Back to Journals » Drug Design, Development and Therapy » Volume 12
Clickable prodrugs bearing potent and hydrolytically cleavable nicotinamide phosphoribosyltransferase inhibitors
Authors Sadrerafi K ![]() , Mason EO
, Mason EO ![]() , Lee Jr MW
, Lee Jr MW ![]()
Received 27 September 2017
Accepted for publication 31 January 2018
Published 24 April 2018 Volume 2018:12 Pages 987—995
DOI https://doi.org/10.2147/DDDT.S152685
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Keivan Sadrerafi, Emilia O Mason, Mark W Lee Jr
Department of Chemistry, University of Missouri, Columbia, MO, USA
Purpose: Our previous study indicated that carborane containing small-molecule 1-(hydroxymethyl)-7-(4′-(trans-3″-(3‴-pyridyl)acrylamido)butyl)-1,7-dicarbadodecaborane (hm-MC4-PPEA), was a potent inhibitor of nicotinamide phosphoribosyltransferase (Nampt). Nampt has been shown to be upregulated in most cancers and is a promising target for the treatment of many different types of cancers, including breast cancers.
Patients and methods: To increase the selectivity of hm-MC4-PPEA toward cancer cells, three prodrugs were synthesized with different hydrolyzable linkers: ester, carbonate, and carbamate. Using click chemistry a fluorophore was attached to these prodrugs to act as a model for our conjugation strategy and to serve as an aid for prodrug stability studies. The stabilities of these drug conjugates were tested in phosphate-buffered saline (PBS) at normothermia (37°C) using three different pH levels, 5.5, 7.5, and 9.5, as well as in horse serum at physiological pH. The stability of each was monitored using reversed-phase HPLC equipped with both diode array and fluorescence detection. The inhibitory activity of hm-MC4-PPEA was also measured using a commercially available colorimetric assay. The biological activities of the drug conjugates as well as those of the free drug (hm-MC4-PPEA), were evaluated using the MTT assay against the human breast cancer cell lines T47D and MCF7, as well as the noncancerous, transformed, Nampt-dependent human breast epithelium cell line 184A1.
Results: hm-MC4-PPEA showed to be a potent inhibitor of recombinant Nampt activity, exhibiting an IC50 concentration of 6.8 nM. The prodrugs showed great stability towards hydrolytic degradation under neutral, mildly acidic and mildly basic conditions. The carbamate prodrug also showed to be stable in rat serum. However, the carbonate and the ester prodrug release at various rates in serum presumably owing to the presence of several different classes of esterase. The biological activities of the drug conjugates correlate with the stability of their cleavable linkers observed in serum.
Conclusion: The targeted and selective delivery of potent Nampt inhibitors to cancer cells is a potentially new route for the treatment of many cancers. These prodrugs linked to small cancer-associated peptides may be optimum for their use as targetable Nampt inhibitors.
Keywords: carboranes, Nampt, cancer, prodrugs, cleavable linker
Plain language summary
Why was the study done? To develop a platform for targeted delivery of 1-(hydroxymethyl)-7-(4′-(trans-3″-(3‴-pyridyl)acrylamido)butyl)-1,7-dicarbadodecaborane (hm-MC4-PPEA), a potent Nampt inhibitor.
What did the researchers do and find? Three prodrugs were developed, and their stability was tested under various conditions.
What do these results mean? By incorporating a cleavable linker bearing an azide group, these prodrugs may be attached to a variety of delivery vehicles to achieve targeted delivery.
Introduction
Globally, there exists a tremendous unmet need for more efficacious and affordable treatments for nearly every type of cancer. The need for new treatments is even greater for advanced and/or recurrent cancers. A limitation of all small-molecule anticancer agents is toxicity toward healthy cells. The development of such agents is necessarily a compromise between maximizing a drug’s biological activity with its absorption, distribution, metabolism, and excretion. To achieve a clinically meaningful therapeutic effect, most anticancer agents must be administered at, or near, their maximum tolerated dose (MTD). Unfortunately, the drugs clinically used to treat cancer patients today lack the efficacy required to achieve complete disease remission in many patients. Improvements to the therapeutic indices of anticancer agents will come from either 1) increased agent’s potency, which will tend to decrease the minimum effective dose required or 2) an increase in agent selectivity for cancer, which should improve MTD. Ideally, both the potency and selectivity of an agent could be improved.
One possible route toward such improvements is achieved through the conjugation of an anticancer or cytotoxic agent to a tumor-targeting vector, such as a peptide or antibody, producing peptide–drug conjugates (PDCs), and antibody–drug conjugates (ADCs).1–5 The number of molecules of a cytotoxic agent that is necessary to kill a cancer cell must be ideally much less than the maximum number able to be delivered by the targeting vector. This limitation has led to the focus of current research efforts on the use of exceptionally potent small molecules, such as the microtubule agents auristatin and maytansine for the syntheses of new bioconjugates.6–8 However, the cytotoxicity of these molecules is not limited to cancer cells and their delivery to healthy cells must be minimized.
A new and promising target for the treatment of cancer is nicotinamide phosphoribosyltransferase (Nampt; aka visfatin, pre-B-cell colony enhancing factor 1 [PBEF1]). Nampt is the first and rate-limiting enzyme in the mammalian nicotinamide adenine dinucleotide (NAD)+ recycling pathway, catalyzing the conversion of nicotinamide-to-nicotinamide mononucleotide. Over the past several years, it has been revealed that Nampt activity is highly implicated with many diseases, including cancer.9–11 Overexpression of Nampt increases SIRT1 activity.12 SIRT1 and PARP, two NAD+-dependent enzymes, obtain NAD+ primarily from the Nampt-controlled recycling pathway.12 Nampt has been shown to be upregulated in many/most cancers,13–20 and this overexpression is highest in aggressive and refractory cancers.9,21 Nampt overexpression is predictive of poor response to the treatment of various cancers,9,21 including breast cancer.22 Most healthy cells do not depend heavily on the NAD+ from the Nampt-catalyzed recycle pathway.23,24 Given sufficient exposure time, Nampt inhibitors are exceptionally potent when compared with the small-molecule anticancer agents used clinically today.25,26 It must be noted that high levels of Nampt inhibition lead to thrombocytopenia, retinal, and hematopoietic toxicities.27,28 However, the toxic effects may be minimized by lead optimization of the inhibitor and its targeted delivery system.
A small molecule discovered through compound screening designated FK866/APO866 was the first known potent inhibitor of Nampt and has been investigated in a number of Phase I/II clinical trials against several cancers.25 For this class of inhibitors, cell death is preceded by a marked decrease in the concentration of cellular adenine triphosphate following 1–2 days of treatment; cells then remain viable for an additional 2–3 days before sharply declining in numbers.25 While treatment of patients with the drug was determined to be well tolerated and safe at the doses used (Phase II recommended dose: continuous intravenous infusion 0.126 mg/m2/h for 4 consecutive days), the dose-limiting toxicity (thrombocytopenia) prevented the delivery of an efficacious dose.29
Our research group has recently reported a new family of carborane-containing Nampt inhibitors, which exhibit up to 10-fold higher activities than FK866 in several cancer cell lines in vitro and a ~100-fold greater inhibition of Nampt.26 The most potent of these molecules exhibit sub-nanomolar half-maximal inhibitory concentrations (IC50) in cancer cells, in vitro. Recently, these molecules were tested through the National Cancer Institute’s Developmental Therapy Program (DTP) NCI-60 and our molecule 1-(4′-(trans-3″-(3‴-pyridyl)acrylamido)butyl)-1,7-dicarbadodecaborane (MC4-PPEA) was the most potent Nampt inhibitor tested, to date, exhibiting low growth inhibition of 50% of cells (GI50) concentrations in all 60 of the standardized cancer cell lines, representing many different types of cancer. In addition, the sub-nanomolar potency of our agent MC4-PPEA was observed in vivo using small animal experiments.30
We recently reported 1-(hydroxymethyl)-7-(4′-(trans-3″-(3‴-pyridyl)acrylamido)butyl)-1,7-dicarbadodecaborane (hm-MC4-PPEA), a new derivative of MC4-PPEA.31 The structure of these inhibitors is depicted in Figure 1. In vitro assays against three breast cancer cell lines revealed that the presence of the hydroxymethyl group does not greatly diminish the activity of this new molecule and the group provides a convenient linkage for covalent attachment through carbonates, carbamates, esters, etc.
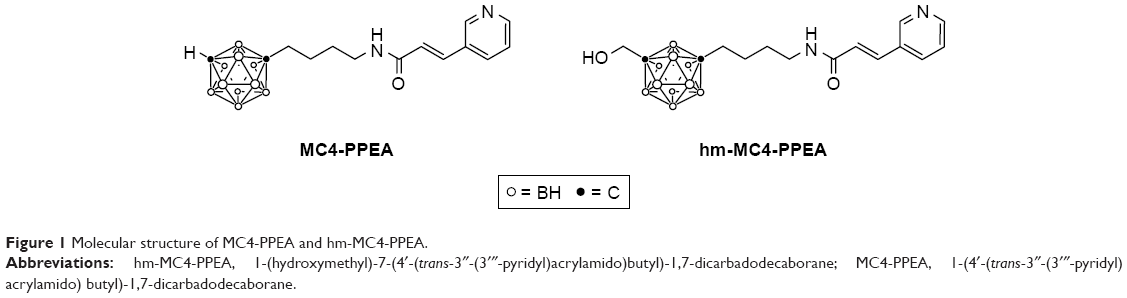 | Figure 1 Molecular structure of MC4-PPEA and hm-MC4-PPEA. |
In the present study, we focus on tailoring a hydrolyzable linker, covalently attached to our therapeutic molecule for possible conjugation with various targeting vectors. Herein, the synthesis of three prodrugs of hm-MC4-PPEA – 5, 7, and 11 each bearing a terminal azide moiety for the attachment to vectors through the facile click-chemistry cycloaddition reaction – is described.32 This scheme promises drug conjugation using mild conditions, while protecting the integrity of both the targeting vector and the cleavable linker. Stability of these prodrugs was tested under various conditions, and the data obtained will be used to prepare future PDCs and ADCs for targeted delivery. A simple fluorophore (14) bearing a terminal alkyne is utilized to click onto our target compounds and monitor the stability of these prodrugs under various conditions. To the best of our knowledge, these new molecules are the first reported conjugatable prodrugs of a potent Nampt inhibitor.
Materials and methods
Materials
All manipulations were performed under nitrogen atmosphere unless stated otherwise. All anhydrous solvents, n-butyllithium (n-BuLi; 2.5 M solution in hexane), paraformaldehyde (PFA), 1-chloro-4-iodobutane (Cl(CH2)4I), HCl (37%), sodium azide (NaN3), sodium iodide (NaI), potassium iodide (KI), triphenylphosphine (PPh3), sodium sulfate (Na2SO4), magnesium sulfate (MgSO4), sodium thiosulfate (Na2S2O3), trans-3-(3′-pyridyl)acrylic acid (C5NH4(CH)2COOH), thionyl chloride (SOCl2), Hünig’s base or N, N-diisopropylethylamine (DIPEA), pyridine (C5H5N), chloromethyl chloroformate (ClCH2CO2Cl), diethylene glycol, tosyl chloride (TsCl), silver oxide (Ag2O), propargyl bromide (CHCCH2Br), chromium trioxide (CrO3), and dansyl chloride were purchased from Aldrich and used as received. All solvents were dried using 3 Å molecular sieves (20%, m/v).33 Silica gel was purchased from Sorbent Technologies (60 Å, 63–200 μm).
Instrumentation
Nuclear magnetic resonance spectra were recorded on Bruker DRX300, DRX500, Bruker Ultrashield 400, and Ultrashield 500 Plus Nuclear Magnetic Spectrometers and analyzed using TopSpin. Chemical shifts (δ, ppm) for 1H and 13C were referenced to residual solvent peaks. Mass spectra were obtained on an ABI Q-Star under both positive and negative modes, reported in m/z, and analyzed using the Mariner Biospectrometry Workstation by PerSeptive Biosystems. HPCORE ChemStation was used for data analysis. Triply distilled water was used as the eluent with 5%–95% gradient of high-performance liquid chromatography (HPLC) grade acetonitrile.
Prodrug stability studies
Samples for stability tests were prepared by dissolving each analyte in a 50:50 solution of acetonitrile and the appropriate buffer. For serum stability experiments, a 5 μL solution of each prodrug dissolved in acetonitrile was added to 200 μL of horse serum and sonicated briefly in a water bath. At time intervals, which were dependent on the apparent stability of each prodrug under the test conditions, samples were injected into an Agilent 1100 Series HPLC System equipped with both a UV–visible detector and a fluorescence detector. Agilent ChemStation was used for both data collection and analysis. Triply distilled water was used as the eluent with a 5%–95% gradient of HPLC grade acetonitrile. The retention time and the percent area of each peak were recorded. The retention time of the free drug was measured using a sample containing pure compound (hm-MC4-PPEA). This sample produced a single peak on the UV chromatogram, while each prodrug was observed as single peak on both the UV and fluorescence chromatograms. The cleavage of each prodrug resulted in the appearance of two peaks, one of these corresponding with free drug (hm-MC4-PPEA) and the other associated with the liberated fluorescent linker.
Cell cultures and treatment
All cell lines were obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). An MTT assay kit was purchased from Promega Corporation (Fitchburg, WI, USA). FK866 was purchased from Enzo Life Sciences (Farmingdale, NY, USA). T47D and MCF-7 cells were maintained in DMEM containing 350 μL/L gentamicin and 25 mL/L fetal bovine serum. 184A1 cells were maintained in HuMEC media containing 175 μL/L gentamicin, HuMEC supplement, and bovine pituitary extract. The cells were grown to confluency in either T-12.5 or T-25 cm2 tissue culture flasks using an incubator maintained at 5% CO2 humid atmosphere at 37°C. The MTT assay34,35 was carried out as follows: T47D, MCF-7, and 184A1 cells were plated in 96-well plates at a density of 10,000 cells/well for overnight at 37°C. The cells were then treated with different concentrations of the test agent and incubated for 72 h. The MTT reagent was added to the cells for 4 h for developing formazan crystals. Following this, solubilization buffer was added to the wells and the absorbance was measured at 570 nm. Each experiment was repeated four times, and the standard deviations are reported.
Results and discussion
Hydrolyzable linkers
Three different hydrolyzable groups were investigated to attach hm-MC4-PPEA with an azido-linker, such as an ester, carbonate, and carbamate. These moieties have been previously utilized for the preparation of drug conjugates.36 They range in stability and are known to hydrolyze under varying conditions, including those found in the acidic environments of many solid tumors.36
To prepare the carbamate prodrug 5, 4-nitrophenyl chloroformate was first reacted with the hydroxyl group of hm-MC4-PPEA, forming the amine reactive intermediate 4 (Scheme 1).
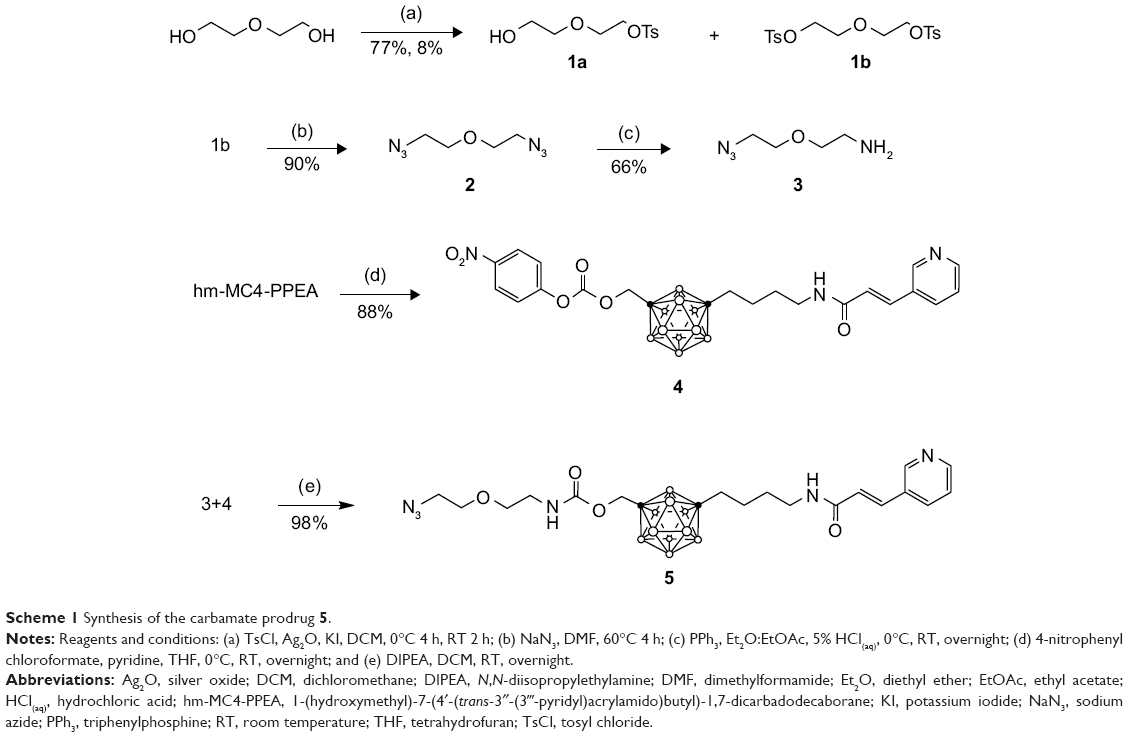 | Scheme 1 Synthesis of the carbamate prodrug 5. |
The carbonate prodrug 7 was prepared in two steps by first reacting hm-MC4-PPEA with 2-chloroethyl chloroformate to form the intermediate 6, followed by the displacement of the chloro group with an azide in a SN2 type reaction (Scheme 2). In a similar fashion, chloroformate derivatized polyethylene glycol linkers of varying lengths may be synthesized using phosgene and PEGs bearing a single terminal halide.
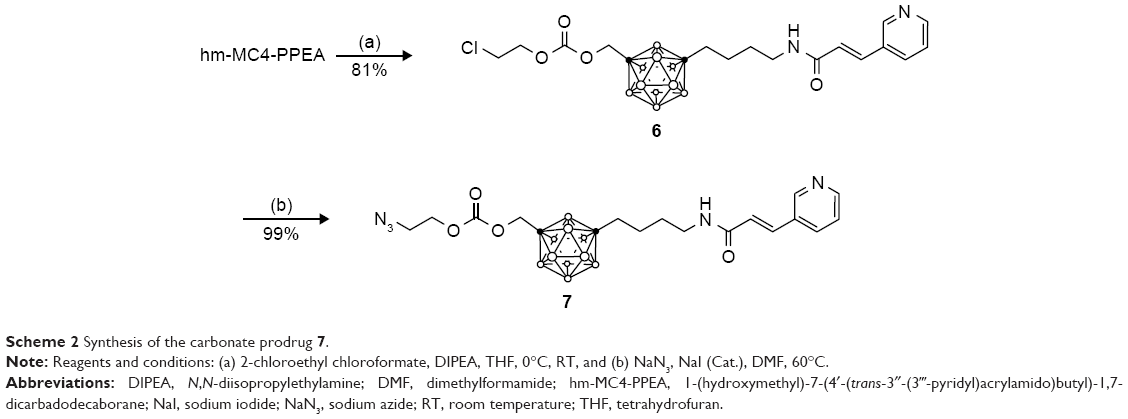 | Scheme 2 Synthesis of the carbonate prodrug 7. |
The ester prodrug 11 was prepared by coupling an acid chloride derivatized linker with 10. Azido-diethylene glycol 8 was first oxidized using Jones reagent. Postoxidation, the newly formed carboxylic acid 9 was treated with thionyl chloride to make the acid chloride 10. This intermediate, in turn, was reacted with the hydroxyl group present on hm-MC4-PPEA to form 11 in high yield (Scheme 3). To increase the solubility of the prodrug, different length of PEG linkers may be utilized in place of diethylene glycol.
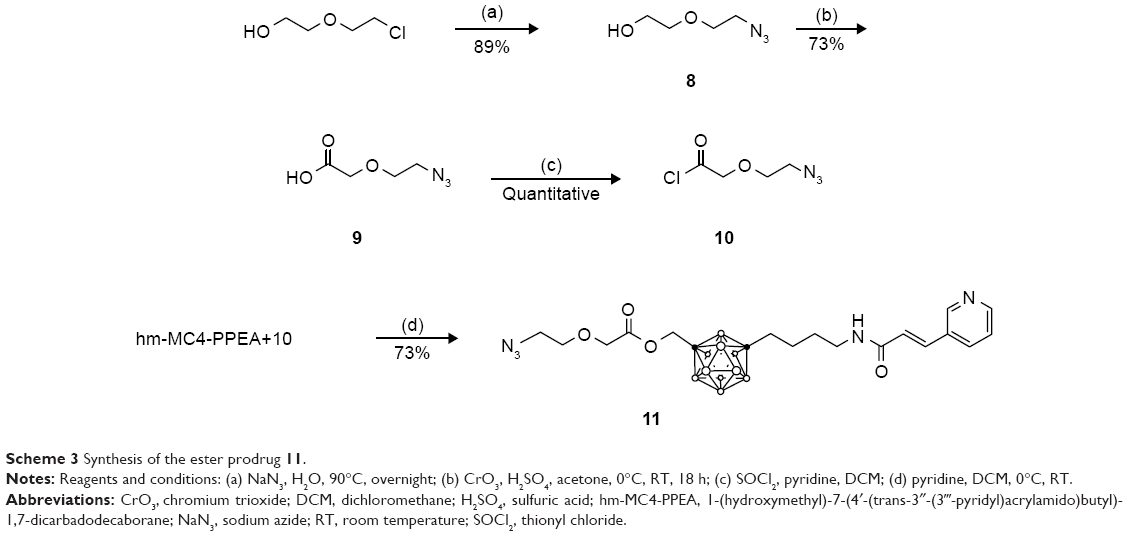 | Scheme 3 Synthesis of the ester prodrug 11. |
Clickable fluorophore
To act as a model for our conjugation strategy using click chemistry and to serve as an aid for prodrug stability studies, a fluorophore bearing an alkyne was synthesized. Diethylene glycol was modified with an amine at one terminal end and an alkyne at the other end. The amine was then reacted with dansyl chloride to afford the stable sulfonamide bond of compound 14 (Scheme 4).
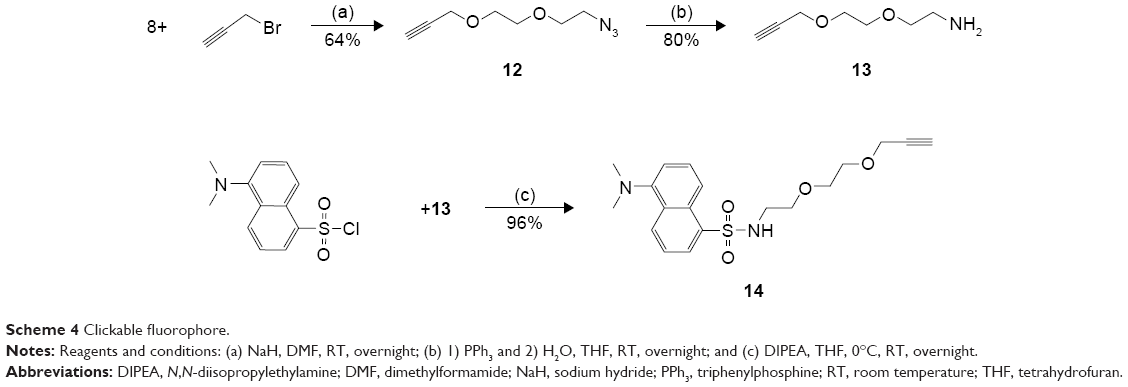 | Scheme 4 Clickable fluorophore. |
The azide containing prodrugs 5, 7, and 11 were covalently bound to the alkyne containing fluorophore 14 through click type chemistry in high yields. Anhydrous dimethyl sulfoxide was degassed by bubbling nitrogen gas through the solvent to minimize the presence of molecular oxygen and subsequent oxidation of the metal catalyst. Copper(I) iodide was used as the catalyst, and N, N, N′, N′, N″-pentamethyldiethylenetriamine was used as a tridentate chelating ligand to stabilize copper(I) and prevent its oxidation.32 DIPEA was used as a base, and the reaction mixture was sonicated in a water bath for 1 h at room temperature. The drug conjugates 15–17 (Scheme 5) were synthesized using the same conditions in high yield (75%, 99%, and 60%, respectively). Proton and carbon NMR spectra of compounds 15–17 can be found in the Supplementary materials. It is noteworthy that these reactions were carefully optimized to achieve high yields in just under an hour.
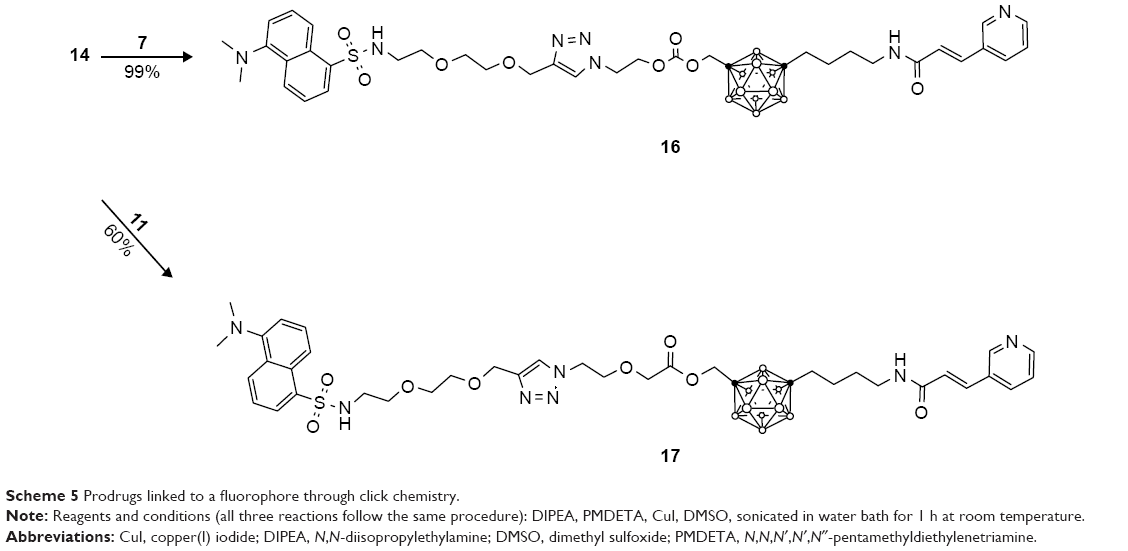 | Scheme 5 Prodrugs linked to a fluorophore through click chemistry. |
Linker stability test
The stabilities of drug conjugates (15–17) were tested in phosphate-buffered saline (PBS) at normothermia (37°C) using three different pH levels, 5.5, 7.5, and 9.5, as well as in horse serum at physiological pH. The stability of each was monitored using reversed-phase HPLC equipped with both diode array and fluorescence detection, allowing a quantitative measurement of the disappearance of each conjugate, as well as the emergence of the hydrolytic degradation products, including the appearance of a peak corresponding with that of the free drug (hm-MC4-PPEA). Graphs depicting the stability of 15–17 over several days for each test condition are shown in Figures S1–S3.
A marked difference was observed between the stability of the prodrug linkers incubated in PBS of varying acidity and those measured in serum. While the carbamate linkage was stable under all conditions tested, including in serum, the ester and carbonates were far more labile in serum than in PBS. Human blood serum is known to contain several classes of esterase enzymes, including albumin, butyrylcholinesterase, and paraoxonase.37 The horse serum that was utilized contained cholinesterase that resembles those found in human blood serum and is responsible for the hydrolysis of many aliphatic esters.38 The liability of the ester and carbamate moieties is presumably owing to the presence of these classes of esterases.
The quantity of prodrug remaining in each test sample was measured over a period of several hours by integrating the area under the curve for the analyte peak in the fluorescence chromatogram. The half-lives (t1/2) were calculated by fitting the data to an equation and extrapolating the time for which 50% of the prodrug remained. The results of the stability test for prodrug–fluorophore conjugates (15–17) are summarized in Table 1. As stated, the carbamate prodrug (15) was observed to be stable under all test conditions. The half-lives of 16 in pH buffers 5.5, 7.4, and 9.5 were >600 (extrapolated), 18.9, and 1.7 days, respectively, while the half-life of 16 in serum was 4.6 hours. The ester prodrug (17) was observed to be the least stable, exhibiting half-lives of 200 (extrapolated), 5.4, and 1.5 days, respectively, at pH 5.5, 7.4, and 9.5. In serum, the half-life of 17 was 1.0 hours.
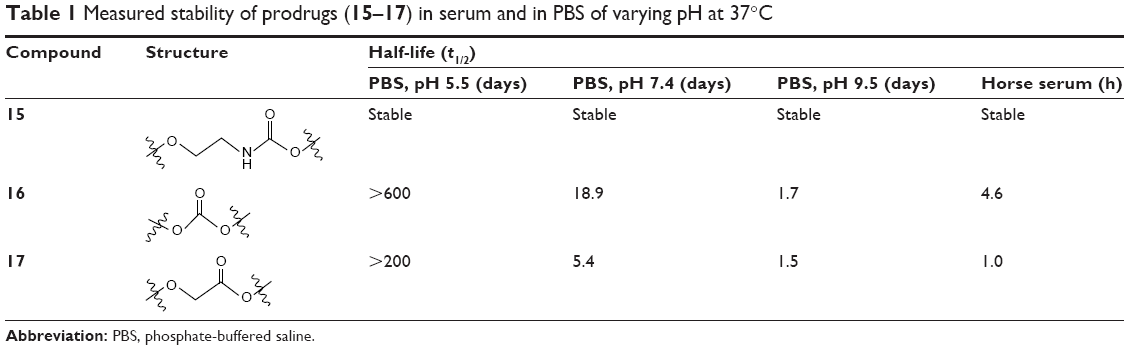 | Table 1 Measured stability of prodrugs (15–17) in serum and in PBS of varying pH at 37°C |
Recombinant Nampt inhibition assay, MTT assay, and IC50 measurements
Previously, we reported that the unsubstituted parent molecule MC4-PPEA inhibited Nampt, (IC50 =4.2 nM) demonstrating a ~100-fold higher inhibitory activity than FK866.26 We measured the inhibitory activity of hm-MC4-PPEA using a commercially available colorimetric assay and found that this molecule is also a potent inhibitor of recombinant Nampt activity, exhibiting an IC50 concentration of 6.8 nM. Protocol for the recombinant Nampt inhibition assay as well as a graph illustrating percentage inhibition of control vs inhibitor concentration (Figure S4) is found in the Supplementary materials.
The biological activities of 15–17, as well as those of the free drug (hm-MC4-PPEA), were evaluated using the MTT assay against the human breast cancer cell lines T47D and MCF7, as well as the noncancerous, transformed, Nampt-dependent human breast epithelium cell line 184A1 (Figures S5–S7).34 The concentration-dependent cell viability exhibited by 15–17 against these cell lines is summarized in Table 2.
 | Table 2 Measure of IC50 for agents hm-MC4-PPEA and 15–17 against the human breast cancer cell lines T47D, MCF7, and 184A1 (noncancerous) |
Compound 15 exhibited a cell-line-dependent IC50 of 32.0±0.4, >200, and 9.1±0.9 nM for the T47D, MCF7, and 184A1 cell lines, respectively. Compound 16 exhibited an IC50 of 2.9±0.3, 81.0±2.5, and 4.7±1.0 nM for T47D, MCF7, and 184A1 cell lines, respectively, while compound 17 exhibited an IC50 of 5.5±0.5, 34.0±1.3, and 1.8±0.5 nM for these same cell lines. For each prodrug, the biological activities correlate with the stabilities observed in serum. The carbamate prodrug (15), which was observed to be stable under all conditions tested, exhibited the lowest biological activity of the three, with an ~5-fold decrease in activity below the parent compound (hm-MC4-PPEA). The carbonate prodrug (16), which exhibited a half-life of 4.6 h in serum, possessed higher activity than the carbamate prodrug (15) in each of the cell lines, presumably due to its cleavage and the resultant release of (hm-MC4-PPEA) by esterases, which are present in the fetal bovine serum containing cell culture assay medium. The ester prodrug (17) exhibited the highest cell inhibitory activity and was nearly equal to that of the free drug (hm-MC4-PPEA) in each cell line (IC50 4.9±0.3, 34.0±1.5, and 1.9±0.4 nM for T47D, MCF7, and 184A1 cell lines, respectively). These results are consistent with the observed 1.0 h half-life of 17 in serum; during the cell viability assay, essentially all the prodrug would be cleaved, releasing an equal molar amount of hm-MC4-PPEA.
Conclusion
The targeted and selective delivery of potent Nampt inhibitors to cancer cells is a potentially new route for the treatment of many different cancers, including breast cancer. The three new prodrugs of our potent Nampt inhibitor (hm-MC4-PPEA) described herein, each bearing an azide moiety, may be readily conjugated with alkyne-bearing linkers for the preparation of future ADCs and/or PDCs using facile click chemistry and in excellent yields. In principle, different cancer types may be targeted through conjugation using antibodies or peptides associated with a specific cancer. To our knowledge, these are the first conjugatable Nampt inhibitor prodrugs.
The ester, carbamate, and carbonate covalent linkers investigated herein are quite stable toward hydrolytic degradation under neutral, mildly acidic, and mildly basic conditions. However, the ester and carbonate containing derivatives are readily cleaved in serum, presumably owing to the presence of several different classes of esterase. Experiments are now underway to prepare PDCs using these three prodrugs linked to small cancer-associated peptides and to test their stability using different classes of serum esterases.
Acknowledgments
The authors are grateful to Dr Raghuraman Kannan and Dr Ajit Prakash Zambre for their assistance with cell studies. This article was presented at the 49th Midwest Regional Meeting of the American Chemical Society as a poster presentation with interim findings. The poster is available online at researchgate.net: https://www.researchgate.net/publication/283462753_Development_of_a_Nampt_Inhibitor_Prodrug_and_the_Stability_of_its_Cleavable_Linkers.
Disclosure
The authors report no conflicts of interest in this work.
References
Chari RVJ, Miller ML, Widdison WC. Antibody–drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed. 2014;53(15):3796–3827. | ||
Sievers EL, Senter PD. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64(1):15–29. | ||
Alley SC, Okeley NM, Senter PD. Antibody–drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol. 2010;14(4):529–537. | ||
Casi G, Neri D. Antibody–drug conjugates: basic concepts, examples and future perspectives. J Controll Release. 2012;161(2):422–428. | ||
Hughes B. Antibody–drug conjugates for cancer: poised to deliver? Nat Rev Drug Discov. 2010;9(9):665–667. | ||
Bouchard H, Viskov C, Garcia-Echeverria C. Antibody–drug conjugates – A new wave of cancer drugs. Bioorg Med Chem Lett. 2014;24(23):5357–5363. | ||
Perez HL, Cardarelli PM, Deshpande S, et al. Antibody–drug conjugates: current status and future directions. Drug Discov Today. 2014;19(7):869–881. | ||
Trail P. Antibody drug conjugates as cancer therapeutics. Antibodies. 2013;2(1):113. | ||
Khan JA, Forouhar F, Tao X, Tong L. Nicotinamide adenine dinucleotide metabolism as an attractive target for drug discovery. Expert Opin Ther Targets. 2007;11(5):695–705. | ||
Gallí M, Van Gool F, Rongvaux A, Andris F, Leo O. The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res. 2010;70(1):8–11. | ||
Garten A, Petzold S, Korner A, Imai S, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab. 2009;20(3):130–138. | ||
Revollo JR, Grimm AA, Imai S-I. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279(49):50754–50763. | ||
Hufton SE, Moerkerk PT, Brandwijk R, de Bruïne AP, Arends J-W, Hoogenboom HR. A profile of differentially expressed genes in primary colorectal cancer using suppression subtractive hybridization. FEBS Lett. 1999;463(1–2):77–82. | ||
van Beijnum JR, Moerkerk PTM, Gerbers AJ, et al. Target validation for genomics using peptide-specific phage antibodies: a study of five gene products overexpressed in colorectal cancer. Int J Cancer. 2002;101(2):118–127. | ||
Nahimana A, Attinger A, Aubry D, et al. The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood. 2009;113:3276–3286. | ||
Reddy PS, Umesh S, Thota B, et al. PBEF1/NAmPRTase/Visfatin: a potential malignant astrocytoma/glioblastoma serum marker with prognostic value. Cancer Biol Ther. 2008;7(5):663–668. | ||
Nakajima TE, Yamada Y, Hamano T, et al. Adipocytokine levels in gastric cancer patients: resistin and visfatin as biomarkers of gastric cancer. J Gastroenterol. 2009;44(7):685–690. | ||
Nakajima TE, Yamada Y, Hamano T, et al. Adipocytokines as new promising markers of colorectal tumors: adiponectin for colorectal adenoma, and resistin and visfatin for colorectal cancer. Cancer Sci. 2010;101(5):1286–1291. | ||
Yang H, Yang T, Baur JA, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130(6):1095–1107. | ||
Patel ST, Mistry T, Brown JEP, et al. A novel role for the adipokine visfatin/pre-B cell colony-enhancing factor 1 in prostate carcinogenesis. Peptides. 2010;31(1):51–57. | ||
Bi T, Che X. Nampt/PBEF/visfatin and cancer. Cancer Biol Ther. 2010;10(2):119–125. | ||
Lee Y-C, Yang Y-H, Su J-H, Chang H-L, Hou M-F, Yuan S-SF. High Visfatin expression in breast cancer tissue is associated with poor survival. Cancer Epidemiol Biomarkers Prev. 2011;20(9):1892–1901. | ||
Wosikowski K, Mattern K, Schemainda I, Hasmann M, Rattel B, Löser R. WK175, a novel antitumor agent, decreases the intracellular nicotinamide adenine dinucleotide concentration and induces the apoptotic cascade in human leukemia cells. Cancer Res. 2002;62(4):1057–1062. | ||
Muruganandham M, Alfieri AA, Matei C, et al. Metabolic signatures associated with a NAD synthesis inhibitor–induced tumor apoptosis identified by 1H-decoupled-31P magnetic resonance spectroscopy. Clin Cancer Res. 2005;11(9):3503–3513. | ||
Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63(21):7436–7442. | ||
Lee MW Jr, Sevryugina YV, Khan A, Ye SQ. Carboranes increase the potency of small molecule inhibitors of nicotinamide phosphoribosyltranferase. J Med Chem. 2012;55(16):7290–7294. | ||
Zabka TS, Singh J, Dhawan P, et al. Retinal toxicity, in vivo and in vitro, associated with inhibition of nicotinamide phosphoribosyltransferase. Toxicol Sci. 2015;144(1):163–172. | ||
Tarrant JM, Dhawan P, Singh J, et al. Preclinical models of nicotinamide phosphoribosyltransferase inhibitor-mediated hematotoxicity and mitigation by co-treatment with nicotinic acid. Toxicol Mech Methods. 2015;25(3):201–211. | ||
Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Invest New Drugs. 2008;26(1):45–51. | ||
Huang P, Lee MW Jr, Sadrerafi K, et al. MC-PPEA as a new and more potent inhibitor of CLP-induced sepsis and pulmonary inflammation than FK866. Drug Des Devel Ther. 2017;11:629–641. | ||
Sadrerafi K, Zargham EO, Lee MW Jr. Improved synthesis of MC4-PPEA and the biological evaluation of its hydroxymethyl derivative. Bioorg Med Chem Lett. 2016;26(2):618–621. | ||
Meldal M, Tornøe CW. Cu-catalyzed azide-alkyne cycloaddition. Chem Rev. 2008;108(8):2952–3015. | ||
Williams DBG, Lawton M. Drying of organic solvents: quantitative evaluation of the efficiency of several desiccants. J Org Chem. 2010;75(24):8351–8354. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Campling BG, Pym J, Galbraith PR, Cole SP. Use of the MTT assay for rapid determination of chemosensitivity of human leukemic blast cells. Leuk Res. 1988;12(10):823–831. | ||
Binderup E, Bjorkling F, Hjarnaa PV, et al. EB1627: a soluble prodrug of the potent anticancer cyanoguanidine CHS828. Bioorg Med Chem Lett. 2005;15(10):2491–2494. | ||
Li B, Sedlacek M, Manoharan I, et al. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem Pharmacol. 2005;70(11):1673–1684. | ||
Sturge LM, Whittaker VP. The esterases of horse blood. 1. The specificity of horse plasma cholinesterase and ali-esterase. Biochem J. 1950;47(5):518–525. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.