Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Clinical Development of AKT Inhibitors and Associated Predictive Biomarkers to Guide Patient Treatment in Cancer Medicine
Authors Coleman N, Moyers JT, Harbery A, Vivanco I, Yap TA
Received 17 September 2021
Accepted for publication 3 November 2021
Published 25 November 2021 Volume 2021:14 Pages 1517—1535
DOI https://doi.org/10.2147/PGPM.S305068
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Niamh Coleman,1 Justin T Moyers,1,2 Alice Harbery,3 Igor Vivanco,4 Timothy A Yap1,5,6
1Department of Investigational Cancer Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 2Division of Hematology and Oncology, Department of Medicine, University of California, Irvine, Orange, CA, USA; 3Division of Cancer Therapeutics, Institute of Cancer Research, London, SM2 5NG, UK; 4Institute of Pharmaceutical Sciences, School of Cancer and Pharmaceutical Sciences, King’s College London, London, UK; 5Institute for Applied Cancer Science, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 6Khalifa Institute for Personalized Cancer Therapy, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Correspondence: Timothy A Yap
Investigational Cancer Therapeutics (Phase I Program), The University of Texas MD Anderson Cancer Center, 1400 Holcombe Boulevard, Houston, TX, 77030, USA
Email [email protected]
Abstract: The serine/threonine kinase AKT is a critical effector of the phosphoinositide 3-kinase (PI3K) signaling cascade and has a pivotal role in cell growth, proliferation, survival, and metabolism. AKT is one of the most commonly activated pathways in human cancer and dysregulation of AKT-dependent pathways is associated with the development and maintenance of a range of solid tumors. There are multiple small-molecule inhibitors targeting different components of the PI3K/AKT pathway currently at various stages of clinical development, in addition to new combination strategies aiming to boost the therapeutic efficacy of these drugs. Correlative and translational studies have been undertaken in the context of clinical trials investigating AKT inhibitors, however the identification of predictive biomarkers of response and resistance to AKT inhibition remains an unmet need. In this review, we discuss the biological function and activation of AKT, discuss its contribution to tumor development and progression, and review the efficacy and toxicity data from clinical trials, including both AKT inhibitor monotherapy and combination strategies with other agents. We also discuss the promise and challenges associated with the development of AKT inhibitors and associated predictive biomarkers of response and resistance.
Keywords: AKT, PI3K, m-TOR, AKT inhibitor
Introduction
The PI3K–AKT–mTOR signaling pathway is one of the most frequently dysregulated pathways in human cancer.1 AKT is a key effector of this signaling cascade, which controls a number of key cellular processes, such as metabolism, motility, growth, and proliferation, and supports the survival, expansion and dissemination of cancer cells.2 Dysregulation of AKT-dependent pathways is associated with the development and maintenance of numerous solid tumors,3–5 and as a result, AKT has been relentlessly pursued as a therapeutic target in modern cancer drug development for more than a decade. A number of small-molecule inhibitors targeting various nodes in the PI3K–AKT–mTOR pathway have been investigated in solid tumors and are at various stages of clinical development.
Broadly, there are two major classes of small-molecule AKT inhibitors being investigated in the clinic: allosteric and ATP-competitive inhibitors.6 Allosteric inhibitors (such as miransertib (ARQ 092) and MK-2206)6 interfere with PH-domain mediated membrane recruitment (the first step in AKT activation) and inhibit AKT kinase activation and AKT phosphorylation. In contrast, ATP-competitive inhibitors of AKT (such as ipatasertib and capivasertib) bind to the active kinase, in which the PH-domain has shifted from the kinase domain and exposed the ATP-binding pocket site, thus inhibiting ATP binding. Targeting AKT with small-molecule inhibitors may enhance approved or investigational anticancer treatments, and several allosteric and ATP-competitive AKT inhibitors have now been tested in clinical trials.
Clinically, the development of AKT inhibitors in solid tumors has been complex, however there have been recent successes in select populations using combination strategies, such as capivasertib in combination with fulvestrant in AKT1 E17K mutant hormone receptor positive metastatic breast cancer.7 AKT is a significant downstream effector of PI3K signaling; interestingly, there are three different PI3K inhibitors FDA-approved for the treatment of follicular lymphoma, namely idelalisib, copanlisib, and duvelisib, and while these drugs vary in regard to the isoforms that they target, small-molecule inhibitors of AKT have not yet been explored in this context.
In this review, we discuss the biological function of AKT, discuss its contribution to tumor development and progression, and review up-to-date efficacy of clinically relevant AKT inhibitors in solid tumors, as monotherapy and in combination with other agents. We discuss the issues and limitations involved with targeting the AKT–PI3K–mTOR pathway, associated predictive biomarkers and predict future challenges for these novel anticancer agents.
Biology: AKT Structure and Function
AKT comprises a family of serine/threonine kinases, consisting of three isoforms (AKT1, AKT2 and AKT3), regulated upstream by the activation of PI3K, following growth factor stimulation. Several downstream substrates of activated AKT play a major role in the regulation of cell size, cell cycle progression, glucose metabolism, genome stability, transcription, protein synthesis and inhibition of pro-apoptotic proteins.8–10 The three AKT isoforms are encoded by different genes with high sequence homology and display a conserved protein structure (Figure 1).11 All three AKT proteins have three functional domains: a N-terminal fragment with a pleckstrin-homology (PH) domain, a central kinase domain (KD) and a C-terminal fragment with a regulatory region (RR) containing a hydrophobic motif (Figure 1). Enhanced activation of all the isoforms can be implicated in tumor development and progression, though interestingly their functional spectrum shows some variety: AKT1 has a suggested role in cell proliferation and survival, while AKT2 exercises its control over metabolism, regulates cytoskeleton dynamics; AKT3 has a more restricted expression pattern, with increased expression in brain tissue, and is implicated in mediating cell growth processes along with AKT1.12
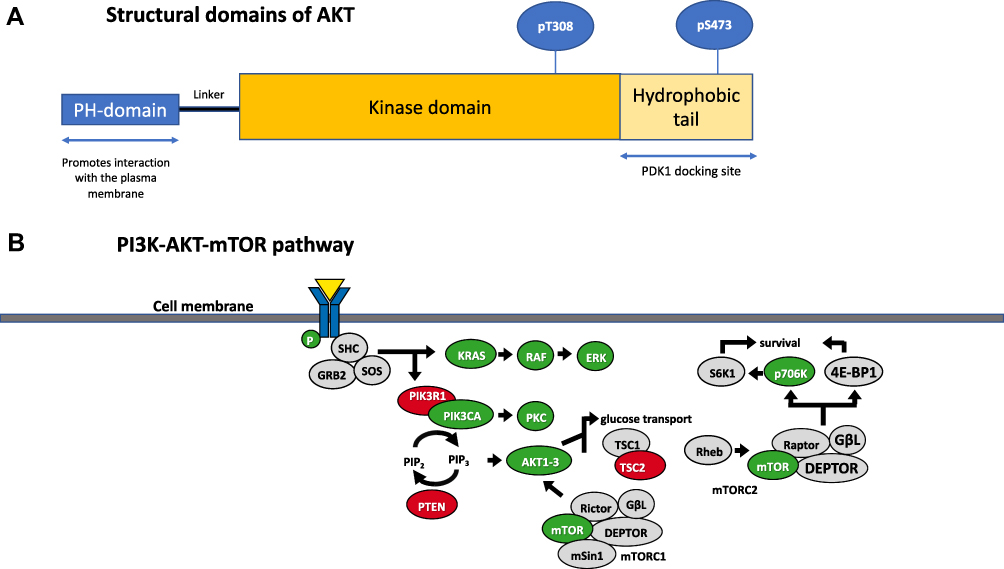 |
Figure 1 (A) Structural domains of AKT. AKT1, AKT2 and AKT3 share common domain structure with other members of the cAMP‐dependent, cGMP‐dependent and protein kinase C (AGC) subfamily of protein kinases: this consists of an N‐terminus pleckstrin homology (PH) domain, a large central kinase domain and a C‐terminus hydrophobic domain. (B) The PI3K-AKT-mTOR pathway. A selection of the downstream targets of AKT is shown. |
AKT Activation
Initiating signaling through the PI3K/Akt/mTOR pathway occurs through a number of different mechanisms, and typically begins from the plasma membrane, initiated by receptor tyrosine kinases, G-protein coupled receptors and immune receptors.13 These receptors are usually activated by ligand binding, which lead to signals that classically activate class I PI3K, leading to the production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PI45P2). PIP3 and PI34P2 then bind to the PH domain of AKT which leads to the re-localization of AKT to the plasma membrane, which triggers membrane recruitment and potentiates signals that ultimately lead to the activation of AKT. Phosphatidylinositol-3,4-bisphosphate (PI34P2) can also be produced by either dephosphorylation of PIP3 by 5-phosphatases or by activation of class II PI3Ks that phosphorylate phosphatidylinositol-4-phosphate (PI4P).13
Once at the membrane, AKT undergoes further conformational changes, resulting in the phosphorylation of Thr308 residue by phosphoinositide-dependent kinase-1 (PDK1) and phosphorylation of Ser473 by target of rapamycin complex 2 (mTORC2) of the mammalian target of rapamycin (mTOR) kinase (Figure 1).14 In addition to mTORC2, multiple different kinases, including DNA-PK (DNA-dependent protein kinase) which is a PI3K-like kinase (PIKK), have been shown under certain conditions to phosphorylate AKT on Ser473.15 Through phosphorylation, activated AKT exerts control over a vast number (>100) of protein substrates distributed throughout the cell including the plasma membrane, various endomembrane compartments, the mitochondria, cytosol, and the nucleus.13
mTOR, is a key downstream node in PI3K/AKT signaling, and a key cell metabolism regulator; activated mTOR phosphorylates ribosomal protein S6 kinase (p70S6K) and eIF4E binding-protein-1 (4E-BP1) to promote protein synthesis. Tuberous sclerosis complex 1 and 2 (TSC1 and TSC2) tumor suppressors, which are negative regulators of mTOR/S6K pathway, are phosphorylated by AKT, resulting in their inhibition.16,17 AKT phosphorylates the proline-rich AKT substrate of 40 kDa (PRAS40) at Thr246 and the yes-associated protein (YAP) at Ser127 to induce their interaction with 14–3-3, which correlates with their inactivation.18–20 Further negative regulators of the PI3K/AKT signaling pathway, include tumor suppressor genes and phosphatases PTEN (phosphatase and tensin homolog), PP2A (protein phosphatase 2A) and PHLPP (PH domain and leucine rich repeat protein phosphatase), which dephosphorylate PIP3, AKT pThr308 and AKT pSer473 respectively. PIP3 functions as a second messenger in the PI3K pathway that binds and activates proteins that have a pleckstrin homology domain, such as AKT1, and triggers their activation and localization to the plasma membrane, promoting cellular proliferation and survival.21 Consequently, loss of PTEN, a frequent aberration in human cancer (frequency of PTEN loss by IHC 30%, PTEN mutations 6%)22 leads to PI3K/AKT pathway activation.
AKT also exerts its effect on cell cycle progression by phosphorylating and inhibiting cyclin dependent kinase (CDK) inhibitors, p21 and p27, which function as G1 checkpoints to arrest the cell cycle.17 AKT regulates apoptosis through inhibition of BAD, BCL-2-like protein 11 (BIM), caspase 9, and forkhead box protein O1 (FoxO1).23–25 It also phosphorylates MDM2, allowing its entry into the nucleus, which results in p53 degradation.26,27 In reality, there are numerous AKT substrates, which have been reported in the literature and been extensively validated, involving multiple cellular processes functions, including cell growth, proliferation and survival, cellular metabolism, glucose uptake and angiogenesis.13
Deregulation of AKT Pathway and Role in Oncogenesis
AKT signaling plays a key role in multiple pathways involved in tumorigenesis, and hyperactivation of the pathway is a common event in cancer. There is significant overlap and crosstalk between substrates and substrate functions of AKT, and AKT regulates many of its cellular functions through phosphorylation.13 Although the status of all the activators, modulators and downstream effectors of AKT play a critical role in tumor development, aberrant AKT activation itself is highly oncogenic.
Aberrant activation of AKT in cancer can occur through a number of mechanisms, due to amplification and/or mutation of AKT, or via genomic changes at various levels in the pathway. PI3K/AKT pathway aberrations have been identified in up to 40% of all tumor types: PTEN loss by immunohistochemistry occurs most frequently (30%), followed by mutations in PIK3CA (13%), PTEN (6%) and AKT (1%).22 AKT1 is the most frequently mutated isoform in solid tumors, though its frequency varies by cancer type: the overall frequency of AKT1 hotspot mutations is low (<1%) in cancer, but the overall frequency of PI3K pathway activation is high. For example, in breast cancer, mutations of the PI3K catalytic alpha subunit (PIK3CA) are common events, occurring in 9–45% of breast cancer according to the subtype, PTEN loss of function occurs in 13–35% and AKT substitutions (2–4%) or amplification (5–10%) have also been described.28 Frequency of pathway activation then differs again by cancer subtype, where up to 50% of HR+ breast cancer demonstrates PI3K/AKT pathway hyperactivation (predominantly via by PIK3CA point mutations in HR + tumors), 5–10% of ER+ metastatic breast cancer tumors harbor somatic PTEN mutations29 and an estimated 7% of ER+ breast cancers, pathway activation can occur through mutation in AKT1, predominantly AKT1E17K (∼80%).30 Combined activating mutations in PIK3CA and AKT1, with inactivating PTEN mutations occur in ∼25%‒30% of advanced triple negative breast cancer (TNBC).31 The pathway is also deregulated in human epidermal growth factor receptor 2 (HER2)-enriched breast cancer where it is involved in the development of resistance toward anti-HER2 agents.32 Given the frequency of dysregulation, AKT inhibitors are an important and promising class of antitumor agents for targeting breast cancer.
AKT2 amplifications occur more frequently than mutations: analysis of pan-cancer MSK-IMPACT Clinical Sequencing Cohort (MSKCC, Nat Med 2017) on (10,336 patients/10,945 samples) on the cBioPortal genomic database demonstrate that AKT2 amplification is found in up to 4% of endometrial cancer (0.5% frequency of mutation), 4% of ovarian (0.5% mutation frequency) and 3.6% of small cell lung cancer (Figure 2).33,34 In the same cohort, AKT1 mutations are more common than amplification (Figure 2), with alterations in AKT occurring in 4.6% of endometrial and 5% of breast cancer, as well as approximately 1.5% of lung, bladder, ovarian and colorectal cancers. AKT3 alterations are also infrequent and analysis of the pan-cancer MSK-IMPACT Clinical Sequencing Cohort shows that AKT3 alterations are found rarely in breast cancer (0.45% frequency of mutation; 1.06% frequency of amplification), endometrial cancer (3.21% frequency of mutation; 1.83% frequency of amplification), and somewhat more frequently in melanoma (6.08% frequency of mutation) and tumors of the peripheral nervous system (1.25% frequency of amplification, 3.75% frequency of deep deletion) (Figure 2). In pre-clinical breast cancer models, AKT3 expression has been shown to be markedly upregulated to the AKT Inhibitor MK2206 in Breast Cancer MK2206-resistant cells, which may provide a rationale for developing therapeutics targeting AKT3 to overcome acquired resistance in breast cancer.
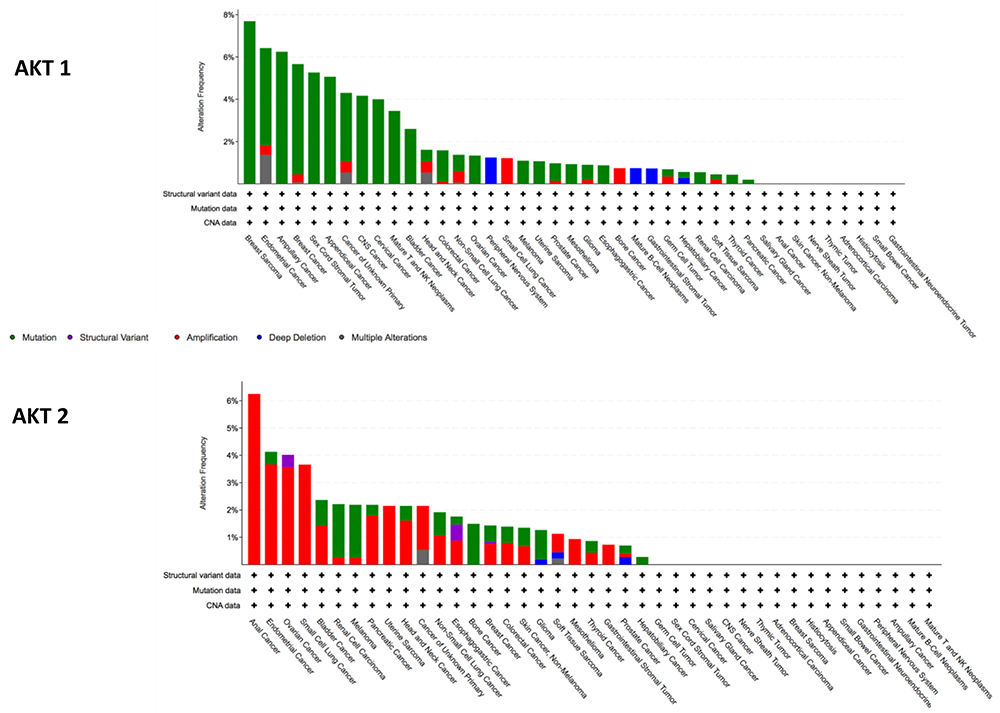 |
Figure 2 Frequency of AKT1 and AKT2 alterations from analysis of MSK-IMPACT Clinical Sequencing Cohort (MSKCC, Nat Med 2017), cBioPortal genomic database.33,34 |
Overexpression or activating mutations upstream of AKT in the PI3K/AKT pathway can lead to AKT activation, as may loss of tumor suppressor proteins, such as PTEN, PHLPP, PP2A. Germline PTEN loss-of-function mutations can lead to dominant AKT activation as a driving oncogenic event, for example in Cowden syndrome, which leads to an increased risk of breast, thyroid and endometrial cancer.35 Indeed, exceptional durable responses to AKT Inhibition have been demonstrated in patients with breast cancer and germline PTEN mutations.35
Potential Biomarkers of AKT Activity
In the era of precision medicine, identifying predictive biomarkers of response has become a key goal of translational cancer research. Accordingly, the aim is to establish biomarkers that predict “addiction” to the PI3K–AKT pathway to match patients with appropriate inhibitors. Multiple translational studies have been extensively undertaken in the context of clinical trials, however results are still inconclusive. For instance, investigating the phosphorylation levels of downstream effectors of AKT in order to determine whether AKT inhibition effectively downregulates PI3K hyperactivation has been pursued, albeit with mixed outcomes. In the STAKT trial, a two-stage, double-blind, randomized, placebo-controlled, “window-of-opportunity” study in patients with newly diagnosed ER+ invasive breast cancer, post-treatment phosphorylation of downstream AKT effects were significantly decreased from baseline versus placebo (n = 11) in pGSK3β, pPRAS40, pS6, while pAKT (and nuclear FOXO3a) also increased in accordance with the mechanism of capivasertib.36 Another window-of-opportunity trial which investigated MK-2206 demonstrated only a modest effect on downstream effectors, such as pS6 and PTEN phosphorylation.37 Comprehensive phosphoproteomic analysis was undertaken in patients with early HR-negative/ HER2-positive breast cancer and triple negative breast cancer (TNBC) of the I-SPY2 trial, which was a multicenter, Phase II randomized neoadjuvant platform trial that screened experimental therapies to identify potential predictive biomarker signatures.38 In this study, in the HER2-positive subset, increased levels of pAKT, pSGK, pmTOR, and pTSC2 levels before neoadjuvant treatment with MK-2206 and standard therapy correlated positively with pathological complete responses.38 In contrast, in the TNBC cohort, responding patients had lower levels of AKT pathway phospho-proteins, such as pAKT, pmTOR, and pTSC2 and pathway mutations did not appear to account for these associations.38
In the PAKT and LOTUS phase II trials discussed further below,39,40 which combined either capivasertib or ipatasertib, respectively, with paclitaxel, combination treatment resulted in improved PFS in patients with TNBC, with the benefit more pronounced in the subgroup of patients with PIK3CA/AKT/PTEN alterations. Interestingly, the same benefit was not observed in LOTUS among PTEN-low patients.39 In addition, in the molecularly selected Phase III trial investigating ipatasertib in combination with paclitaxel,41 patients with PIK3CA/AKT1/PTEN-altered TNBC did not achieve improved PFS, results which differ from findings in both randomized phase II trials of AKT inhibitors in TNBC (LOTUS and PAKT). A unifying theme in these biomarker-driven breast cancer trials is a clear lack of correlation between PI3K/AKT pathway alterations and efficacy of AKT inhibitors.42–44 There are clearly elements in this intricate pathway that the currently designed clinical trials are not taking into account. The complex network of the PI3K-AKT pathway, including crosstalk and negative feedback loops, is certainly a potential cause for the disappointing efficacy using AKT inhibitors, such as capivasertib in the PI3K/AKT/PTEN altered population.
An exception to these inconsistencies, appears to be the AKT1 E17K mutation, which predicts for response to most ATP-competitive inhibitors, but not allosteric AKT inhibitors. Aside from this alteration, there do not appear to be other robust predictive biomarkers of AKT inhibitor response. Recent successes in targeting the AKT1 E17K mutation in solid tumors only underscore the importance of understanding the impact each PI3K/AKT pathway alteration has on binding to a specific molecularly targeted agent. Novel biomarkers are clearly urgently needed to increase the impact of AKT inhibitors as part of biomarker-driven clinical trials. These studies may incorporate other AKT mutations, AKT amplification, and other non-AKT biomarkers (such as PTEN loss) and will likely be critical in uncovering potential biomarkers of response and resistance. Given the complexity of the pathway, it is likely that multiple context-specific biomarkers to better predict response are needed.
The Clinical Development of AKT Inhibitors
Inhibiting PI3K/AKT signaling has long been an attractive therapeutic approach in oncology. Numerous compounds that inhibit the pathway at all levels are now in clinical development, in both monotherapy and combination strategies.45 Notably, there are currently several ATP-competitive inhibitors of AKT at early stages of clinical development. All ATP-competitive inhibitors in clinical development are able to target all three AKT isoforms (ie, AKT1, AKT2 and AKT3), though with varying degrees of potency; allosteric inhibitors generally spare AKT3. Here, we describe progress to date, including most recent developments in strategies used to target AKT in solid tumors.
Monotherapy with AKT Inhibitors
Allosteric Inhibitors
Until recently, AKT inhibitors utilized as monotherapy agents, had failed to build on initial promise demonstrated in pre-clinical models. For instance, the following allosteric AKT inhibitors have been all been tested in phase I/II trials, including MK-2206, ARQ 092, ARQ 751 and BAY1125976, and while some of these agents have demonstrated manageable safety profiles,46–49 limited clinical activity in phase II monotherapy trials has been observed.50–54 Allosteric AKT inhibitor MK-2206 for example, is predominantly an AKT1/2 inhibitor with reduced potency against AKT3, shown to have single-agent activity against a range of cell lines harboring RTK activation, PTEN loss/mutation and/or AKT2 amplification.55 MK2206 was previously the most clinically advanced allosteric inhibitor of AKT: a phase I trial of MK-2206 monotherapy in patients with solid tumors demonstrated a MTD of 60mg on alternate days with DLTs of skin rash and stomatitis; drug-related AEs included rash (51.5%), nausea (36.4%), pruritus (24.2%), hyperglycemia (21.2%), and diarrhea (21.2%).56 While single-agent MK-2206 showed evidence of significant AKT pathway blockade (on-target PD tumor effects such as a decrease in AKT pSer473 were observed), its toxicity profile was challenging, although more intermittent dosing schedules seemed to be better tolerated.49 Given the limited antitumor activity seen with MK-2206 monotherapy in phase II trials across a range of tumor types, attention then switched to combination trials. The allosteric inhibitor ARQ 092 has also been tested in both solid and hematological cancers, and demonstrated acceptable tolerability, though with limited activity and few partial responses clinically.47,57 ARQ 751 is currently being tested in a Phase 1 study for solid tumors with PIK3CA/AKT/PTEN mutations (NCT02761694), both as monotherapy and in combination with other anti-cancer agents.
ATP-Competitive Inhibitors
Multiple ATP-competitive AKT inhibitors have also undergone clinical testing in phase I trials, including GSK2141795, GSK690693 and LY2780301.58,59 The antitumor activity of these agents as monotherapy in molecularly unselected patient populations has been generally disappointing: GDC-0068 and AZD5363 have been tested in a number of monotherapy trials with limited activity; GDC-0068 has also demonstrated modest antitumor activity (30% stable disease) across tumor types in a phase I study.60 Recently, capivasertib (AZD5363) a selective ATP-competitive pan-AKT kinase inhibitor has shown clinically meaningful activity in patients with various tumor histologies who have AKT1 E17K mutations or PTEN mutations.61,62
The AKT1 E17K mutation plays a crucial role in cancer development and is mutually exclusive to PIK3CA mutations and PTEN loss,30,63,64 suggesting that the mutational activation of the phosphatidylinositol 3-kinase pathway by any one of these means may be biologically equivalent. Significant antitumor responses were initially reported using capivasertib in patients with heavily pretreated AKT1 E17K mutant solid tumors in a basket trial (including 20 patients with ER-positive (ER+) metastatic breast cancer, where the objective response rate (ORR) was 20% and median progression-free survival (PFS) was 5.5 months.65
Recently, the results of a subprotocol of the National Cancer Institute Molecular Analysis for Therapy Choice (NCI-Match) trial reported promising ORR with capivasertib in AKT1 E17K-mutated tumors.61 Among 35 treated patients, the objective response rate was 28.6%, the median PFS was 5.5 months, with a median duration of response of 4.4 months among responders; one patient achieved complete response (CR).61 Of those patients with confirmed PR, 7 had HR-positive/ERBB2-negative breast cancer, 1 had uterine leiomyosarcoma, and 1 had oncocytic parotid gland carcinoma.61 The majority of patients treated had either breast cancer (51%) or gynecologic cancers (31%), and while responses were seen across different subgroups, it may be difficult to extrapolate these findings to non-breast and non-gynecology cancers due to the small numbers treated. In addition, the documented adverse event profile in this study was higher than that reported in similar studies of capivasertib, as there were 26% of patients with grade 3 or higher hyperglycemia, and 11% with grade 3 rash.61
Capivasertib plus fulvestrant has also shown antitumor activity in heavily pretreated patients with PTEN-mutated ER+ metastatic breast cancer, particularly in patients with prior progression on fulvestrant.61 Five to ten percent of ER+ metastatic breast cancer tumors harbor somatic PTEN mutations and loss of function of this tumor-suppressor gene defines a highly aggressive, treatment-refractory disease.29 In this study, capivasertib was deemed tolerable and clinically active: the 24-week clinical benefit rate was 42% and ORR was 21% in the 19 fulvestrant pre-treated patients.
Ipatasertib (GDC-0068), is another potent and selective ATP-competitive pan-AKT kinase inhibitor where impressive durable antitumor response in combination with fulvestrant has been reported in heavily pre-treated patients with AKT1 E17K mutant metastatic breast cancer.60,66 Ipatasertib is currently being investigated in a monotherapy treatment arm as part of NCI MATCH.67
Combination Strategies
There is strong preclinical rationale for AKT inhibitor combination strategies in multiple settings, including acquired resistance to other anti-cancer agents, adaptive de novo resistance due to relief of negative feedback, and both chemo- and radio-resistance. AKT activation is implicated as mechanisms of resistance in all of these scenarios. Thus, AKT inhibitors are currently being tested in combination with a range of chemotherapeutic and targeted agents.
Chemotherapy Combinations
Activation of AKT has been linked to resistance in both chemotherapy and radiotherapy. DNA-PK is one of the apical kinases involved in DNA double strand break repair and is essential for classical non-homologous end-joining, the predominant repair pathway of DSBs in human cells.68 AKT is activated in a DNA-PK–dependent manner following the generation of DNA DSBs, and promotes survival through anti-apoptotic mechanisms.69,70 AKT inhibition has also been shown, in pre-clinical models, to sensitize cells to a number of chemotherapeutic agents.71–73
Until recently, the most encouraging data in combination chemotherapy data was in metastatic TNBC, based upon randomized phase II trials that demonstrated an improvement in clinical outcomes (progression-free survival (PFS) and overall survival (OS)) with paclitaxel in combination with AKT inhibition versus paclitaxel alone.39,74 A recently reported randomized phase III trial in the first-line setting for TNBC has however dampened enthusiasm for this combination strategy. Presented at the 2020 San Antonio Breast Cancer Symposium, the analysis of IPATunity130 Cohort A assessed the combination of ipatasertib with paclitaxel chemotherapy versus paclitaxel chemotherapy alone, in patients with metastatic TNBC, and showed no difference in both PFS and OS.75 This trial selected TNBC patients with PIK3CA/AKT1 or PTEN pathway alterations, and given the recently updated survival benefit shown in the in the LOTUS phase II trial in TNBC with these alterations,41 the lack of clinical benefit in this biomarker-selected group of TNBC patients is disappointing.
Prostate cancer is another notable area of development in the AKT field. In the PROCAID trial, capivasertib (AZD5363) was combined with docetaxel and prednisolone in a Phase I trial for patients with metastatic castration-resistant prostate cancer (CRPC): the combination was well tolerated and a recommended Phase II dose was determined for this combination.76 In the recently reported Phase II trial, the addition of capivasertib to chemotherapy did not extend PFS in metastatic CRPC, irrespective of PI3K/AKT/PTEN pathway activation status.77 However, a significant improvement in OS, a secondary endpoint, was observed (median OS was 31.15 months in the capivasertib group vs 20.27 months in the placebo group (HR = 0.54, 95% CI = 0.34–0.88, P = 0.01), which will need to be validated prospectively in future studies.
Targeted Therapy Combinations
Increasingly, AKT inhibitor combination strategies have involved molecularly targeted agents. These data are summarized below and in Table 1.78
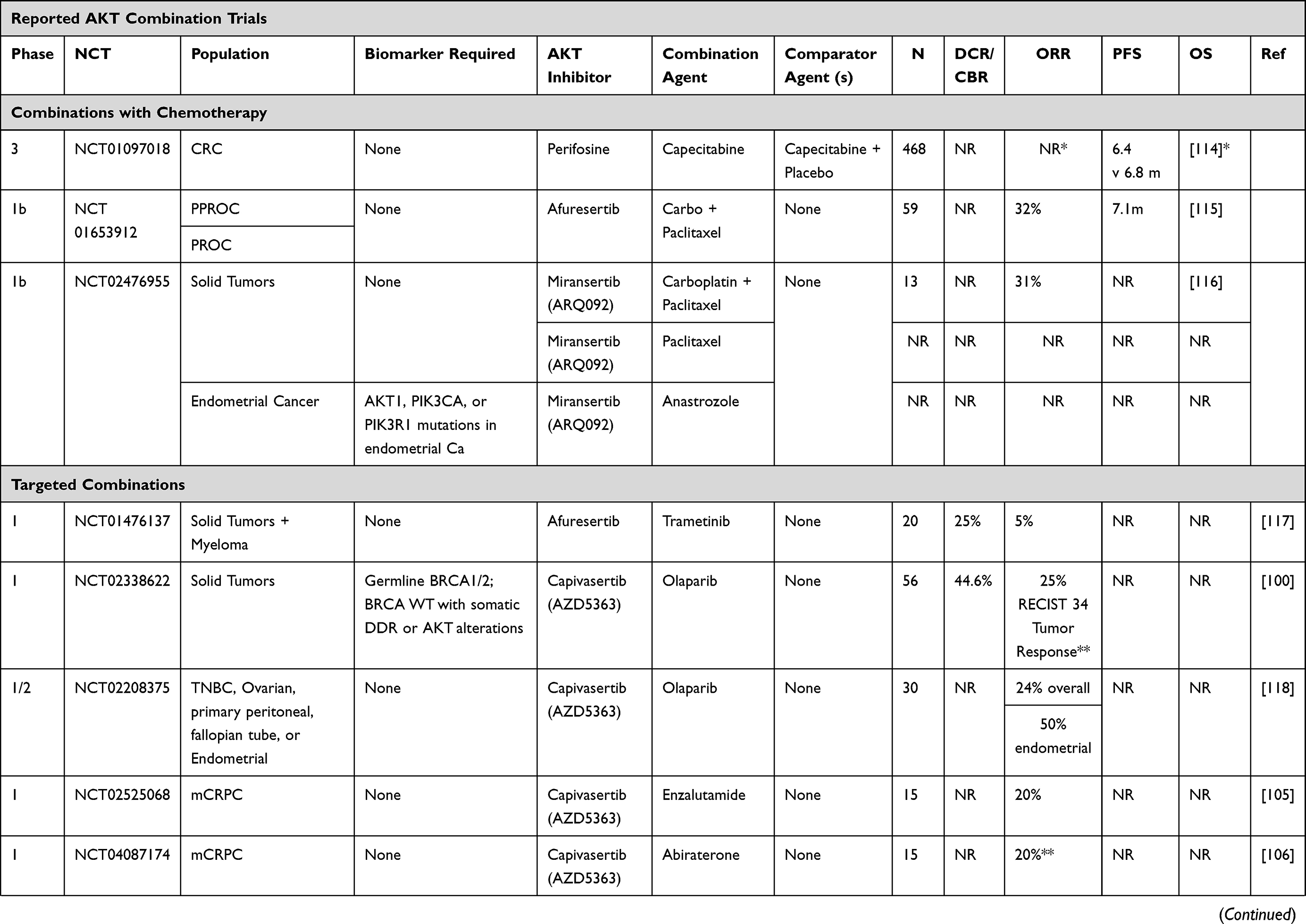 | 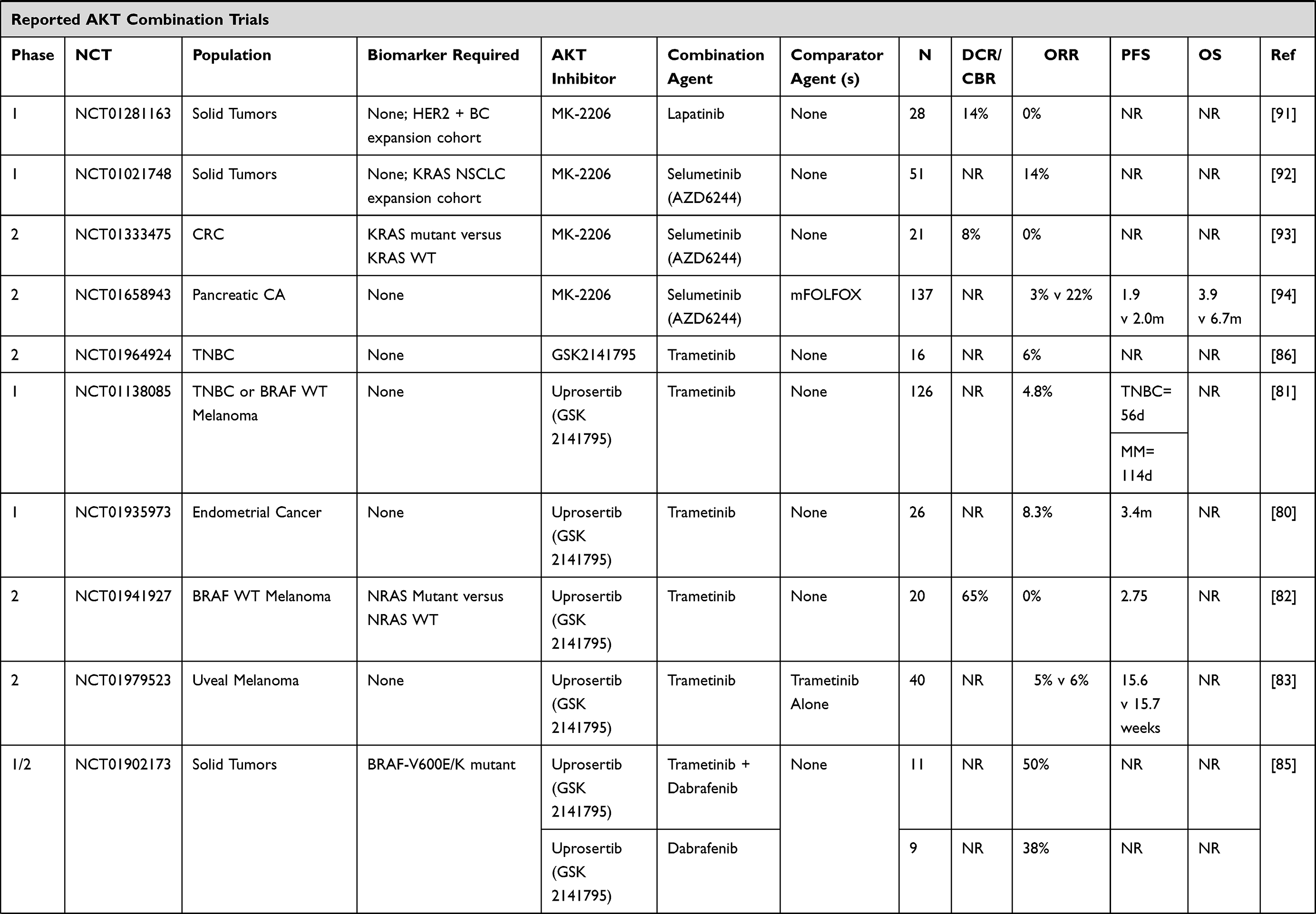 | 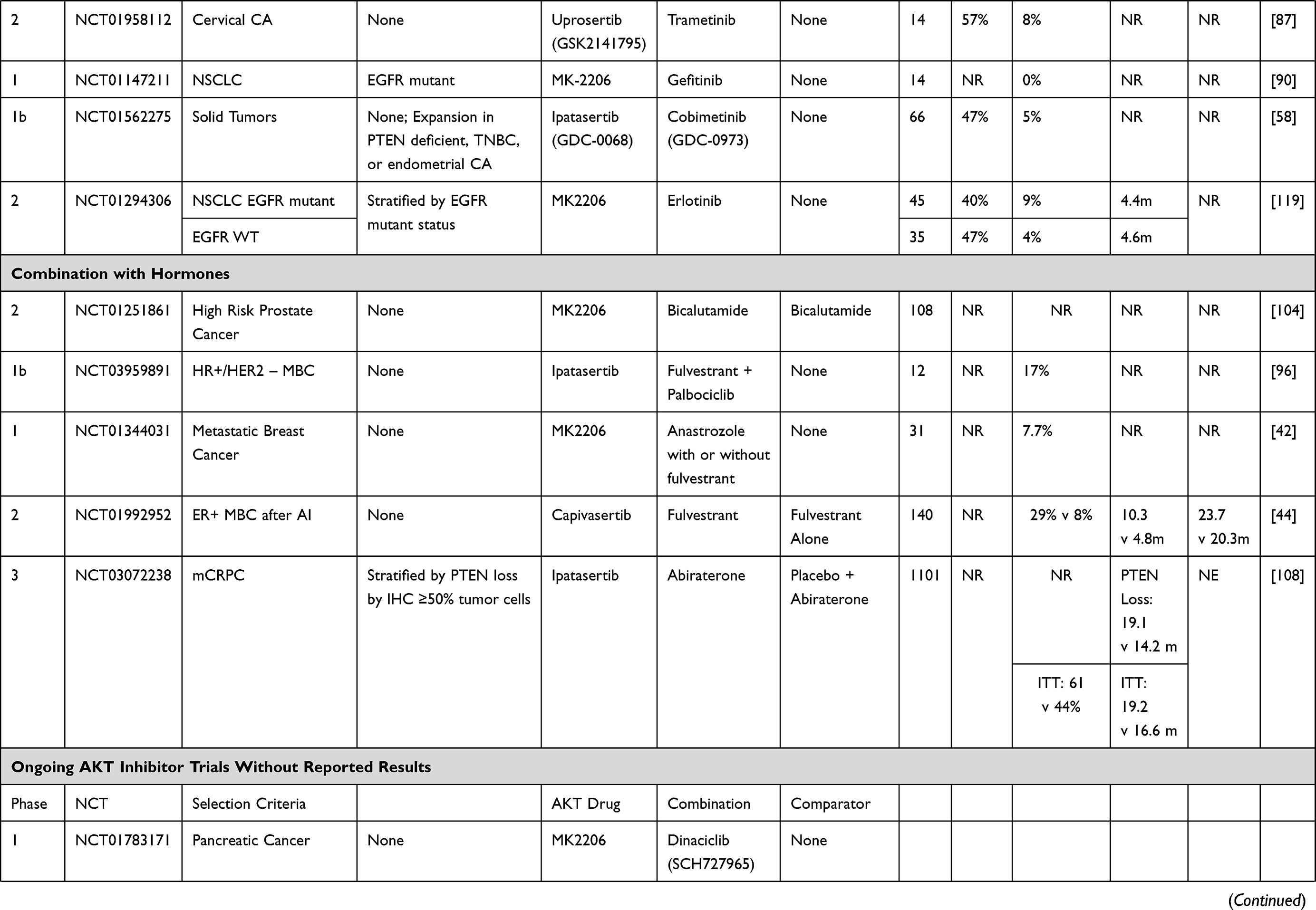 | 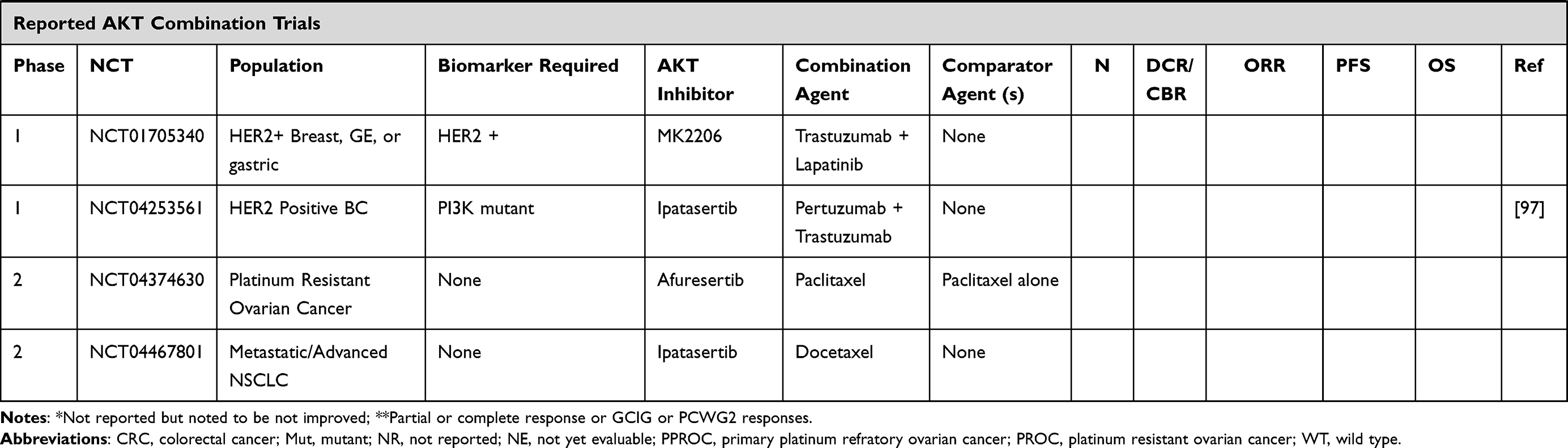 |
Table 1 Trials Including Combination Strategies with AKT Inhibitors |
Uprosertib (GSK2141795)
Alterations in the PI3K and RAS pathways have been implicated in tumorigenesis of endometrial cancer and are associated with poor treatment outcomes.79 Treatment with single-agent AKT inhibitors has, however, led to modest results. In a phase I/II study, the ATP-competitive AKT inhibitor uprosertib (GSK2141795) and MEK1/2 inhibitor trametinib were assessed in refractory or persistent endometrial cancer. The combination was poorly tolerated and clinical benefit was limited with only one response observed in 12 (8.3%) patients. The median PFS was only 3.4 months and the Phase II study was not initiated due to toxicity and a lack of efficacy.80 Another phase I trial in TNBC and BRAF-WT melanoma investigated the combination of uprosertib and trametinib.81 This was poorly tolerated, with over half of all patients experiencing DLTs and preventing dose escalation to dose levels predicted to have clinical activity. The ORR amongst 126 treated patients was <5%. In a phase II study of the same combination in BRAF WT melanoma including 24 NRAS WT and 24 NRAS mutant melanomas, 65% of patients achieved stable disease as their best response, however no objective responses were observed. Median PFS was only 2.75 months with no difference by NRAS mutation status.82 The combination was also found to add no PFS or ORR benefit in a separate Phase 2 trial in uveal melanoma when compared to trametinib alone.83
In BRAF-V600E/K mutant advanced solid tumors including melanoma, aberrant PI3K/AKT signaling may contribute to resistance of MAPK pathway blockade.84 The Phase I/II SWOG S1221 study was conducted to combine uprosertib with dabrafenib with or without trametinib. Both the doublet and triplet combinations were generally well tolerated. In the doublet cohort (n = 8), 2 of 2 treatment-naive patients had PRs, while only 1 of 6 BRAF refractory melanoma patients achieved an objective response. In the triplet cohort, 3 of 6 treatment-naïve patients had PRs. Diarrhea and rash were common dose-limiting toxicities, with 59% of patients experiencing grade 3 toxicities.85
In a two-part single-arm study of metastatic TNBC, trametinib was administered alone in part 1 and in combination with uprosertib upon progression in part 2. In the combination arm, only one of 16 patients experienced an unconfirmed PR and GI toxicities were common.86 Given that PIK3CA and KRAS mutations are common in both adenocarcinoma and squamous cell carcinoma of the cervix, the combination of uprosertib and trametinib has been evaluated in a phase II trial, however, due to manufacturer discontinuation of uprosertib, the trial was halted early. At the time of termination, 14 patients had received study drug with one partial response of 12 evaluable patients.87
MK-2206
The PI3K/AKT signaling pathway is important for epidermal growth factor receptor (EGFR) signaling and may be a mechanism of resistance to EGFR tyrosine kinase inhibitors (TKIs) in non-small cell lung cancer (NSCLC), which is supported by improved preclinical activity with combination blockade.88,89 To this end, MK-2206 was combined with gefitinib in patients with metastatic NSCLC who had progressed following at least 3 months treatment of EGFR TKI. Fourteen patients were treated, two patients had minor tumor regressions, but no objective responses were observed, and hematologic toxicities were dose limiting.90 In a phase I study, MK-2206 was combined with the anti-HER2 agent, lapatinib, in patients with advanced solid tumors. Two of 23 patients in the dose escalation phase maintained stable disease for greater than 4 months, as did 2 of 5 patients in the cohort expansion phase of HER2 positive breast cancers. The overlapping toxicities of rash and diarrhea were typically grade 1–2 and were managed effectively using supportive measures.91 In an effort to undertake dual vertical blockade of the pathway, MK-2206 has also been combined with the MEK inhibitor, selumetinib, in advanced KRAS mutant cancers. Four durable responses were observed in 3 of 13 (23%) in NSCLC and 1 of 2 patients with ovarian cancers. Common dose limiting toxicities included stomatitis, diarrhea, fatigue, and retinal pigment epithelium detachment.92 This combination was subsequently assessed in advanced colorectal cancer patients and stratified by KRAS mutations, however no objective responses were observed among twenty-one enrolled patients and the pre-specified target response criteria of 70% inhibition of phosphorylated ERK and AKT levels in paired tumor biopsies was not met.93
In attempts to target KRAS through downstream inhibition of MEK and PI3K/AKT pathways in pancreatic cancer, 137 patients with metastatic pancreatic cancer who progressed following gemcitabine-based chemotherapy were randomized to MK-2206 with selumetinib or mFOLFOX. Only two responses out of 39 patients were observed in the experimental doublet-targeted therapy arm and overall survival was not improved compared to mFOLFOX (3.9 v 6.7, HR 1.38, p = 0.15).94
Ipatasertib (GDC-0068)
Preclinical studies have indicated that CDK4/6 resistance is driven through AKT1 activation, providing rationale for the combination of an AKT inhibitor and CDK4/6 inhibitors.95 The ongoing phase 1b TAKTIC trial combined the AKT inhibitor ipatasertib (GDC-0068) with fulvestrant, or used a triplet combination of fulvestrant with ipatasertib and palbociclib for the treatment of HR+ HER2 negative metastatic breast cancer, following at least 1 prior therapy and up to 2 lines of chemotherapy. Preliminary results of the triplet combination have shown two partial responses amongst 12 patients, and the combination was well tolerated with no dose limiting toxicities at reported dose levels.96
A phase Ib trial of cobimetinib and ipatasertib was however poorly tolerated and all 66 patients who received study drug reported an adverse event, with 53% experiencing drug-related AE of grade 3 or worse severity, though none were dose-limiting toxicities. Although biomarker analyses indicated blockade of combined RAS/MEK/ERK and pI3K/AKT/mTOR pathways in paired tumor biopsies, objective responses were only observed in two patients with endometrial cancer and one with ovarian cancer.58 A phase I trial has been registered to begin enrollment in HER2 positive metastatic breast cancers with PI3K pathway mutations utilizing the triplet combination of ipatasertib with anti-HER2 agents pertuzumab and trastuzumab (NCT04253561).97
Capivasertib (AZD5363)
Preclinical studies have demonstrated synergy between PARP and PI3K/AKT pathway inhibitors in BRCA1 and BRCA2 (BRCA1/2)–deficient and BRCA1/2-proficient tumors.98,99 Consequently, the combination is being explored in clinical trials. In a phase I trial, PARP inhibitor olaparib in combination with capivasertib was well tolerated and antitumor activity was observed as twenty-five (44.6%) of 56 evaluable patients achieved clinical benefit (RECIST complete response/partial response or stable disease ≥4 months), including patients with tumors harboring germline BRCA1/2 mutations and BRCA1/2 wild-type cancers with or without DDR and PI3K–AKT pathway alterations.100 Five (71.4%) of 7 patients with germline BRCA1/2-mutant breast cancers achieved clinical benefit, and antitumor responses were also observed in patients who had previously developed disease progression on PARP and PI3K pathway inhibitors.100 Although based on small numbers, this study supports the combination of capivasertib with olaparib as a rational strategy to potentially improve patient benefit beyond that of single-agent olaparib.
Endocrine Therapy Combinations
Hyperactivation of the PI3K-AKT pathway is associated with endocrine resistance in estrogen receptor-positive breast cancer.101,102 In a Phase II (NCT01776008) trial in stage II or III ER+ and HER2-negative breast cancer with PI3KCA mutations, MK-2206 was combined anastrozole, an aromatase inhibitor, in the neoadjuvant setting. The primary endpoint was the pathologic complete response rate at the time of surgery. A cycle 1 day 17 biopsy was performed and if Ki67 was greater than 10%, treatment would be discontinued. Fifty-one patients were preregistered and 16 of 22 patients with PIK3CA mutant tumors received study treatments. Two patients discontinued study due to high Ki67 and 1 due to toxicity. Of 13 patients who completed neoadjuvant therapy, no pathologic complete responses were observed.103
A Phase 1 study of MK-2206 combined with hormonal therapies evaluated 31 postmenopausal women with ER positive MBC. Grade 3 rash was the main dose-limiting toxicity, which was ameliorated with prophylactic prednisone. The overall response rate was however only 7.7% and did not correlate with PIK3CA mutation status. In the combination with anastrozole, two partial responses were observed in 13 patients, with no partial responses noted in the combination with fulvestrant, or with anastrozole (n = 6) and fulvestrant (n = 6).42 The FAKTION trial evaluated the addition of capivasertib to fulvestrant in a double-blind placebo-controlled phase 2 trial. Following disease progression on an aromatase inhibitor, patients with HER2-negative metastatic or locally advanced breast cancer were enrolled to receive 400mg of capivasertib or matching placebo with IM fulvestrant. A total of 140 patients were enrolled. PFS was improved to 10.3 months in the capivasertib group versus 4.8 months in the placebo group, with HR of 0.58 (95% CI 0.39–0.84, p = 0.0018). For the combination therapy, PFS was improved in non-PI3K/PTEN pathway mutated cancers (HR = 0.56, p = 0.035) and non-significantly improved in PI3K/PTEN pathway mutated tumors (HR = 0.59, p = 0.064). Compared to placebo, the most common adverse events in combination were hypertension, diarrhea, and infection.44
In castration-resistant prostate cancer (CRPC), activation of the PI3K/AKT/mTOR pathway through loss of phosphatase and tensin homolog (PTEN) occurs in approximately 50% of patients with metastatic CRPC; the combination of androgen deprivation with AKT inhibition is thus postulated to improve PSA responses. A phase II trial of bicalutamide combined with MK-2206 versus bicalutamide alone was conducted in men with high risk localized prostate cancer after local therapy with PSA recurrence and fast doubling time with a primary endpoint of PSA ≤0.2. A non-statistically significant improvement in PSA response of 35.6% (16/45) was noted in the combination arm versus 20.4% (10/49) in the bicalutamide alone arm (p = 0.08).104 In patients with metastatic CRPC progressing on abiraterone and/or enzalutamide, a phase I trial investigated escalating doses of capivasertib with continuous enzalutamide dosing.105 The combination was safe and well tolerated and antitumor activity was observed in patients with PTEN loss or activating mutations in AKT, low or absent AR-V7 expression, as well as those with an increase in phosphorylated extracellular signal-regulated kinase (pERK) in post-exposure samples.105 Similarly, interim results of a phase 1 study in metastatic CRPC combining abiraterone and capivasertib in 15 patients reported acceptable safety and tolerability (rash, diarrhea, and hyperglycemia as the most common adverse events). Five patients had reduced PSA levels (>20%) on treatment, with 3 patients showing sustained falls in PSA over 12 weeks.106
Consistent with encouraging phase I data, a randomized Phase II trial demonstrated that ipatasertib in combination with abiraterone, results in prolonged radiographic progression-free survival (rPFS) over placebo with abiraterone.107 The effect on rPFS was even greater in patients with PTEN-loss in tumors compared to those with intact PTEN. As a result, the IPATential150 randomized, Phase III trial of abiraterone with ipatasertib 400 mg PO QD vs abiraterone with placebo was developed and recently reported data are encouraging.108 Radiological PFS was improved in the ipatasertib plus abiraterone group over the placebo plus abiraterone group (median rPFS 18.5 vs 16.5 months, respectively), with a stratified hazard ratio [HR] 0.77; 95% confidence interval [CI] 0.61–0.98, p = 0.0335, statistical significance set at α=0.05). Post-hoc analysis of rPFS in the next-generation sequencing defined PTEN-loss population was also significant with a median rPFS 19.1 vs 14.2 months, respectively (HR 0.65; 95% CI 0.45–0.95, p = 0.0206), while secondary endpoints of confirmed ORR, PSA response, and time to PSA progression all favored the ipatasertib arm. While overall survival data are not yet mature, these data are encouraging and may allow us to better learn how to identify and define the PTEN-loss population.
Toxicity Profile of AKT Inhibitors
There are a number of well-defined toxicities related to the pharmacological inhibition of the AKT/PI3K pathway,109 and safety and tolerability issues have likely hampered the development of some AKT inhibitors (eg, MK2206). The toxicity spectrum of AKT inhibitors becomes increasingly important in the context of developing combination strategies and in novel clinical trial design. Dermatological toxicities are typically observed with all AKT inhibitors; for example, in the phase I trial of MK-2206 monotherapy, DLTs were skin rash and stomatitis, and drug-related skin AEs were frequent; these included rash (51.5%) and pruritus (24.2%).56 Skin toxicities were also frequent in Phase I–II trials of capivasertib or ipatasertib, although the overall incidence of grade ≥3 adverse events, comparably, was lower with these compounds.7,110,111 Diarrhea is another well-known and frequent toxicity, and a DLT in early dose-finding trials,50,56 even though it was for the majority, mild or moderate (8–23% incidence of grade 3 or higher). Diarrhea tends to occur early in onset of treatment, but is reversible following treatment discontinuation and optimum management with anti-diarrheal agents, such as loperamide. Hyperglycemia is a well-defined class toxicity of AKT/PI3K inhibitors,42,50,61 as insulin-mediated glucose homeostasis, appears to largely depend on PI3K signaling via GSK3β and FOXO,109 though the incidence of hyperglycemia of any grade in clinical trials of AKT inhibitors has varied, for example from 21.2% in the monotherapy MK2206 phase I trial,56 to 92% with MK-2206 and anastrozole trial,42 and as low as 4% with ipatasertib and paclitaxel.41 Phase I trials of capivasertib and ipatasertib have also shown considerable grade 3 or higher incidence of hyperglycemia, albeit with no documented reports of hyperosmolar coma or ketoacidosis,50,65 though increasingly, it is clear that improved patient selection and aggressive patient optimization, including dietary intervention, glucose lowering medications, and treatment interruption with or without dose modifications can help with this class-specific toxicity.
To date, the close homology between the three AKT isoforms has hampered the development of isoform-specific AKT inhibitors, though perhaps the development of isoform-specific AKT inhibitors could avoid some of the toxicities that pan-AKT inhibitors cause and could be a strategy which may improve their side-effect profile as a class.
Future Directions
Moving forward, a more detailed understanding of the biology of AKT, including perhaps the development of isoform-specific AKT inhibitors, and the continued use of innovative clinical trial design will be important. Due to the rarity of some of the alterations in AKT, recently adapted novel trial designs, such as the basket trial of AKT inhibitor capivasertib in AKT1 E17K mutant solid tumors, and adaptive platform trials (such as the Tumor-Agnostic Precision Immuno-Oncology and Somatic Targeting Rational for You (TAPISTRY) Platform Study, NCT04589845) will likely be important. Novel AKT inhibitors are also under investigation: for example borussertib, a first-in-class covalent-allosteric AKT inhibitor has recently been developed and is showing anti-proliferative activity in cell lines and xenograft models when combined with the MEK inhibitor trametinib.112 Interestingly, a nanoparticle-encapsulated version of capivasertib has also been developed and tested in radio-resistant models of head and neck squamous cell carcinoma which may have the potential to achieve better treatment outcomes than the free drug combination.113
Conclusions
AKT is a key factor in cell survival and growth, with dysregulation of AKT implicated in a number of solid tumors. AKT remains an attractive target for cancer therapy as it is a central node along a key oncogenic pathway, where it diverges and integrates with signals from other important pathways. There is a veritable plethora of studies that support the role of AKT as a well-validated target for drug development, and while the translation of AKT inhibition into therapeutic benefit has been complex, recent clinical successes using combination strategies, particularly in breast cancer, while modest, are driving enthusiasm in the field. The multiple layers of AKT pathway regulation clearly impair our ability to more accurately predict efficacy to AKT inhibitors and only by delving more into the biology of the complex web of AKT pathway interactions will progress be made.
Looking to the future, the continued incorporation of tumor biopsies and analyses of ctDNA as part of biomarker-driven clinical trials will be critical in uncovering potential biomarkers of response and resistance to AKT inhibitors. To address the issues of suboptimal drug exposures due to toxicities, the development of such predictive biomarkers to optimally select patients, together with proof of target modulation are likely to be vital in the development of AKT inhibitors. Overcoming these challenges will enable genuine progress to be made in the development of AKT inhibitors for patients with cancer on a personalized basis, to improve outcomes and transform patient care.
Acknowledgments
The MD Anderson Cancer Center Support grant (NIH/NCI P30 CA016672), Clinical Translational Science Award 1UL1 TR003167 and the Cancer Prevention Research Institute of Texas (CPRIT) Precision Oncology Decision Support Core (RP150535), and Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy.
Disclosure
TAY: Research support (to Institution): AstraZeneca, Artios, Bayer, Beigene, Clovis, Constellation, Cyteir, Eli Lilly, EMD Serono, Forbius, F-Star, GlaxoSmithKline, Genentech, ImmuneSensor, Ipsen, Jounce, Karyopharm, Kyowa, Merck, Novartis, Pfizer, Ribon Therapeutics, Repare, Regeneron, Sanofi, Scholar Rock, Seattle Genetics, Tesaro, and Vertex Pharmaceuticals. Consultancies: Almac, Aduro, AstraZeneca, Atrin, Axiom, Bayer, Bristol Myers Squibb, Calithera, Clovis, Cybrexa, EMD Serono, F-Star, Guidepoint, Ignyta, I-Mab, Jansen, Merck, Pfizer, Repare, Roche, Rubius, Schrodinger, Seattle Genetics, Varian and Zai Labs. IV: Non-financial support from Arqule Therapeutics, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi:10.1038/nature12912
2. Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15(5):273–291. doi:10.1038/nrclinonc.2018.28
3. Clark AS, West K, Streicher S, Dennis PA. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol Cancer Ther. 2002;1(9):707–717. doi:10.1158/1535-7163.MCT-11-0712
4. Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016;82(4):943–956. doi:10.1111/bcp.13021
5. Hinz N, Jücker M. Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Communication and Signaling. 2019;17(1):1–29. doi:10.1186/s12964-019-0450-3
6. Lazaro G, Kostaras E, Vivanco I. Inhibitors in AKTion: ATP-competitive vs allosteric. Biochem Soc Trans. 2020;48(3):933–943. doi:10.1042/BST20190777
7. Smyth LM, Tamura K, Oliveira M, et al. Capivasertib, an AKT kinase inhibitor, as monotherapy or in combination with fulvestrant in patients with AKT1 E17K -mutant, ER-positive metastatic breast cancer. Clin Cancer Res. 2020;26(15):3947–3957. doi:10.1158/1078-0432.CCR-19-3953
8. Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005. doi:10.1016/S0065-230X(05)94002-5
9. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi:10.1038/nrc839
10. Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J. 2008;415:333–344. doi:10.1042/BJ20081056
11. Matheny RW, Adamo ML. Current perspectives on akt akt-ivation and akt-ions. Exp Biol Med. 2009;234(11):1264–1270. doi:10.3181/0904-MR-138
12. Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8(16):2502–2508. doi:10.4161/cc.8.16.9335
13. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405.
14. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. doi:10.1126/science.1106148
15. Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279(39):41189–41196. doi:10.1074/jbc.M406731200
16. A Altomare D, R Khaled A. Homeostasis and the Importance for a balance between AKT/mTOR activity and intracellular signaling. Curr Med Chem. 2012;19(22):3748–3762. doi:10.2174/092986712801661130
17. Jacinto E, Facchinetti V, Liu D, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–137. doi:10.1016/j.cell.2006.08.033
18. Li XJ, Leem S-H, Park MH, Kim SM. Regulation of YAP through an Akt-dependent process by 3, 3′-diindolylmethane in human colon cancer cells. Int J Oncol. 2013;43(6):1992–1998. doi:10.3892/ijo.2013.2121
19. Xu Q, Liu L-Z, Qian X, et al. MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 2012;40(2):761–774. doi:10.1093/nar/gkr730
20. Morrison DK. The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009;19(1):16–23. doi:10.1016/j.tcb.2008.10.003
21. Miller TW, Pérez-Torres M, Narasanna A, et al. Loss of phosphatase and tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69(10):4192–4201. doi:10.1158/0008-5472.CAN-09-0042
22. Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19 784 diverse solid tumors. JAMA Oncol. 2016;2(12):1565. doi:10.1001/jamaoncol.2016.0891
23. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91(2):231–241. doi:10.1016/S0092-8674(00)80405-5
24. Sangawa A, Shintani M, Yamao N, Kamoshida S. Phosphorylation status of Akt and caspase-9 in gastric and colorectal carcinomas. Int J Clin Exp Pathol. 2014;7(6):3312.
25. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta Mol Cell Res. 2011;1813(11):1978–1986. doi:10.1016/j.bbamcr.2011.03.010
26. Ogawara Y, Kishishita S, Obata T, et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277(24):21843–21850. doi:10.1074/jbc.M109745200
27. Park S, Kim D, Dan HC, et al. Identification of Akt interaction protein PHF20/TZP that transcriptionally regulates p53. J Biol Chem. 2012;287(14):11151–11163. doi:10.1074/jbc.M111.333922
28. Brown JS, Banerji U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol Ther. 2017;172:101–115. doi:10.1016/j.pharmthera.2016.12.001
29. Bertucci F, Ng CKY, Patsouris A, et al. Genomic characterization of metastatic breast cancers. Nature. 2019;569(7757):560–564. doi:10.1038/s41586-019-1056-z
30. Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. doi:10.1038/nature05933
31. Pascual J, Turner NC. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann Oncol. 2019;30(7):1051–1060. doi:10.1093/annonc/mdz133
32. Nagata Y, Lan K-H, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127. doi:10.1016/j.ccr.2004.06.022
33. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
34. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269). doi:10.1126/scisignal.2004088
35. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24(50):7455–7464. doi:10.1038/sj.onc.1209085
36. Robertson JFR, Coleman RE, Cheung K-L, et al. Proliferation and AKT activity biomarker analyses after capivasertib (AZD5363) treatment of patients with ER + invasive breast cancer (STAKT). Clin Cancer Res. 2020;26(7):1574–1585. doi:10.1158/1078-0432.CCR-19-3053
37. Kalinsky K, Hong F, Ck M, et al. AZD5363 in Patients (Pts) with tumors with AKT mutations: NCI-MATCH subprotocol EAY131-Y, A trial of the ECOG-ACRIN Cancer Research Group (EAY131-Y). Eur J Cancer. 2018.
38. Wolf DM, Yau C, Wulfkuhle J, et al. Mechanism of action biomarkers predicting response to AKT inhibition in the I-SPY 2 breast cancer trial. NPJ Breast Cancer. 2020;6(1). doi:10.1038/s41523-020-00189-2
39. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360–1372. doi:10.1016/S1470-2045(17)30450-3
40. Schmid P, Abraham J, Chan S, et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: the PAKT trial. J Clin Oncol. 2020;38(5):423–433. doi:10.1200/JCO.19.00368
41. Dent R, Oliveira M, Isakoff SJ, et al. 139O final results of the double-blind placebo (PBO)-controlled randomised phase II LOTUS trial of first-line ipatasertib (IPAT) + paclitaxel (PAC) for inoperable locally advanced/metastatic triple-negative breast cancer (mTNBC). Ann Oncol. 2020;31:S64–S65. doi:10.1016/j.annonc.2020.03.240
42. Ma CX, Sanchez C, Gao F, et al. A Phase I study of the AKT inhibitor MK-2206 in combination with hormonal therapy in postmenopausal women with estrogen receptor-positive metastatic breast cancer. Clin Cancer Res. 2016;22(11):2650–2658. doi:10.1158/1078-0432.CCR-15-2160
43. Turner NC, Alarcón E, Armstrong AC, et al. BEECH: a dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA. Ann Oncol. 2019;30(5):774–780. doi:10.1093/annonc/mdz086
44. Jones RH, Casbard A, Carucci M, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020;21(3):345–357. doi:10.1016/S1470-2045(19)30817-4
45. Yap TA, Garrett MD, Walton MI, et al. Targeting the PI3K–AKT–mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8(4):393–412. doi:10.1016/j.coph.2008.08.004
46. Falchook GS, Kurzrock R, Hm A, et al. First-in-man phase i trial of the selective MET inhibitor tepotinib in patients with advanced solid tumors. Clin Cancer Res. 2020;26(6):1237–1246. doi:10.1158/1078-0432.CCR-19-2860
47. Harb W, Saleh MN, Papadopoulos KP, et al. Clinical trial results from the subgroup of lymphoma/CLL in a Phase 1 Study of ARQ 092, a novel pan AKT-inhibitor. Blood. 2015;126(23):5116. doi:10.1182/blood.V126.23.5116.5116
48. Pant S, Subbiah V, Rodon J, et al. Abstract CT024: results of a phase I dose escalation study of ARQ 751 in adult subjects with advanced solid tumors with AKT1, 2, 3 genetic alterations, activating PI3K mutations, PTEN-null, or other known actionable PTEN mutations. Cancer Res. 2018. doi:10.1158/1538-7445.am2018-ct024
49. Yap TA, Yan L, Patnaik A, et al. Interrogating two schedules of the AKT inhibitor MK-2206 in patients with advanced solid tumors incorporating novel pharmacodynamic and functional imaging biomarkers. Clin Cancer Res. 2014;20(22):5672–5685. doi:10.1158/1078-0432.CCR-14-0868
50. Banerji U, Dean EJ, Pérez-Fidalgo JA, et al. A Phase I Open-Label Study to identify a dosing regimen of the pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA -mutated breast and gynecologic cancers. Clin Cancer Res. 2018;24(9):2050–2059. doi:10.1158/1078-0432.CCR-17-2260
51. Ahn DH, Li J, Wei L, et al. Results of an abbreviated phase-II study with the Akt inhibitor MK-2206 in patients with advanced biliary cancer. Sci Rep. 2015;5(1). doi:10.1038/srep12122
52. Ramanathan RK, McDonough SL, Kennecke HF, et al. Phase 2 study of MK-2206, an allosteric inhibitor of AKT, as second-line therapy for advanced gastric and gastroesophageal junction cancer: a SWOG cooperative group trial (S1005). Cancer. 2015;121(13):2193–2197. doi:10.1002/cncr.29363
53. Dasari A, Overman MJ, Fogelman DR, et al. A phase II and co-clinical study of an AKT inhibitor in patients (pts) with biomarker-enriched, previously treated metastatic colorectal cancer (mCRC). J Clin Oncol. 2016;34(15_suppl):3563. doi:10.1200/JCO.2016.34.15_suppl.3563
54. Xing Y, Lin NU, Maurer MA, et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019;21(1). doi:10.1186/s13058-019-1154-8
55. Lu W, Defeo-Jones D, Davis L, et al. Abstract #3714: in vitro and in vivo antitumor activities of MK-2206, a new allosteric Akt inhibitor. Cancer Res. 2009;69:3714.
56. Yap TA, Yan L, Patnaik A, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29(35):4688–4695. doi:10.1200/JCO.2011.35.5263
57. Tolcher A, Harb W, Sachdev J, et al. 338 results from a phase 1 study of ARQ 092, a novel pan AKT-inhibitor, in subjects with advanced solid tumors or recurrent malignant lymphoma. Eur J Cancer. 2015;51:S66. doi:10.1016/s0959-8049(16)30201-5
58. Shapiro GI, LoRusso P, Cho DC, et al. A phase Ib open-label dose escalation study of the safety, pharmacokinetics, and pharmacodynamics of cobimetinib (GDC-0973) and ipatasertib (GDC-0068) in patients with locally advanced or metastatic solid tumors. Invest New Drugs. 2021;39(1):163–174. doi:10.1007/s10637-020-00975-6
59. Burris HA, Siu LL, Infante JR, et al. Safety, pharmacokinetics (PK), pharmacodynamics (PD), and clinical activity of the oral AKT inhibitor GSK2141795 (GSK795) in a phase I first-in-human study. J Clin Oncol. 2011;29(15_suppl):3003. doi:10.1200/jco.2011.29.15_suppl.3003
60. Saura C, Roda D, Roselló S, et al. A first-in-human phase I study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 2017;7(1):102–113. doi:10.1158/2159-8290.CD-16-0512
61. Smyth LM, Batist G, Meric-Bernstam F, et al. Selective AKT kinase inhibitor capivasertib in combination with fulvestrant in PTEN-mutant ER-positive metastatic breast cancer. NPJ Breast Cancer. 2021;7(1). doi:10.1038/s41523-021-00251-7
62. Kalinsky K, Hong F, McCourt CK, et al. Effect of capivasertib in patients with an AKT1 E17K -mutated tumor. JAMA Oncol. 2021;7(2):271. doi:10.1001/jamaoncol.2020.6741
63. Bleeker FE, Felicioni L, Buttitta F, et al. AKT1E17K in human solid tumours. Oncogene. 2008;27(42):5648–5650. doi:10.1038/onc.2008.170
64. Kehr EL, Jorns JM, Ang D, et al. Mucinous breast carcinomas lack PIK3CA and AKT1 mutations. Hum Pathol. 2012;43(12):2207–2212. doi:10.1016/j.humpath.2012.03.012
65. Hyman DM, Smyth LM, Donoghue MTA, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol. 2017;35(20):2251–2259. doi:10.1200/JCO.2017.73.0143
66. Bose S, Kalinsky K. Durable clinical activity to the AKT inhibitor ipatasertib in a heavily pretreated patient with an AKT1 E17K mutant metastatic breast cancer. Clin Breast Cancer. 2021;21(3):e150–e153. doi:10.1016/j.clbc.2020.10.002
67. Flaherty KT, Gray RJ, Chen AP, et al. Molecular landscape and actionable alterations in a genomically guided cancer clinical trial: national cancer institute molecular analysis for therapy choice (NCI-MATCH). J Clin Oncol. 2020;38(33):3883–3894. doi:10.1200/JCO.19.03010
68. Jette N, Lees-Miller SP. The DNA-dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol. 2015;117(2–3):194–205. doi:10.1016/j.pbiomolbio.2014.12.003
69. Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBα/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell. 2008;30(2):203–213. doi:10.1016/j.molcel.2008.02.024
70. Wendel H-G, Stanchina ED, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428(6980):332–337. doi:10.1038/nature02369
71. Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9(7):1956–1967. doi:10.1158/1535-7163.MCT-09-1012
72. VanderWeele DJ, Zhou R, Rudin CM. Akt up-regulation increases resistance to microtubule-directed chemotherapeutic agents through mammalian target of rapamycin. Mol Cancer Ther. 2004;3(12):1605–1613.
73. Davies BR, Greenwood H, Dudley P, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11(4):873–887. doi:10.1158/1535-7163.MCT-11-0824-T
74. Schmid P, Abraham J, Chan S, et al. AZD5363 plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (PAKT): a randomised, double-blind, placebo-controlled, phase II trial. J Clin Oncol. 2018;36(15_suppl):1007. doi:10.1200/jco.2018.36.15_suppl.1007
75. Dent R, Kim SB, Oliveira M, et al. Abstract GS3-04: double-blind placebo (PBO)-controlled randomized phase III trial evaluating first-line ipatasertib (IPAT) combined with paclitaxel (PAC) for PIK3CA/AKT1/PTEN-altered locally advanced unresectable or metastatic triple. Cancer Res. 2021;81:
76. Crabb SJ, Birtle AJ, Martin K, et al. ProCAID: a phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Invest New Drugs. 2017;35(5):599–607. doi:10.1007/s10637-017-0433-4
77. Crabb SJ, Griffiths G, Marwood E, et al. Pan-AKT inhibitor capivasertib with docetaxel and prednisolone in metastatic castration-resistant prostate cancer: a randomized, placebo-controlled phase II trial (procaid). J Clin Oncol. 2021;39(3):190–201. doi:10.1200/JCO.20.01576
78. Yap TA, Omlin A, De Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013;31(12):1592–1605. doi:10.1200/JCO.2011.37.6418
79. Wang L-E, Ma H, Hale KS, et al. Roles of genetic variants in the PI3K and RAS/RAF pathways in susceptibility to endometrial cancer and clinical outcomes. J Cancer Res Clin Oncol. 2012;138(3):377–385. doi:10.1007/s00432-011-1103-0
80. Westin SN, Sill M, Coleman RL, et al. Limited access safety lead-in of the MEK inhibitor trametinib in combination with GSK2141795, an AKT inhibitor, in patients with recurrent or persistent endometrial cancer: a Gynecologic Oncology Group study. Gynecol Oncol. 2016;141:4–5. doi:10.1016/j.ygyno.2016.04.038
81. Tolcher AW, Kurzrock R, Valero V, et al. Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother Pharmacol. 2020;85(4):673–683. doi:10.1007/s00280-020-04038-8
82. Algazi AP, Muthukumar AH, O’Brien K, et al. Phase II trial of trametinib in combination with the AKT inhibitor GSK 2141795 in BRAF wild-type melanoma. J Clin Oncol. 2015;33(15_suppl):9068. doi:10.1200/jco.2015.33.15_suppl.9068
83. Shoushtari AN, Kudchadkar RR, Panageas K, et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J Clin Oncol. 2016;34(15_suppl):9511. doi:10.1200/JCO.2016.34.15_suppl.9511
84. Irvine M, Stewart A, Pedersen B, et al. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis. 2018;7(9):72. doi:10.1038/s41389-018-0081-3
85. Algazi AP, Moon J, Chmielowski B, et al. SWOG S1221: a phase 1 dose escalation study co-targeting MAPK-dependent and MAPK-independent BRAF inhibitor resistance in BRAF mutant advanced solid tumors with dabrafenib, trametinib, and GSK2141795 (ClinicalTrials.gov NCT01902173). J Clin Oncol. 2017;35(15_suppl):2578. doi:10.1200/JCO.2017.35.15_suppl.2578
86. Ramaswamy B, Mrozek E, Lustberg M, et al. Abstract LB-216: NCI 9455: phase II study of trametinib followed by trametinib plus AKT inhibitor, GSK2141795 in patients with advanced triple negative breast cancer. Cancer Res. 2016;76:
87. Liu JF, Gray KP, Wright AA, et al. Results from a single arm, single stage phase II trial of trametinib and GSK2141795 in persistent or recurrent cervical cancer. Gynecol Oncol. 2019;154(1):95–101. doi:10.1016/j.ygyno.2019.05.003
88. Bokobza SM, Jiang Y, Weber AM, Devery AM, Ryan AJ. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget. 2014;5(13):4765–4778. doi:10.18632/oncotarget.2017
89. Li H, Schmid-Bindert G, Wang D, et al. Blocking the PI3K/AKT and MEK/ERK signaling pathways can overcome gefitinib-resistance in non-small cell lung cancer cell lines. Adv Med Sci. 2011;56(2):275–284. doi:10.2478/v10039-011-0043-x
90. Lin -C-C, Yu C-J, Ho -C-C, et al. P1.12 - a Phase I Dose Defining Study for MK-2206 combined with gefitinib in NSCLC population enriched with EGFR mutation. Ann Oncol. 2015;26:ii16. doi:10.1093/annonc/mdv090.12
91. Wisinski KB, Tevaarwerk AJ, Burkard ME, et al. Phase I Study of an AKT inhibitor (MK-2206) combined with lapatinib in adult solid tumors followed by dose expansion in advanced HER2 + breast cancer. Clin Cancer Res. 2016;22(11):2659–2667. doi:10.1158/1078-0432.CCR-15-2365
92. Tolcher AW, Khan K, Ong M, et al. Antitumor activity in RAS -driven tumors by blocking AKT and MEK. Clin Cancer Res. 2015;21(4):739–748. doi:10.1158/1078-0432.CCR-14-1901
93. Do K, Speranza G, Bishop R, et al. Biomarker-driven phase 2 study of MK-2206 and selumetinib (AZD6244, ARRY-142886) in patients with colorectal cancer. Invest New Drugs. 2015;33(3):720–728. doi:10.1007/s10637-015-0212-z
94. Chung V, McDonough S, Philip PA, et al. Effect of selumetinib and MK-2206 vs oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 2017;3(4):516–522. doi:10.1001/jamaoncol.2016.5383
95. Costa C, Wang Y, Ly A, et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer Discov. 2020;10(1):72–85. doi:10.1158/2159-8290.CD-18-0830
96. Wander SA, Juric D, Supko JG, et al. Phase Ib trial to evaluate safety and anti-tumor activity of the AKT inhibitor, ipatasertib, in combination with endocrine therapy and a CDK4/6 inhibitor for patients with hormone receptor positive (HR+)/HER2 negative metastatic breast cancer (MBC) (TAKTI. J Clin Oncol. 2020;38(15_suppl):1066. doi:10.1200/JCO.2020.38.15_suppl.1066
97. Oliveira M, Villagrasa P, Ciruelos E, et al. Abstract OT-35-01: solti-1507 A phase ib study of ipatasertib and anti-her2 therapy in her2-positive advanced breast cancer with pik3ca mutation (ipather). Cancer Res. 2021;81:
98. Ibrahim YH, García-García C, Serra V, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–1047. doi:10.1158/2159-8290.CD-11-0348
99. Rehman FL, Lord CJ, Ashworth A. The promise of combining inhibition of PI3K and PARP as cancer therapy. Cancer Discov. 2012;2(11):982–984. doi:10.1158/2159-8290.CD-12-0433
100. Yap TA, Kristeleit R, Michalarea V, et al. Phase I trial of the PARP inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2 - and Non– BRCA1/2 -mutant cancers. Cancer Discov. 2020;10(10):1528–1543. doi:10.1158/2159-8290.CD-20-0163
101. Ma CX, Crowder RJ, Ellis MJ. Importance of PI3-kinase pathway in response/resistance to aromatase inhibitors. Steroids. 2011;76(8):750–752. doi:10.1016/j.steroids.2011.02.023
102. Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. 2015;15(5):261–275. doi:10.1038/nrc3920
103. Ma CX, Suman V, Goetz MP, et al. A Phase II trial of neoadjuvant MK-2206, an AKT inhibitor, with anastrozole in clinical stage II or III PIK3CA-mutant ER-positive and HER2-negative breast cancer. Clin Cancer Res. 2017;23:6823–6832.
104. Ferrari AC, Chen Y-H, Hudes GR, et al. E2809: androgen receptor (AR) modulation by bicalutamide (Bic) and MK-2206 (MK) in men with rising PSA at high-risk of progression after local prostate cancer (PC) treatment.. J Clin Oncol. 2016;34(2_suppl):9. doi:10.1200/jco.2016.34.2_suppl.9
105. Kolinsky MP, Rescigno P, Bianchini D, et al. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann Oncol. 2020;31:619–625. doi:10.1016/j.annonc.2020.01.074
106. Shore ND, Mellado B, Shah S, et al. A phase I study of capivasertib in combination with Abiraterone acetate in patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2021;39:85. doi:10.1200/JCO.2021.39.6_suppl.85
107. De Bono JS, De Giorgi U, Rodrigues DN, et al. Randomized phase II study evaluating AKT blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res. 2019;25(3):928–936. doi:10.1158/1078-0432.CCR-18-0981
108. de Bono JS, Bracarda S, Sternberg CN, et al. LBA4 IPATential150: phase III study of ipatasertib (ipat) plus Abiraterone (abi) vs placebo (pbo) plus abi in metastatic castration-resistant prostate cancer (mCRPC). Ann Oncol. 2020;31:S1153–S1154. doi:10.1016/j.annonc.2020.08.2250
109. Zhang Y, Yan H, Xu Z, et al. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin Drug Metab Toxicol. 2019;15(9):767–774. doi:10.1080/17425255.2019.1663169
110. Chien AJ, Cockerill A, Fancourt C, et al. A phase 1b study of the Akt-inhibitor MK-2206 in combination with weekly paclitaxel and trastuzumab in patients with advanced HER2-amplified solid tumor malignancies. Breast Cancer Res Treat. 2016;155(3):521–530. doi:10.1007/s10549-016-3701-7
111. Chien AJ, Tripathy D, Albain KS, et al. MK-2206 and standard neoadjuvant chemotherapy improves response in patients with human epidermal growth factor receptor 2–positive and/or hormone receptor–negative breast cancers in the I-SPY 2 trial. J Clin Oncol. 2020. doi:10.1200/JCO.19.01027
112. Weisner J, Landel I, Reintjes C, et al. Preclinical efficacy of covalent-allosteric AKT inhibitor borussertib in combination with trametinib in KRAS-mutant pancreatic and colorectal cancer. Cancer Res. 2019:
113. Lang L, Shay C, Zhao X, et al. Simultaneously inactivating Src and AKT by saracatinib/capivasertib co-delivery nanoparticles to improve the efficacy of anti-Src therapy in head and neck squamous cell carcinoma. J Hematol Oncol. 2019;12(1). doi:10.1186/s13045-019-0827-1
114. Bendell, J. C. et al. Results of the X-PECT study: A phase III randomized double-blind, placebo-controlled study of perifosine plus capecitabine (P-CAP) versus placebo plus capecitabine (CAP) in patients (pts) with refractory metastatic colorectal cancer (mCRC). J Clin Oncol. 2012;30:LBA3501–LBA3501.
115. Blagden, S. P. et al. Phase IB dose escalation and expansion study of akt inhibitor afuresertib with carboplatin and paclitaxel in recurrent platinum-resistant ovarian cancer. Clin Cancer Res. 2019. doi:10.1158/1078-0432.CCR-18-2277
116. Lakhani, N. et al. Results of a phase Ib study of ARQ 092 in combination with carboplatin (C) plus paclitaxel (P), or with P in patients (pts) with solid tumors. J Clin Oncol. 2017. doi:10.1200/jco.2017.35.15_suppl.2524.
117. Tolcher, A. W. et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol. 2015;75:183–189.
118. Westin, S.et al. Phase I expansion of olaparib (PARP inhibitor) and AZD5363 (AKT inhibitor) in recurrent ovarian, endometrial and triple negative breast cancer. Ann Oncol. 2017;8:v130–v131.
119. LaraJr., P. N.et al. Phase II Study of the AKT Inhibitor MK-2206 plus Erlotinib in Patients with Advanced Non-Small Cell Lung Cancer Who Previously Progressed on Erlotinib. Clin Cancer Res. 2015;21:4321–4326.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.