. Grade IV lymphopenia (absolute lymphocyte count <200/mm3) was seen in 1% of patients on siponimod whereas it is observed in up to 18% of patients on fingolimod.45,46 Moreover, after discontinuation of siponimod, lymphocyte count return to normal levels within 10 days in 90% of patients, while it typically takes 14–21 days and can reach 2 months in fingolimod after treatment discontinuation.47
The pre-treatment work-up includes a baseline ECG, ophthalmologic examination, CBC and liver function tests (within 6 months before starting therapy), a pregnancy test, counseling on the use of effective contraception in females of reproductive, and VZV serology or confirmation of prior exposure, along with the CYP2C9 genetic test.48 Respiratory function tests are recommended for patients with pulmonary symptoms because siponimod may cause a decline in respiratory function.49 First-dose cardiac observation is only mandatory in high-risk patients (those with sinus bradycardia, first- or second-degree [Mobitz type I] AV block, or a history of myocardial infarction or heart failure). In general, routine vaccination is recommended prior to treatment start, specifically the VZV vaccine in patients with a negative VZV serology and older individuals.50
CYP2C9 Status
Siponimod is metabolized in the liver via the Cytochrome P450 (CYP) 2C9 enzyme, which contributes to 79.3% of the metabolism, and to a lesser extend via the CYP3A4 enzyme. Siponimod is excreted in the feces. The pharmacokinetics, more specifically the elimination rates of siponimod, differs according to CYP2C9 genotypes. Serum concentration of siponimod increases by twofold with CYP2C9*2/*3 and fourfold with CYP2C9*3/*3 genotype compared to CYP2C9*1/*1 (counts for up to 65% of genotypes of white population). The elimination half-life was also prolonged with CYP2C9*2/*3 and CYP2C9*3/*3 genotypes (reaching 126 hours for the latter compared with 28 hours for CYP2C9 *1/*1)51 Gene testing for CYP2C9 is hence required prior to initiation siponimod to adjust both titration and maintenance dosages (Table 2).
Siponimod is contraindicated in CYP2C9 *3/*3 genotype,48 CYP2C9 *3/*3 is present only in 0.3–0.4% of white individuals.51 CYP2C9/3A4 inhibitors like fluconazole interact with siponimod elimination,51,52 and are not recommended to use concomitantly with siponimod.48 On the other hand, CYP2C9/3A4 inducers can decrease serum drug concentration. For instance, the dual strong CYP3A4/moderate CYP2C9 inducers reduce siponimod exposure by up to 78%, and are to be avoided for all CYP2C9 genotypes. Moderate/strong CYP3A4 inducers are not recommended for patients with CYP2C9*1/*3 and *2/*3 genotypes.52
Extension Studies and Latest Case Reports and Case Series
Long-term data regarding safety and efficacy of siponimod is lacking to date, since this medication was only recently approved and used. The AMASIA study as discussed above will be helpful to inform us on real-world safety in the next few years.38 To date, one case of breakthrough disease after switching from fingolimod to siponimod without a washout period was reported in a 49-year-old female patient.53
Safety Monitoring
To mitigate the risk of adverse events, a series of follow-up tests should be performed as seen in Table 3.
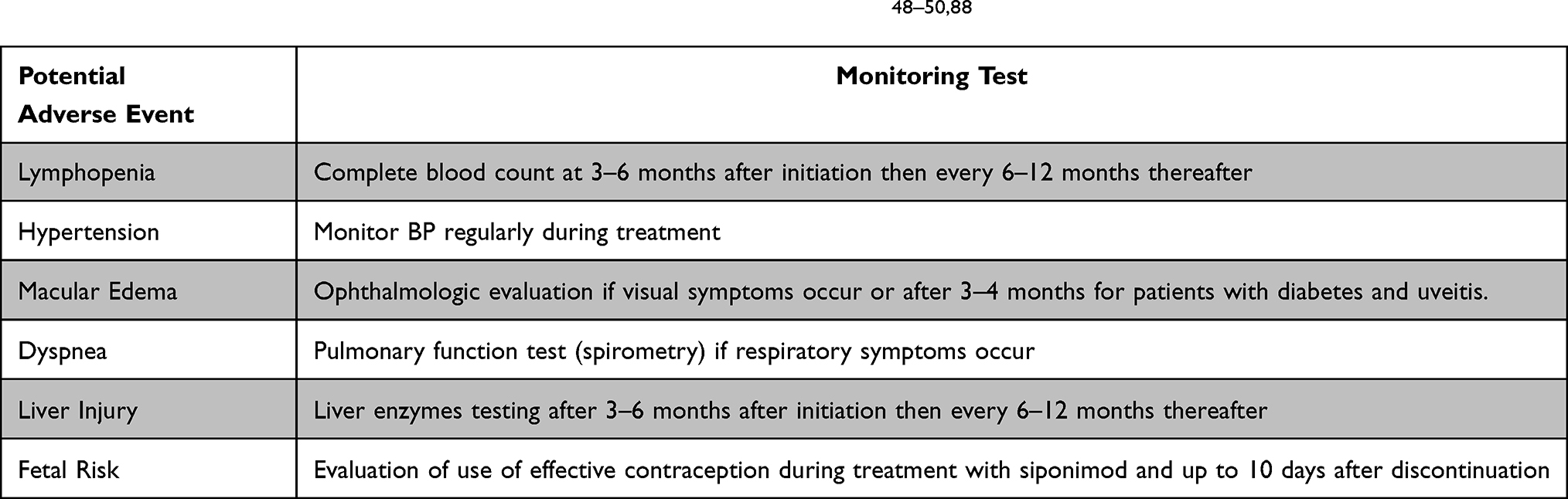 |
Table 3 Monitoring of Patients on Siponimod per FDA Recommendations.48–50,88 |
When to Discontinue Siponimod?
For all DMTs, it remains unclear when to discontinue DMTs. In the current practice, experts recommend discontinuing DMTs in patients with progressive disease without evidence of clinical or radiological activity who continue to worsen despite treatment. For example, The Canadian agency for drugs and technologies in health (CADTH) recommends the discontinuation of siponimod if the EDSS score reaches 7 (equivalent to being wheelchair-bound), of if there is a worsening of the T25W test of ≥20% while on siponimod.54
It is important to mention that no cases of rebound inflammatory activity after siponimod discontinuation are reported to date. However, a potential risk exists since it has been described with fingolimod. Thus, close monitoring after siponimod discontinuation is warranted.
COVID-19 and Siponimod
In the Coronavirus disease-19 (COVID-19) era, the vulnerability to the SARS-CoV-2 viral infection itself and the COVID-19 vaccine response with different DMTs is in the spotlight. Recent data suggest that the risk of serious COVID-19 infection is not increased in patients on S1PRM (particularly fingolimod), however the vaccine response is significantly blunted.45,55 An observational study of 54 SPMS patients (mean age of 54 years) treated with siponimod with confirmed SARS-CoV-2 infection, showed that 85.7% of those patients were asymptomatic, or had mild-to-moderate infection, 17% were hospitalized (compared to 14% in overall population), and 6% died (mortality of 5% in the general population). The slight increase in hospitalization and death may be due to older age and disability in the siponimod population, making this group of patient more prone to severe infection independently of treatment.56,57
The benefit of COVID-19 vaccination for patients on DMTs often outweigh the risk of not taking the vaccine and the risk of stopping DMTs abruptly to receive the vaccine; however, this decision should be done in a case-by-case approach. A recent study reported significantly lower post-vaccination reactive IgG rates in patients receiving anti-CD20 therapies and S1PRM.45 Even though S1PRM can reduce vaccine efficacy, vaccination is nevertheless encouraged in all patients.56,57,58
A Phase 4, multicentric, prospective trial evaluating the development of functional anti-SARS-CoV-2 antibodies and T-cell titers following SARS-CoV-2 modRNA vaccines is ongoing, and siponimod-treated SPMS patients will be included. The study started in March 2021, and should be extended over 14 months (NCT04792567).
Cost-Effectiveness
In the USA (according to the Institute for Clinical and Economic Review) and Canada (according to the National Institute for Health and Care Excellence), siponimod was considered a non-cost-effective agent.59 In Canada, a reduction of 63% of its price was suggested for this drug to be cost-effective as compared to best supportive care with a threshold of $50,000 per quality-adjusted life-year (QALY).54 In the UK, the National Institute for Health and Care Excellence (NICE) considered siponimod a non-cost effective in June 2020 but after price reduction by the campaigners, this decision had been cancelled. In Switzerland, siponimod, compared to beta interferon drugs in treatment of active SPMS, was labeled as cost effective in April 2021.60
Patient Adherence and Expectations
Patient adherence to DMT is crucial for optimal response to treatment. It has previously been reported that a level of adherence above 80% in terms of proportion of days covered (PDC) contributes to the reduction of costs in MS management (including risk of hospitalization and outpatient visits).61 In the only study reporting adherence to siponimod in MS,62 a PDC above 80% was observed in 81.1% of patients with a follow-up duration of more than 6 months, and in 82.4% of patients with a follow-up duration of more than 12 months.62 Similarly, a meta-analysis reported PDC above 80% in 75.6% of patients on oral DMT (not including siponimod) during a follow-up of one year.63 Some demographic characteristics increase the risk of non-adherence, such as age and disease duration. In fact, non-adherence is higher in patients with pediatric-onset MS and adolescents with MS and increases with disease duration.64 Of note, siponimod is not approved for use in this population. Numerous psychosocial factors also drive non-adherence, including cognitive impairment and psychiatric comorbidities, poor socio-economic status, lack of peer support, and higher level of physical disability.64–66
Factoring patient preferences in treatment choices is a key element in optimizing adherence,67 as recommended in the AAN and the EAN guidelines for MS management.25,26 While data on patient expectations regarding siponimod in SPMS is lacking, patient expectations from other DMTs have been the subject of recent studies.68–73 Among the patient concerns, the method of administration, safety profiles, and efficacy of therapy are the 3 main themes of interest.68–73 Concerning the preferred route of administration, prior studies suggest that oral DMT is favored by patients,68,69,72,73 whereas in a newer study, infusions seem to be gaining popularity, particularly when less than three injections per year are required.70 Regarding safety, liver toxicity, severe adverse events, and common side effects were the most important for the patients.74 A few studies evaluated whether patients prioritize safety or efficacy. The results mainly differed between treatment-naïve patients with shorter disease duration and patients who have been exposed to DMTs with a longer time since diagnosis. Patients within five years of the diagnosis were more concerned about adverse events than treatment efficacy, whereas patients who were diagnosed for >5 years had opposite preferences.69 These results were later confirmed by Bauer et al with a cut-off of ten years since diagnosis.70
When comparing the patient and the provider goals and concerns of treatment, patients seem to focus on specific symptoms (ability to walk, vision loss, cognitive dysfunction, body strength), whereas physicians were more concerned about the overall progression of the disease.71 Among reported physician goals in MS treatment, preventing disability ranked first, followed by efficient DMT choice, and quality of life improvement.71 Previous studies have suggested that demographic factors such as sex and age, the level of disability and prior use of DMT can predict adherence to treatment.75,76 Physician can rely on several factors that can predict patient adherence namely treatment goals, past treatments, disease course and demographics.
Future Directions
Several challenges in the care and treatment of patients with SPMS are to be mentioned. First, there is no clear consensus on the definition of SPMS and biomarkers of transition from RRMS to a SPMS are lacking. In phase 3 SPMS trials, a definition of SPMS is used in the inclusion criteria, yet medications such as siponimod and cladribine are only approved for active SPMS. Some studies have worked on finding an objective and accurate definitions of SPMS, both active and inactive, to enable comparability of studies and guide clinical decisions.13
The lack of consensus on the definition of SPMS in clinical trials is also shared in clinical practice. Clinical progression independent of relapse activity can be very difficult to capture in clinical practice, which makes the diagnosis of SPMS retrospective and delayed by about 3 years after a period of clinical uncertainty.14 Most definitions take into account worsening on the EDSS; however, this instrument is not consistently used in clinical practice and lacks sensitivity to evaluate functional systems other than ambulation such as upper extremity and cognitive functions and could fail to capture “progression” in other important quality of life indicators for MS patients.77 To mitigate this limitation, modern trials have used a variety of outcome measures to assess treatment efficacy. The AMASIA trial is an example, and will evaluate the EDSS coupled to the SDMT for cognitive functioning.78,79 The latter composite outcome has been validated for use in MS.80 Other outcome measures focused on longitudinally and objectively capturing progression independent of relapses are needed. Technology-enabled tools to capture such data and analyse them at the individual level can be incorporated in clinical practice and have emerged in the past few years. Standardized definition and technology-enabled tests will hopefully fill the needed gap to better identify the onset of clinical progression.81
Recent research efforts to better understand the heterogeneity of SPMS and of therapeutic responses are increasing. For example, data from the AMSIA trial will be integrated to the MSD-3D82 virtual platform designed to store and compare patients’ data from various clinical studies, and could help the MS community to compare data from different studies. Possible examples include the PANGAEA 2.0 EVOLUTION study, comparable to AMASIA in its methodology, and designed to assess patients with SPMS or high-risk RRMS treated according to the standard of care before the approval of siponimod in SPMS.83
Conclusion
The extensive therapeutic options favor a patient-centered approach in MS management. That said, therapeutic success is not solely measured via objective tools such as clinical and radiological response to treatments but by equally important criteria such as patient adherence to treatment, satisfaction with therapeutic regimen and effect on quality of life. This holistic aspect of patient care is underrepresented in treatment efficacy evaluation. An important benchmark of clinical meaningfulness for patients would be employment. Ocrelizumab was found to positively impact employment in MS and this outcome should be evaluated in other trials.84 Patient-centred outcomes in SPMS trials will help to shape future care of this population. Future studies may include a consensus-driven definition of active SPMS and also focus on patient’s goals and expectations to enhance long-term quality of life of those living with MS.
Disclosure
Sajida Sabsabi, Georges Jalkh, and Elio Mikhael have nothing to disclose.
Gabrielle Macaron has served on advisory boards for Merck, Genentech, and Roche, participated in educational programs for John Hopkins e-Literature review, Neurology Live, and Novartis, participated in lectures for Novartis, Roche, and Biologix, and received fellowship funding from the National Multiple Sclerosis Society Institutional Clinician Training Award ICT 0002 and from Biogen Fellowship Grant 6873-P-FEL (2017–2019).
Dr. Mary Rensel has received research funding (PPD, Biogen, Genentech, CBJ Foundation and NMSS), patient education funds (Genzyme) she also served on DSMC for Biogen was a speaker or consultant for (Serono, Novartis, Genentech, Genzyme, Horizon, TG, Improve Consulting, Kijia and MSAA) and is founder of Brain Fresh and has a joint venture; Brain Ops Group.
References
1. Swallow E, Patterson-Lomba O, Yin L, et al. Comparative safety and efficacy of ozanimod versus fingolimod for relapsing multiple sclerosis. J Comp Eff Res. 2020;9(4):275–285. doi:10.2217/cer-2019-0169
2. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö BL. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338(5):278–285. doi:10.1056/nejm199801293380502
3. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018;391(10130):1622–1636. doi:10.1016/S0140-6736(18)30481-1
4. Cree BAC, Gourraud PA, Oksenberg JR, et al. Long-term evolution of multiple sclerosis disability in the treatment era. Ann Neurol. 2016;80(4):499–510. doi:10.1002/ana.24747
5. Eriksson M, Andersen O. Long-term follow up of patients with clinically isolated syndromes, relapsing-remitting and secondary progressive multiple sclerosis. Mult Scler J. 2003;9:260–274.
6. Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain. 2006;129(3):606–616. doi:10.1093/brain/awl007
7. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis. Neurology. 2014;83(3):278–286. doi:10.1212/WNL.0000000000000560
8. Brown JWL, Coles A, Horakova D, et al. Association of initial disease-modifying therapy with later conversion to secondary progressive multiple sclerosis. J Am Med Assoc. 2019;321(2):175–187. doi:10.1001/jama.2018.20588
9. Buron MD, Chalmer TA, Sellebjerg F, et al. Initial high-efficacy disease-modifying therapy in multiple sclerosis: a nationwide cohort study. Neurology. 2020;95(8):e1041–e1051. doi:10.1212/WNL.0000000000010135
10. Spelman T, Magyari M, Piehl F, et al. Treatment escalation vs immediate initiation of highly effective treatment for patients with relapsing-remitting multiple sclerosis: data from 2 different national strategies. JAMA Neurol. 2021;78(10):1197–1204. doi:10.1001/jamaneurol.2021.2738
11. Iaffaldano P, Lucisano G, Caputo F, et al. Long-term disability trajectories in relapsing multiple sclerosis patients treated with early intensive or escalation treatment strategies. Ther Adv Neurol Disord. 2021;14:175628642110195. doi:10.1177/17562864211019574
12. Khurana V, Sharma H, Medin J. Estimated prevalence of secondary progressive multiple sclerosis in the USA and Europe: results from a systematic literature search (P2.380). Neurology. 2018;90(15Supplement):
13. Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain. 2016;139(9):2395–2405. doi:10.1093/brain/aww173
14. Sand IK, Krieger S, Farrell C, Miller AE. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult Scler J. 2014;20(12):1654–1657. doi:10.1177/1352458514521517
15. Kappos L, Wolinsky JS, Giovannoni G, et al. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020;77(9):1132–1140. doi:10.1001/jamaneurol.2020.1568
16. Davies F, Wood F, Brain KE, et al. The transition to secondary progressive multiple sclerosis: an exploratory qualitative study of health professionals’ experiences. Int J MS Care. 2016;18(5):257–264. doi:10.7224/1537-2073.2015-062
17. Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol. 2014;27(3):271–278. doi:10.1097/WCO.0000000000000094
18. Cree BAC, Hollenbach JA, Bove R, et al. Silent progression in disease activity–free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653–666. doi:10.1002/ana.25463
19. Deibel F, Edwards M, Edwards A. Patients’, carers’ and providers’ experiences and requirements for support in self-management of multiple sclerosis: a qualitative study. Eur J Pers Centered Healthc. 2013;1(2):457. doi:10.5750/ejpch.v1i2.687
20. Fox RJ, Chataway J. Advancing trial design in progressive multiple sclerosis. Mult Scler. 2017;23(12):1573–1578. doi:10.1177/1352458517729768
21. Mills EA, Begay JA, Fisher C, Mao-Draayer Y. Impact of trial design and patient heterogeneity on the identification of clinically effective therapies for progressive MS. Mult Scler J. 2018;24(14):1795–1807. doi:10.1177/1352458518800800
22. Tur C, Montalban X. Progressive MS trials: lessons learned. Mult Scler. 2017;23(12):1583–1592. doi:10.1177/1352458517729460
23. Ontaneda D, Cohen JA, Amato MP. Clinical outcome measures for progressive MS trials. Mult Scler. 2017;23(12):1627–1635. doi:10.1177/1352458517729465
24. Giovannoni G, Cutter G, Pia-Sormani M, et al. Is multiple sclerosis a length-dependent central axonopathy? The case for therapeutic lag and the asynchronous progressive MS hypotheses. Mult Scler Relat Disord. 2017;12:70–78. doi:10.1016/j.msard.2017.01.007
25. Montalban X, Gold R, Thompson AJ, et al. ECTRIMS/EAN Guideline on the pharmacological treatment of people with multiple sclerosis. Mult Scler. 2018;24(2):96–120. doi:10.1177/1352458517751049
26. Rae-Grant A, Day GS, Marrie RA, et al. Practice guideline recommendations summary: disease-modifying therapies for adults with multiple sclerosis: report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology. Neurology. 2018;90(17):777–788. doi:10.1212/WNL.0000000000005347
27. Macaron G, Cohen JA. Integrating multiple sclerosis guidelines into practice. Lancet Neurol. 2018;17(8):658–660. doi:10.1016/S1474-4422(18)30248-5
28. Subei AM, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs. 2015;29(7):565–575. doi:10.1007/S40263-015-0261-Z
29. Behrangi N, Fischbach F, Kipp M. Mechanism of siponimod: anti-inflammatory and neuroprotective mode of action. Cells. 2019;8(1):24. doi:10.3390/cells8010024
30. Pan S, Gray NS, Gao W, et al. Discovery of BAF312 (Siponimod), a potent and selective S1P receptor modulator. ACS Med Chem Lett. 2013;4(3):333–337. doi:10.1021/ml300396r
31. Kipp M. Does siponimod exert direct effects in the central nervous system? Cells. 2020;9(8):1771. doi:10.3390/cells9081771
32. Forrest M, Sun SY, Hajdu R, et al. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309(2):758–768. doi:10.1124/jpet.103.062828
33. Dimarco JP, O’Connor P, Cohen JA, et al. First-dose effects of fingolimod: pooled safety data from three phase 3 studies. Mult Scler Relat Disord. 2014;3(5):629–638. doi:10.1016/j.msard.2014.05.005
34. Faissner S, Gold R. Progressive multiple sclerosis: latest therapeutic developments and future directions. Ther Adv Neurol Disord. 2019;12:1–11. doi:10.1177/1756286419878323
35. Olsson T, Boster A, Fernández Ó, et al. Oral ponesimod in relapsing-remitting multiple sclerosis: a randomised Phase II trial. J Neurol Neurosurg Psychiatry. 2014;85(11):1198–1208. doi:10.1136/jnnp-2013-307282
36. Colombo E, Bassani C, De Angelis A, et al. Siponimod (BAF312) activates Nrf2 while hampering nfκb in human astrocytes, and protects from astrocyte-induced neurodegeneration. Front Immunol. 2020;11. doi:10.3389/fimmu.2020.00635
37. Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263–1273. doi:10.1016/S0140-6736(18)30475-6
38. Ziemssen T, Hoffmann O, Klotz L, Schreiber H, Weber MS, Rauser B. Gaining first insights on secondary progressive multiple sclerosis patients treated with siponimod in clinical routine: protocol of the noninterventional study Amasia. JMIR Res Protoc. 2020;9(7):e19598. doi:10.2196/19598
39. Benedict RHB, Tomic D, Cree BA, et al. Siponimod and cognition in secondary progressive multiple sclerosis: EXPAND secondary analyses. Neurology. 2021;96(3):e376–e386. doi:10.1212/WNL.0000000000011275
40. Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–1084. doi:10.1016/S0140-6736(15
41. Samjoo IA, Worthington E, Haltner A, et al. Indirect comparisons of siponimod with fingolimod and ofatumumab in multiple sclerosis: assessing the feasibility of propensity score matching analyses. Curr Med Res Opin. 2021;37(11):1933–1944. doi:10.1080/03007995.2021.1968362
42. Vermersch P. Sphingosine-1-phosphate receptor modulators in multiple sclerosis. Eur Neurol Rev. 2017;13(1):25–30. doi:10.17925/ENR.2018.13.1.25
43. Legangneux E, Shakeri-Nejad K, Aslanis V, et al. Cardiac effects of siponimod (BAF312) re-initiation after variable periods of drug discontinuation in healthy subjects. Clin Ther. 2016;38(3):631–645.e1. doi:10.1016/j.clinthera.2016.01.021
44. Selmaj K, Li DKB, Hartung HP, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, Phase 2 study. Lancet Neurol. 2013;12(8):756–767. doi:10.1016/S1474-4422(13
45. Cohen JA, Bermel RA, Grossman CI, et al. Immunoglobulin G immune response to SARS-CoV-2 vaccination in people living with multiple sclerosis within multiple sclerosis partners advancing technology and health solutions. Mult Scler J. 2022:135245852110613. doi:10.1177/13524585211061343.
46. Full prescribing information 1 indications and usage Gilenya; 2012. Available from: https://www.novartis.us/sites/www.novartis.us/files/gilenya.pdf.
47. Schweitzer F, Laurent S, Fink GR, Barnett MH, Hartung HP, Warnke C. Effects of disease-modifying therapy on peripheral leukocytes in patients with multiple sclerosis. J Neurol. 2021;268(7):2379–2389. doi:10.1007/s00415-019-09690-6
48. Novartis Pharmaceuticals Corporation. MAYZENT® (siponimod). Mayzent FDA label; 2019. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/209884s000lbl.pdf.
49. Highlights of prescribing information. Metabolism: Clinical and Experimental; 2008. Available from: www.fda.gov/.
50. Jalkh G, Abi Nahed R, Macaron G, Rensel M. Safety of newer disease modifying therapies in multiple sclerosis. Vaccines. 2021;9(1):1–30. doi:10.3390/vaccines9010012
51. Gardin A, Ufer M, Legangneux E, et al. Effect of fluconazole coadministration and CYP2C9 genetic polymorphism on siponimod pharmacokinetics in healthy subjects. Clin Pharmacokinet. 2019;58(3):349–361. doi:10.1007/s40262-018-0700-3
52. Huth F, Gardin A, Umehara K, He H. Prediction of the impact of cytochrome P450 2C9 genotypes on the drug–drug interaction potential of siponimod with physiologically-based pharmacokinetic modeling: a comprehensive approach for drug label recommendations. Clin Pharmacol Ther. 2019;106(5):1113–1124. doi:10.1002/cpt.1547
53. Senzaki K, Ochi H, Ochi M, Okada Y, Miura S, Ohyagi Y. Disease reactivation in a patient with secondary progressive multiple sclerosis after switching treatment from fingolimod to siponimod. eNeurologicalSci. 2021;23:100346. doi:10.1016/j.ensci.2021.100346
54. Canadian Agency for Drugs and Technologies in Health (CADTH). Drug reimbursement recommendation siponimod (Mayzent). Canada; 2019.
55. Simpson-Yap S, De Brouwer E, Kalincik T, et al. Associations of disease-modifying therapies with COVID-19 severity in multiple sclerosis. Neurology. 2021;97(19):e1870–e1885. doi:10.1212/wnl.0000000000012753
56. Sullivan R, Kilaru A, Hemmer B, et al. COVID-19 infection in fingolimod- or siponimod-treated patients. Neurol Neuroimmunol Neuroinflammation. 2022;9(1):e1092. doi:10.1212/nxi.0000000000001092
57. Skoric MK, Rogic D, Lapic I, Dragana Segulja MH. Humoral immune response to COVID-19 vaccines in people with secondary progressive multiple sclerosis treated with siponimod. Mult Scler Relat Disord J. 2020;57:103435. doi:10.1016/j.msard.2021.103435
58. National Multiple Sclerosis Society. COVID-19 vaccine guidance for people living with MS; 2021. Available from: https://www.nationalmssociety.org/coronavirus-covid-19-information/multiple-sclerosis-and-coronavirus/covid-19-vaccine-guidance.
59. Schur N, Gudala K, Vudumula U, et al. Cost effectiveness and budget impact of siponimod compared to interferon beta-1a in the treatment of adult patients with secondary progressive multiple sclerosis with active disease in Switzerland. Pharmacoeconomics. 2021;39(5):563–577. doi:10.1007/s40273-021-01023-8
60. Chaplin S. Siponimod for treating secondary progressive multiple sclerosis. NICE. 2020;31(11–12):34–35. doi:10.1002/psb.1882
61. Kołtuniuk A, Rosińczuk J. Adherence to disease-modifying therapies in patients with multiple sclerosis. Patient Prefer Adherence. 2018;12:1557–1566. doi:10.2147/PPA.S175095
62. Deshpande C, Wang M, Shah R, et al. Preferred features of oral treatments and predictors of non-adherence: two web-based choice experiments in multiple sclerosis patients. Interact J Med Res. 2015;4(1):e3776.
63. Nicholas JA, Edwards NC, Edwards RA, Dellarole A, Grosso M, Phillips AL. Real-world adherence to, and persistence with, once- and twice-daily oral disease-modifying drugs in patients with multiple sclerosis: a systematic review and meta-analysis. BMC Neurol. 2020;20(1):1–15. doi:10.1186/s12883-020-01830-0
64. Thannhauser JE, Mah JK, Metz LM. Adherence of adolescents to multiple sclerosis disease-modifying therapy. Pediatr Neurol. 2009;41(2):119–123. doi:10.1016/j.pediatrneurol.2009.03.004
65. Tremlett H, Van der Mei I, Pittas F, et al. Adherence to the immunomodulatory drugs for multiple sclerosis: contrasting factors affect stopping drug and missing doses. Pharmacoepidemiol Drug Saf. 2008;17(6):565–576. doi:10.1002/pds.1593
66. McKay KA, Tremlett H, Patten SB, et al. Determinants of non-adherence to disease-modifying therapies in multiple sclerosis: a cross-Canada prospective study. Mult Scler. 2017;23(4):588–596. doi:10.1177/1352458516657440
67. Ben-Zacharia A, Adamson M, Boyd A, et al. Impact of shared decision making on disease-modifying drug adherence in multiple sclerosis. Int J MS Care. 2018;20(6):287–297. doi:10.7224/1537-2073.2017-070
68. Jonker MF, Donkers B, Goossens LMA, et al. Summarizing patient preferences for the competitive landscape of multiple sclerosis treatment options. Med Decis Mak. 2020;40(2):198–211. doi:10.1177/0272989X19897944
69. Garcia-Dominguez JM, Muñoz D, Comellas M, Gonzalbo I, Lizán L, Sánchez CP. Patient preferences for treatment of multiple sclerosis with disease-modifying therapies: a discrete choice experiment. Patient Prefer Adherence. 2016;10:1945–1956. doi:10.2147/PPA.S114619
70. Bauer B, Brockmeier B, Devonshire V, Charbonne A, Wach D, Hendin B. An international discrete choice experiment assessing patients’ preferences for disease-modifying therapy attributes in multiple sclerosis. Neurodegener Dis Manag. 2020;10(6):369–382. doi:10.2217/nmt-2020-0034
71. Col NF, Solomon AJ, Springmann V, et al. Whose preferences matter? A patient-centered approach for eliciting treatment goals. Med Decis Mak. 2018;38(1):44–55. doi:10.1177/0272989X17724434
72. Arroyo R, Sempere AP, Ruiz-Beato E, et al. Conjoint analysis to understand preferences of patients with multiple sclerosis for disease-modifying therapy attributes in Spain: a cross-sectional observational study. BMJ Open. 2017;7(3):e014433. doi:10.1136/bmjopen-2016-014433
73. Bottomley C, Lloyd A, Bennett G, Adlard N. A discrete choice experiment to determine UK patient preference for attributes of disease modifying treatments in multiple sclerosis. J Med Econ. 2017;20(8):863–870. doi:10.1080/13696998.2017.1336099
74. Wicks P, Brandes D, Park J, Liakhovitski D, Koudinova T, Sasane R. Preferred features of oral treatments and predictors of non-adherence: two web-based choice experiments in multiple sclerosis patients. Interact J Med Res. 2015;4(1):e6. doi:10.2196/ijmr.3776
75. Lanzillo R, Prosperini L, Gasperini C, et al. A multicentRE observational analysiS of PErsistenCe to Treatment in the new multiple sclerosis era: the RESPECT study. J Neurol. 2018;265(5):1174–1183. doi:10.1007/s00415-018-8831-x
76. Moccia M, Palladino R, Carotenuto A, et al. Predictors of long-term interferon discontinuation in newly diagnosed relapsing multiple sclerosis. Mult Scler Relat Disord. 2016;10:90–96. doi:10.1016/j.msard.2016.09.011
77. van Munster CEP, Uitdehaag BMJ. Outcome measures in clinical trials for multiple sclerosis. CNS Drugs. 2017;31(3):217–236. doi:10.1007/s40263-017-0412-5
78. Rhodes JK, Schindler D, Rao SM, et al. Multiple sclerosis performance test: technical development and usability. Adv Ther. 2019;36(7):1741–1755. doi:10.1007/S12325-019-00958-X
79. Højsgaard Chow H, Schreiber K, Magyari M, et al. Progressive multiple sclerosis, cognitive function, and quality of life. Brain Behav. 2018;8(2):2. doi:10.1002/BRB3.875
80. Kappos L, Vermersch P, Cree B, et al. A novel functional composite endpoint to characterize disease progression in patients with secondary progressive multiple sclerosis (S12.006); 2019.
81. Macaron G, Moss BP, Li H, et al. Technology-enabled assessments to enhance multiple sclerosis clinical care and research. Neurol Clin Pract. 2020;10(3):222. doi:10.1212/CPJ.0000000000000710
82. Schultheiß T, Kempcke R, Kratzsch F, et al. [Multiple sclerosis management system 3D. Moving from documentation towards management of patients]. Nervenarzt. 2012;83(4):450–457. German. doi:10.1007/S00115-011-3376-6
83. Schulze-Topphoff U. PANGAEA 2.0 EVOLUTION: state of the art multiple sclerosis patient. ECTRIMS Online Library; 2019 Nov 30-1: 279599. Available from: https://onlinelibrary.ectrims-congress.eu/ectrims/2019/stockholm/279599/ulf.schulzetopphoff.pangaea.2.0.evolution.state.of.the.art.multiple.sclerosis.html.
84. Neuberger EE, Abbass IM, Jones E, Engmann NJ. Work productivity outcomes associated with ocrelizumab compared with other disease-modifying therapies for multiple sclerosis. Neurol Ther. 2021;10(1):183–196. doi:10.1007/s40120-020-00224-1
85. Quarta S, Camprubí-Robles M, Schweigreiter R, et al. Sphingosine-1-phosphate and the S1P3 receptor initiate neuronal retraction via RhoA/ROCK associated with CRMP2 phosphorylation. Front Mol Neurosci. 2017;10:1–13. doi:10.3389/fnmol.2017.00317
86. Xiong Y, Hla T. S1P control of endothelial integrity. Curr Top Microbiol Immunol. 2014;378:85–105. doi:10.1007/978-3-319-05879-5_4
87. Yanagida K, Liu CH, Faraco G, et al. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc Natl Acad Sci USA. 2017;114(17):4531–4536. doi:10.1073/pnas.1618659114
88. Accorinti M, Okada AA, Smith JR, Gilardi M. Epidemiology of macular edema in uveitis. Ocul Immunol Inflamm. 2019;27(2):169–180. doi:10.1080/09273948.2019.1576910
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.