Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 5
Clinical and experimental studies of multiple sclerosis in Russia: experience of the leading national research centers
Authors Zavalishin I, Belogurov Jr A, Lomakin Y, Ponomarenko N, Morozova S, Suslina Z, Piradov M, Illarioshkin S, Gabibov A
Received 21 October 2014
Accepted for publication 21 May 2015
Published 6 August 2015 Volume 2015:5 Pages 83—90
DOI https://doi.org/10.2147/DNND.S46023
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Müller
Igor A Zavalishin,1 Alexey A Belogurov Jr,2–4 Yakov A Lomakin,2 Natalia A Ponomarenko,2 Sofia N Morozova,1 Zinaida A Suslina,1,† Michael A Piradov,1 Sergey N Illarioshkin,1 Alexander G Gabibov2–5
1Research Center of Neurology, 2Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, 3Institute of Fundamental Medicine and Biology, Kazan Federal University, Kazan, Republic of Tatarstan, 4Institute of Gene Biology, RAS, 5Chemistry Department, Moscow State University, Moscow, Russia
†Dr Zinaida A Suslina passed away on June 22, 2014
Abstract: Mechanisms of axonal damage and adaptive capacity in multiple sclerosis (MS), including cortical reorganization, have been actively studied in recent years. The lack of regenerative capabilities and the irreversibility of neurodegeneration in MS are critical factors for the optimization of MS treatment. In this study, we present the results of clinical and basic studies in the field of MS by two leading Russian centers. Clinical and neuroimaging correlations show that spinal damage in MS is accompanied by functional reorganization of the cerebral cortex, which is determined not only by the efferent component but also by the afferent component. Comparative analysis of MS treatment with both interferon β1b (IFN-β1b) and IFN-β1a at a dosage of 22 µg for 3 years through subcutaneous administration and glatiramer acetate showed equally high efficiency in reducing the number of exacerbations in relapsing-remitting MS and secondary-progressive MS. We demonstrate a reduced risk of disability in relapsing-remitting MS and secondary-progressive MS patients in all groups treated with IFN-β1 and glatiramer acetate. MS appears to be a disease that would greatly benefit from the development of personalized therapy; thus, adequate molecular predictors of myelin degradation are greatly needed. Therefore, novel ideas related to the viral hypothesis of the etiology of MS and new targets for therapeutic intervention are currently being developed. In this manuscript, we discuss findings of both clinical practice and fundamental research reflecting challenges and future directions of MS treatment in the Russian Federation.
Keywords: multiple sclerosis, functional MRI, cortical reorganization, disease-modifying therapy, Epstein–Barr virus, autoantibodies, immunoproteasome, personalized medicine
Introduction
Our understanding of the pathology of multiple sclerosis (MS) has significantly changed over past decades. Axonal dysfunction and neurodegeneration are now two widely accepted phenomena of MS along with immune cell activation, blood–brain barrier disruption, multifocal inflammation, demyelination, oligodendrocyte death, and reactive gliosis. Degeneration is considered the primary cause of irreversible neurological deterioration and disability progression in MS. Furthermore, researchers have recently gained interest in the adaptive potential of MS with the goal of inhibiting neurodegeneration and neurological impairment. The compensatory mechanisms of MS have been discussed at different levels: the cellular level (axonal sprouting, changes in synaptic stability, or synapse reorganization); the tissue level (edema resorption, rearrangement of sodium channels along the axonal membrane, and remyelination); the systemic level (eg, changes in excitability of the primary and secondary motor areas in the ipsilateral and contralateral hemisphere); and, finally, the behavioral level (eg, development and training of new movement and cognitive strategies).1
Cortical reorganization affects the recovery of function in MS patients through the formation of myelin lesions and axonal degeneration.2–4 However, the reported data on the dynamics of cortical reorganization during the active stage of MS and during remission are lacking. Furthermore, the effect of spinal pathology on the potential of cortical plasticity is of great interest.
Inflammation and degeneration in multiple sclerosis: functional compensation and the effect of disease-modifying treatment
A dynamic comparison of structural and functional magnetic resonance imaging (MRI) parameters in patients with relapsing MS and motor disorders was conducted at the Research Centre of Neurology, Russian Academy of Medical Sciences (hand paresis was used as a model).5 The study included 25 patients with relapsing-remitting MS (RRMS) in the exacerbation phase, which is characterized by unilateral mild hand paresis, before the initiation of corticosteroid therapy. Dynamic follow-up was performed 3 months later during remission characterized by symptom regression. The control group consisted of 12 individuals without any pathologies of the central nervous system (CNS) or other systems. The study was approved by the local ethics committee. Written informed consent was obtained from all patients. All MS patients underwent neurological examination, including assessment using the Expanded Disability Status Scale (EDSS), dynamometry, and the conventional nine-hole peg test. MRI of the brain and the cervical portion of spine and spinal cord was performed using a Magnetom Avanto MRI unit, 1.5 T (Siemens, Erlangen, Germany). The examination of the brain included both the standard MRI sequences and those used for obtaining functional MRI (fMRI) data and diffusion tensor imaging data. A simple block-designed paradigm6 involving flexion and extension of fingers II–V with a frequency of 1 Hz7 was used to record fMRI data.
The MRI results showed that motor symptom development in all MS patients in the examined group was caused by demyelination of cervical spinal foci; cerebral foci were not found in any of the patients along the corticospinal tract. Bidirectional changes in reorganization of the primary sensorimotor cortex (SM1) during exacerbation were detected: the SM1 activation area (measured in voxels) when moving a paretic hand was smaller in some patients (subgroup 1) and greater in other patients (subgroup 2) compared with that recorded when moving the nonparetic hand. No intergroup differences in the clinical features of MS were identified, except for ipsilateral asymmetric reduction of vibration sensitivity in most of the subgroup 1 patients compared with the subgroup 2 patients (87% and 20%, respectively; P<0.05).
Standard MRI showed that the subgroup 1 patients had a significantly higher total number of foci at the level of the C1–C7 vertebrae. An analysis of the diffusion tensor imaging data showed that there were significant changes in the ipsilateral hemisphere both in the corticospinal tract (at the level of the internal capsule) and in the medial loop in subgroup 1 patients, compared with the control group. In contrast, subgroup 2 patients showed changes only in the corticospinal tract (at the level of the brainstem and the internal capsule).
At the 3-month follow-up, at which time the patients were in the remission phase, all MS patients showed regression of hand paresis. In subgroup 1, asymmetric reduction of vibration sensitivity persisted in 53% of the cases, but it was not observed in subgroup 2. The results of the conventional nine-hole peg test showed significantly less complete recovery of fine motor skills in subgroup 1 compared with the other subgroups.
Also at the 3-month follow-up, at which time patients were in the remission phase, fMRI data for subgroup 1 showed a trend toward an increase in the activation area of the primary SM1 when the patients moved the previously paretic hand. Nevertheless, the level of activation of the primary SM1 was lower than that of the contralateral hemisphere. The changes in diffusion tensor imaging parameters previously observed in subgroup 1 in the corresponding medial loop persisted at the 3-month follow-up. In subgroup 2 patients, the activation area of the primary SM1 when moving the previously paretic hand was found to be reduced to a level comparable to that observed when moving the nonparetic hand.
These data suggest that spinal lesions in the MS patients are accompanied by functional rearrangement of the cerebral cortex. Cortical reorganization in patients with clinically similar paresis is determined by both the efferent and afferent components. Heterogeneity of the detected structural and functional changes during regression of motor symptoms is associated with various initial pathophysiological mechanisms and affects the degree of functional recovery. Finally, these detected differences indicate that a differentiated approach to selecting rehabilitation measures for MS patients with motor disorders should take into account not only the efferent component but also the possible deafferent component.
The exhaustion of recovery potential, the irreversibility of degenerative changes, and the impact of the type of MS disease course during early disease stages on long-term prognosis make personalized optimization of MS therapy necessary. They also necessitate the use of the criteria for suboptimal therapeutic response and a switch in treatment strategy from first-line disease-modifying therapy (DMT) to therapy escalation.8
Researchers at the Research Center of Neurology, Russian Academy of Medical Sciences, have accumulated vast experience in the use of first-line disease-modifying therapies (interferon-β [IFN-β] and glatiramer acetate). Here, we have summarized our own data obtained by comparative analysis of immune modulation therapy of MS over 3 years using IFN-β1b, IFN-β1a (injected subcutaneously at a dose of 22 μg), and glatiramer acetate in the context of their effects on inflammatory and degenerative mechanisms of the pathological process of MS.
The inclusion criteria for MS patients receiving therapy with the aforementioned drugs were as follows: 1) reliable diagnosis of MS; 2) RRMS or secondary-progressive MS (SPMS); 3) at least two acute attacks over the past 2 years for patients with RRMS; 4) severity of neurologic disability as indicated by an EDSS score of 0–5; 5) at least 18 years of age; 6) absence of any other neurological disorder that could potentially cause the existing symptoms; and 7) absence of pregnancy or lactation in female patients. The patients could have received immune modulation therapy or be therapy-naïve. Although an indication for glatiramer acetate therapy is RRMS, we also monitored a relatively small group of patients with SPMS, including those with exacerbations, who received glatiramer acetate therapy. The standard therapy regimen was used for all patients. The dynamic follow-up included examination at the onset of therapy and then for every 3 months. A total of 400 patients were examined: 87 patients received IFN-β1b therapy, 176 patients received IFN-β1a therapy, and 137 patients received glatiramer acetate therapy. At the first examination, all the MS patients had the same relapse rate during the 2 years before the initiation of therapy. A statistically significant increase in relapse rate was observed in each therapeutic group 2 years before the initiation of therapy, which demonstrates an increase in disease activity. Also, during the first year of therapy, the mean relapse rate decreased and subsequently remained at a low level. A reduced relapse rate was also observed in all therapeutic groups of patients with SPMS during the first year of therapy, and this effect persisted during the subsequent therapy. Similar to patients with RRMS, no significant differences in relapse rate at different therapy stages were detected between the therapeutic groups (Table 1).
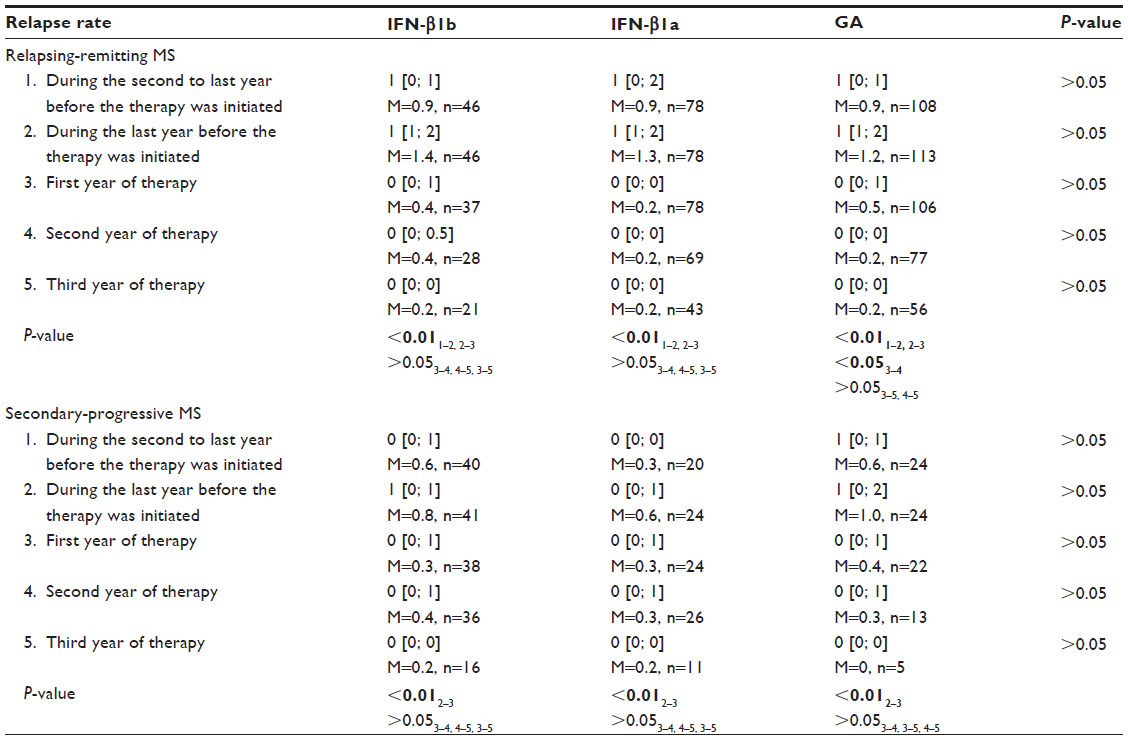 | Table 1 Relapse rate within 2 years before the therapy was initiated and during 3 years of therapy with IFN-β1b, IFN-β1a SC, and glatiramer acetate in MS patients |
When assessing the severity of neurological disability using the EDSS score in patients with RRMS, a higher degree of neurological impairments was initially observed in the subgroup receiving IFN-β1b therapy. Prevention of aggravation of neurological disability was observed during IFN-β1a and glatiramer acetate therapies, while IFN-β1b therapy significantly reduced the EDSS score. Meanwhile, there were no significant differences between the therapeutic groups in the severity of neurological disability at different therapy stages. In patients with SPMS, the severities of neurological disability in the subgroups receiving different therapies were initially identical, but those who received INF-β1b therapy showed a significant increase in EDSS score during the follow-up, while those who received glatiramer acetate and IFN-β1a showed significant prevention of aggravation of neurological disability during the therapy period (Table 2).
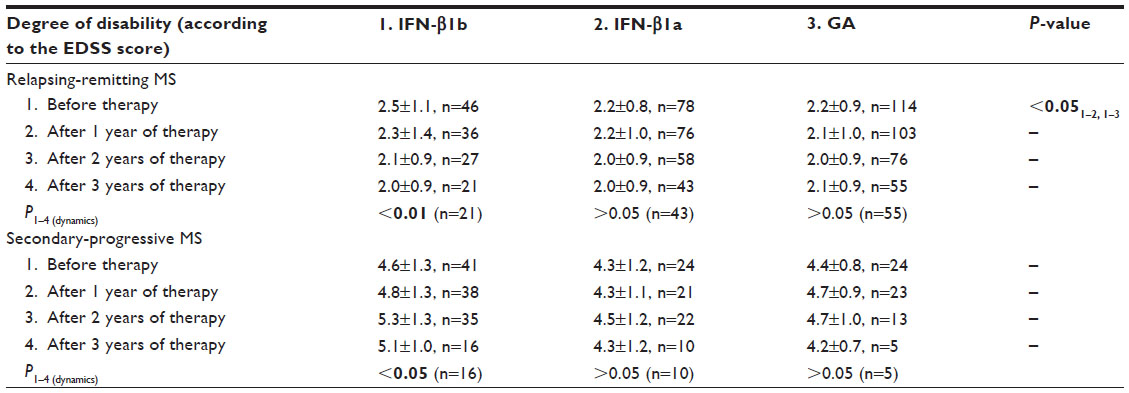 | Table 2 Dynamics of the severity of neurological disability (according to the EDSS score) during 3 years of therapy with IFN-β1b, 22 μg IFN-β1a SC, and glatiramer acetate |
This study is the first to summarize the vast experience in using 22 μg IFN-β1a subcutaneously for MS treatment. The data indicated that this IFN-β dose was effective in patients with RRMS and SPMS. Furthermore, we would like to emphasize the therapeutic effect of glatiramer acetate in patients with SPMS in the context of reducing the relapse rate and preventing the aggravation of severity of neurological disability.
These data are in agreement with the results of multicenter studies in patients with RRMS (REGARD, BEYOND). No significant clinical difference in the effectiveness of high-dose IFN-β and glatiramer acetate therapy was found in those studies; however, advantages of IFN therapy with respect to some neuroimaging parameters were found.9–11 Our clinical data demonstrate the prevention of aggravation of neurological impairments in patients with RRMS by providing therapy with all the three drugs. This effect is most likely due to a reduction of the activity of the immunoinflammatory component of the pathological process (as evidenced by the reduced relapse rate) and, therefore, a reduced risk of secondary axonal damage. Also, the probable neuroprotective effect of these drugs is now being discussed.12–14
Leray et al15 reported that disability progression among the MS patients has two stages: the first stage (EDSS score <3.0) presumably depends on focal inflammation, while the second stage (EDSS score <6.0) is most likely independent of local inflammatory responses. The independence of the duration of the second stage on that of the first stage emphasizes the need for stabilizing neurological impairments at the lower level (at EDSS score <3.0). In Russia, the results of clinical trials conducted at the Research Center of Neurology are an important basis for creating the national standards and recommendations for the therapeutic management of MS. To summarize our experience in treating MS using disease-modifying drugs, our data correspond well with those of previously published studies9–11 that focused on correcting the immune-inflammatory component of MS pathogenesis and on preventing disability aggravation. However, the inevitable exhaustion of the recovery potential and compensatory mechanisms as well as the involvement of both inflammatory and degenerative processes in MS pathogenesis demonstrates the need for using neuroprotective therapeutic approaches.
The environmental hypothesis of multiple sclerosis
The limited knowledge concerning the triggering mechanisms of autoimmunity and, in particular, neurological autoimmunity makes the clinical and fundamental aspects of the “primary event” in MS extremely intriguing.16,17 These investigations are highly necessary, as conceptual drug development approaches in the MS field have rather poor diversity.18 Among the several theories of the mechanism of induction of neuronal autoimmune disorders, the environmental hypothesis is the one that is most well-documented.19 The viral involvement in the triggering of MS and, especially, the Epstein–Barr etiology of MS have gained great interest after numerous epidemiological studies during the first decade of the 21st century.20,21 Recent studies of Epstein–Barr virus (EBV) involvement in the triggering of MS obtained rather controversial data, indicating a complicated relationship between MS and EBV with regard to immunogenicity, immunochemistry, and cellular immunology.21
It is increasingly evident that autoreactive B cells, together with pathogenic T-cells, are among the major players in MS development.22,23 An increased level of immunoglobulin G (IgGs) in the cerebrospinal fluid of MS patients was first detected by Kabat.24 Analyses of myelin neuroantigen-specific antibody repertoires and their possible cross-reactivity against environmental antigens, including viral proteins, could shed light on the mechanism of MS induction and progression. The direct detection of cross-reactive antibodies in blood serum of patients by regular immune assay techniques generates uncertain results due to the high basicity of myelin basic protein (MBP), which is one of the major autoantigens in MS.25–27 It seems appealing to work with broad IgG repertoires from MS patients and to search for potential structural peculiarities in those IgGs that possess cross-reactive features. The analysis of these antibodies by the deep sequencing approach may be useful for the development of prognostic criteria of MS development and may indicate the “viral history” of neurodegeneration triggering in MS.28 To work with antibody repertoires, a phage display library of single-chain variable fragments (scFvs) was constructed from blood lymphocytes of patients with MS.28 Structural alignment of 13 scFvs clones selected against MBP, one of the major myelin antigens, showed high homology within variable regions of cerebrospinal fluid MS-associated antibodies and antibodies toward Epstein–Barr latent membrane protein 1 (LMP1). Three scFv clones showed pronounced specificity for the MBP fragments 65–92 and 130–156, which are similar to the MS antibodies found in the serum. One of these clones in both scFv and full-size human antibody constructs, designated E2, was shown to react with both MBP and LMP1 proteins in vitro, suggesting that it has natural cross-reactivity. Thus, antibodies that are produced against LMP1 during EBV infection might act as an inflammatory trigger by reacting with MBP, which suggests molecular mimicry in the mechanism of MS pathogenesis. The detected “molecular signature” of EBV in the MS antibody repertoire correlates with the environmental hypothesis, which suggests that MS progression is triggered by molecular mimicry. Deep sequencing showed that the two cross-reactive antigens are probably individually recognized by light or heavy chains. According to the high structural homology between selected autoantibodies and a number of various antiviral IgGs, we suggest that a wide range of pathogens, rather than a single virus, are possible triggers of MS.28
Processing of myelin basic antigen as a tool for diagnostic and drug development
During the last two decades, it has been shown that a wide variety of antigens can be processed by autoantibodies.29–33 The discovery of natural catalytic antibodies (abzymes) raised on the background of autoimmune disease revealed their beneficial and pathogenic roles in disease progression. Thus, the conflicting Dr Jekyll and Mr Hyde protective and destructive aspects of catalytic antibodies should be carefully considered in the development of various autoimmune diseases.34 The antibody-mediated processing of MBP was first shown in animal models of the development of experimental autoimmune encephalomyelitis (EAE),35 and, later, the site-specific cleavage of this neuroantigen by autoantibodies from human and mouse sera was clearly documented.36 Human and murine catalytic antibodies have been shown to efficiently cleave MBP but not other polypeptide substrates. The established MS therapeutic agent glatiramer acetate, also known as Copaxone, was found to be a specific abzyme inhibitor. The discovered activity was shown to be the intrinsic property of the IgG molecule. No activity was found in the sera-derived antibody fractions of healthy donors or control mice. Sera of 24 patients with clinically proven MS at different stages of the disease and 20 healthy controls were screened for anti-MBP antibody-mediated proteolytic activity. The activity correlated with the scores on the MS EDSS (r2=0.85, P<0.001). Thus, anti-MBP autoantibody-mediated proteolysis may be regarded as an additional marker of MS disease progression.37 This epitope-specific antibody-mediated degradation of MBP may explain the mechanism of the slow development of neurodegeneration in MS. The sites of MBP cleavage determined by mass spectrometry were localized within immunodominant regions of MBP. The abzymes could also cleave recombinant substrates containing encephalitogenic MBP 85–101 peptide. This finding was used to develop a novel immunochemical diagnostic test for MS progression.37 The MBP-derived recombinant epitope library, which includes all autoantigens, allowed for a precise analysis of the cleavage site of encephalitogenic MBP 85–101 peptide. A number of MBP epitopes specific for the autoantibodies in MS were detected and compared with those from other neuronal disorders (ONDs). ELISA (enzyme-linked immunosorbent assay) and PAGE (polyacrylamide gel electrophoresis)/surface-enhanced laser desorption/ionization mass spectroscopy assays were used to define the epitope binding/cleavage activities of autoantibodies isolated from the sera of 26 MS patients, 22 OND patients, and eleven healthy individuals. The levels of autoantibodies to the MBP fragments 48–70 and 85–170 and to the whole MBP and myelin oligodendrocyte glycoprotein molecules were significantly higher in the sera of MS patients than in those of healthy donors. In contrast, selective reactivity to the MBP fragments 43–68 and 146–170 distinguished the OND patients from the MS patients. Seventy-seven percent of patients with SPMS and 85% of patients with RRMS, but only 9% of patients with OND and no healthy donors, were positive for catalysis, showing pronounced epitope specificity to encephalitogenic MBP peptide 81–103. This peptide retained its substrate properties when flanked with two fluorescent proteins, providing a novel fluorescent resonance energy transfer approach for MS studies. The anti-MBP autoantibody-mediated, epitope-specific binding and cleavage was proposed as a specific characteristic of MS compared with OND and healthy donors and may be recommended as an additional biomarker of MS disease progression.
MBP and MBP-reactive species may not be purely deleterious and may have potential therapeutic applications.38 Multiple MBP-derived peptides were extensively examined as potential drugs for curing MS.39 Unfortunately, the absence of effective suppression of inflammation symptoms and cases of exacerbation sufficiently inhibited the development of peptide-based drugs against MS.40 Recently, newly identified structural parts of MBP with pronounced therapeutic effects toward EAE in Dark Agouti rats were reported.41 Dark Agouti rats with EAE induced by syngeneic spinal cord homogenate in complete Freund’s adjuvant were treated with human MBP peptides 46–62, 81–102, 124–139, and 147–170 and glatiramer acetate through the nasal route. MBP fragments 124–139 and 147–170 displayed only mild therapeutic effects, while MBP fragment 46–62 significantly reduced EAE, as reflected by lower clinical scores and shorter EAE duration compared with controls. These peptides were also examined in preclinical studies,42 where EAE in the Dark Agouti rats was treated with identified immunodominant peptides of the MBP encapsulated in mannosylated small unilamellar vesicles. The liposome-encapsulated MBP fragment 46–62 was shown to be the most effective in reducing maximal disease score during the first attack, while MBP fragments 124–139 and 147–170 completely prevented the development of the exacerbation stage. Both mannosylation of liposomes and encapsulation of peptides are critical for the therapeutic effect of MBP peptides because neither naked peptides nor nonmannosylated liposomes (loaded or empty) have been found to be effective. The liposome-mediated synergistic effect of a mixture of the three MBP peptides mentioned earlier significantly suppressed the progression of protracted EAE, reducing the median cumulative disease score from 22 to 14 points compared with the placebo group; prevented the production of circulating autoantibodies; downregulated the synthesis of Th1 cytokines; and induced the production of brain-derived neurotropic factor in the CNS. Thus, this proposed formulation ameliorates EAE, resulting in a less severe first attack and rapid recovery from exacerbation, and offers a promising therapeutic modality for MS. Currently, this formulation has successfully passed Phase IIa of clinical studies.
New strategies for MS drug discovery may be found in the field of proteosomal machinery of antigen degradation and presentation. Specific proteosomal inhibitors have recently become a powerful tool for curing cancer and some autoimmune disorders.43,44 Recently, it was shown that CNS proteosomal degradation machinery during EAE development may be substantially altered in terms of the upregulation of immunosubunits.45 The absence of ubiquitination control of proteasome-mediated protein degradation has previously been shown for a number of proteins.46 Dr Belogurov et al demonstrated for the first time that a pathophysiologically important autoantigen, ie, MBP, is processed as an ubiquitin-independent substrate of the 26S proteasome.47,48 The discovered lack of ubiquitination control on the degradation of the key neuroantigens may be regarded as one of the causes of inappropriate antigen presentation, which may cause autoimmune dysregulation. Thus, the recently demonstrated therapeutic efficacy of β1-immunoproteasome-targeted inhibitors on EAE progression45 indicates that they may be promising for MS treatment.
Perspectives
MS remains a severe inflammatory demyelinating disease of the CNS affecting over 2.1 million people throughout the world and demanding the development of new drugs and the improvement of existing treatment protocols. Despite substantial progress in MS treatment using glatiramer acetate and IFN-β, the lack of conceptual progress in the development of immune therapy due to MS heterogeneity makes this disease less curable than some distinct forms of cancer.49 Thus, the development of EAE and toxin- or virus-induced demyelination animal models should be continued.50 MS may be regarded as a perspective platform for the development of personalized drug therapy, similar to cancer, as a number of successful antibody-based anticancer approaches have been developed.51–53 The anti-CD20, anti-CD52, and anti-α4-integrin therapies are also under investigation for MS treatment.49 It should be mentioned that the long-term consequences of immunosuppressive treatments are still unknown or, at least, have been poorly investigated because most published clinical trials finish after few years of observation, which is an insufficient period of time to address the long-term consequences of these treatments. Therefore, the development of therapies capable of specific depletion of autoreactive B54 and CD8+ T-cells55 may have potential for personalized treatments of MS. Another important step toward personalized treatment of MS is the development of biomarkers (clinical, immunologic, genetic, etc) that can help predict the course of a disease and its response to a particular medication.
Disclosure
AGG received support from the RFBR Grant 13-04-40277-H and Presidential Grant for the support of leading scientific schools NSh-2064.2014.14 “Chemical Foundations of Biocatalysis”. AAB Jr received support from the Russian Scientific Foundation (project #14-14-00585) and Fellowship of President of Russian Federation (Cπ 2445.2013.4). The authors report no other conflicts of interest in this work.
References
Kesserling J, Comi G, Thompson AJ, editors. Multiple Sclerosis: Recovery of Function and Neurorehabilitation. Cambridge University Press, New York; 2010. | |
Filippi M, Rocca MA. Cortical reorganisation in patients with MS. J Neurol Neurosurg Psychiatry. 2004;75(8):1087–1089. | |
Filippi M, Rocca MA. Functional MR imaging in multiple sclerosis. Neuroimag Clin N Am. 2009;19(1):59–70. | |
Rocca MA, Colombo B, Falini A, et al. Cortical adaptation in patients with MS: a cross-sectional functional MRI study of disease phenotypes. Lancet Neurol. 2005;4(10):618–626. | |
Kulikova SN, Peresedova AV, Krotenkova MV, Bryukhov VV, Trifonova OV, Zavalishin IA. Dynamic study of reorganisation of the cortex and the structure of the conduction pathways in patients with relapsing-remitting multiple sclerosis and hand paralysis. Annaly klinicheskoi i eksperimentalnoi nevrologii. 2014;8(1):22–29. | |
Donaldson DI, Buckner RL. Effective paradigm design. In: Jezzard P, Matthews PM, Smith SM, editors. Functional MRI, an Introduction to Methods. Oxford: Oxford University Press; 2003:177–197. | |
Mancini L, Ciccarelli O, Manfredonia F, et al. Short-term adaptation to a simple motor task: a physiological process preserved in multiple sclerosis. Neuroimage. 2009;45(2):500–511. | |
Freedman MS, Selchen D, Arnold DL, et al. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. 2013;40(3):307–323. | |
Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914. | |
O’Connor P, Filippi M, Arnason B, et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009;8(10):889–897. | |
Filippi M, Rocca MA, Camesasca F, et al. Interferon β-1b and glatiramer acetate effects on permanent black hole evolution. Neurology. 2011;76(14):1222–1228. | |
Arnon R, Aharoni R. Neuroprotection and neurogeneration in MS and its animal model EAE effected by glatiramer acetate. J Neural Transm. 2009;116(11):1443–1449. | |
Arnold DL, Narayanan S, Antel S. Neuroprotection with glatiramer acetate: evidence from the PreCISe trial. J Neurol. 2013;260(7):1901–1906. | |
Kieseier BC, Hartung HP. Interferon-beta and neuroprotection in multiple sclerosis-facts, hopes and phantasies. Exp Neurol. 2007;203(1):1–4. | |
Leray E, Yaouang J, Le Page E, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133(Pt7):1900–1913. | |
Gabibov AG, Belogurov AA Jr, Lomakin YA, et al. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J. 2011;25(12):4211–4221. doi: 10.1096/fj.11-190769. Epub August 22, 2011. | |
Owens GP, Bennett JL. Trigger, pathogen, or bystander: the complex nexus linking Epstein-Barr virus and multiple sclerosis. Mult Scler. 2012;18(9):1204–1208. | |
Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014;275(4):350–363. doi: 10.1111/joim.12203. Epub March 11, 2014. | |
Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev. 1993;6(4):382–427. | |
Levin LI, Munger KL, O’Reilly EJ, et al. Primary infection with the Epstein-Barr virus and the risk of multiple sclerosis. Ann Neurol. 2010;67:824–830.33. | |
Pender MP. Preventing and curing multiple sclerosis by controlling Epstein-Barr virus infection. Autoimmun Rev. 2009;8:563–568. | |
Reindl MP, Kuenz BB, Berger T. B cells and antibodies in MS results. Probl Cell Differ. 2010;51:99–113. | |
Hikada M, Zouali M. Multistoried roles for B lymphocytes in autoimmunity. Nat Immunol. 2010;11:1065–1068. | |
Kabat EA, Glusman M, Knaub V. Quantitative estimation of the albumin and gamma globulin in normal and pathologic cerebrospinal fluid by immunochemical methods. Am J Med. 4:653–662. | |
Chamczuk AJ, Ursell M, O’Connor P, Jackowski G, Moscarello MA. A rapid ELISA-based serum assay for myelin basic protein in multiple sclerosis. J Immunol Methods. 2002;262:21–27. | |
Ponomarenko NA, Durova OM, Vorobiev II, et al. Catalytic activity of autoantibodies toward myelin basic protein correlates with the scores on the multiple sclerosis expanded disability status scale. Immunol Lett. 2006;103(1):45–50. doi:10.1016/j.imlet.2005.10.006. | |
Berger T, Rubner P, Schautzer F, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2003;349(2):139–145. doi:10.1056/NEJMoa022328. | |
Lomakin YA, Zakharova MY, Stepanov AV, et al. Heavy-light chain interrelations of MS-associated immunoglobulins probed by deep sequencing and rational variation. Mol Immunol. 2014;62(2):305–314. doi:10.1016/j.molimm.2014.01.013. | |
Gabibov AG, Ponomarenko NA, Tretyak EB, Paltsev MA, Suchkov SV. Catalytic autoantibodies in clinical autoimmunity and modern medicine. Autoimmun Rev. 2006;5(5):324–330. Epub February 28, 2006. | |
Durova OM, Vorobiev II, Smirnov IV, et al. Strategies for induction of catalytic antibodies toward HIV-1 glycoprotein gp120 in autoimmune prone mice. Mol Immunol. 2009;47(1):87–95. doi:10.1016/j. | |
Mahendra A, Sharma M, Rao DN, et al. Antibody-mediated catalysis: induction and therapeutic relevance. Autoimmun Rev. 2013;12(6):648–652. doi:10.1016/j.autrev.2012.10.009. | |
Doronin VB, Parkhomenko TA, Castellazzi M, et al. Comparison of antibodies hydrolyzing myelin basic protein from the cerebrospinal fluid and serum of patients with multiple sclerosis. PLoS One. 2014;9(9):e107807. | |
Planque SA, Nishiyama Y, Hara M, et al. Physiological IgM class catalytic antibodies selective for transthyretin amyloid. J Biol Chem. 20149;289(19):13243–13258. doi: 10.1074/jbc.M114.557231. Epub March 19, 2014. | |
Belogurov A Jr, Kozyr A, Ponomarenko N, Gabibov A. Catalytic antibodies: balancing between Dr Jekyll and Mr Hyde. Bioessays. 2009;31(11):1161–1171. | |
Ponomarenko NA, Durova OM, Vorobiev II, et al. Catalytic antibodies in clinical and experimental pathology: human and mouse models. J Immunol Methods. 2002;269(1–2):197–211. | |
Ponomarenko NA, Durova OM, Vorobiev II, et al. Autoantibodies to myelin basic protein catalyze site-specific degradation of their antigen. Proc Natl Acad Sci U S A. 2006;103(2):281–286. Epub December 30, 2005. | |
Belogurov AA Jr, Kurkova IN, Friboulet A, et al. Recognition and degradation of myelin basic protein peptides by serum autoantibodies: novel biomarker for multiple sclerosis. J Immunol. 2008;180(2):1258–1267. | |
Nastasijevic B, Wright BR, Smestad J, Warrington AE, Rodriguez M, Maher LJ 3rd. Remyelination induced by a DNA aptamer in a mouse model of multiple sclerosis. PLoS One. 2012;7(6):e39595. doi:10.1371/journal.pone.0039595. | |
Warren KG, Catz I, Ferenczi LZ, Krantz MJ. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II- defined cohort of patients with progressive multiple sclerosis: results of a 24-month double- blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur J Neurol. 2006;13(8):887–895. | |
Fontoura P, Garren H. Multiple sclerosis therapies: molecular mechanisms and future Results Probl Cell Differ. 2010;51:259–285. | |
Belogurov AA Jr, Zargarova TA, Turobov VI, et al. Suppression of ongoing experimental allergic encephalomyelitis in DA rats by novel peptide drug, structural part of human myelin basic protein 46–62. Autoimmunity. 2009;42(4):362–364. | |
Belogurov AA Jr, Stepanov AV, Smirnov IV, et al. Liposome-encapsulated peptides protect against experimental allergic encephalitis. FASEB J. 2013;27(1):222–231. doi:10.1096/fj.12-213975. Epub October 9, 2012. | |
Basler M, Mundt S, Muchamuel T, et al. Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol Med. 2014;6(2):226–238. doi:10.1002/emmm.201303543. | |
Wehenkel M, Ban J-O, Ho Y-K, Carmony KC, Hong JT, Kim KB. A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC-3 cells and suppresses tumour growth in nude mice. Br J Cancer. 2012;107(1):53–62. doi:10.1038/bjc.2012.243. | |
Belogurov A Jr, Kuzina E, Kudriaeva A, et al. Ubiquitin-independent proteosomal degradation of myelin basic protein contributes to development of neurodegenerative autoimmunity. FASEB J. 2015;29(5):1901–1913. | |
Kravtsova-Ivantsiv Y, Ciechanover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci. 2012;125(3):539–548. doi:10.1242/jcs.093567. | |
Belogurov A Jr, Kudriaeva A, Kuzina E, et al. Multiple sclerosis autoantigen myelin basic protein escapes control by ubiquitination during proteasomal degradation. J Biol Chem. 2014;289(25):17758–17766. doi: 10.1074/jbc.M113.544247. Epub April 16, 2014. | |
Kuzina E, Kudriaeva A, Smirnov I, Dubina MV, Gabibov A, Belogurov A Jr. Glatiramer acetate and nanny proteins restrict access of the multiple sclerosis autoantigen myelin basic protein to the 26s proteasome. Biomed Res Int. 2014;2014:926394. doi:10.1155/2014/926394. Epub September 8, 2014. | |
Hohlfeld R, Wekerle H. Autoimmune concepts of multiple sclerosis as a basis for selective immunotherapy: from pipe dreams to (therapeutic) pipelines. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14599–14606. doi:10.1073/pnas.0404874101. | |
Denic A, Johnson AJ, Bieber AJ, Warrington AE, Rodriguez M, Pirko I. The relevance of animal models in multiple sclerosis research. Pathophysiology. 2011;18(1):21–29. | |
Glinka EM, Edelweiss EF, Sapozhnikov AM, et al. A new vector for controllable expression of an anti-HER2/neu mini-antibody-barnase fusion protein in HEK 293T cells. Gene. 2006;366(1):97–103. | |
Deyev SM, Lebedenko EN. Modern technologies for creating synthetic antibodies for clinical application. Acta Naturae. 2009;1(1):32–50. | |
Balandin TG, Edelweiss E, Andronova NV, et al. Antitumor activity and toxicity of anti-HER2 immunoRNase scFv 4D5-dibarnase in mice bearing human breast cancer xenografts Invest New Drugs. 2011;29(1):22–32. | |
Stepanov AV, Belogurov AA Jr, Ponomarenko NA, et al. Design of targeted B cell killing agents. PLoS One. 2011;6(6):e20991. doi: 10.1371/journal.pone.0020991. Epub June 6, 2011. | |
Aleksandar D, Bharath W, Laurie Z, Moses R. Deletion of virus-specific T-cells enhances remyelination in a model of multiple sclerosis. J Neurol Transl Neurosci. 2014;2(1):1032. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.