Back to Journals » International Journal of General Medicine » Volume 14
Classifiers for Predicting Coronary Artery Disease Based on Gene Expression Profiles in Peripheral Blood Mononuclear Cells
Authors Liu J, Wang X, Lin J, Li S, Deng G, Wei J
Received 18 July 2021
Accepted for publication 26 August 2021
Published 15 September 2021 Volume 2021:14 Pages 5651—5663
DOI https://doi.org/10.2147/IJGM.S329005
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Jie Liu,1,2,* Xiaodong Wang,1,2,* Junhua Lin,1,* Shaohua Li,1 Guoxiong Deng,1,2 Jinru Wei1,2
1Department of Cardiology, The Fifth Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, 530022, People’s Republic of China; 2Department of Cardiology, The First People’s Hospital of Nanning, Nanning, Guangxi, 530022, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guoxiong Deng
The Fifth Affiliated Hospital of Guangxi Medical University, 89 Qixing Road, Nanning, Guangxi, 530022, People’s Republic of China
Email [email protected]
Jinru Wei
The Fifth Affiliated Hospital of Guangxi Medical University, 89 Qixing Road, Nanning, Guangxi, 530022, People’s Republic of China
Tel +86 77 12636193
Email [email protected]
Objective: Coronary artery disease (CAD) is a serious global health concern. Current diagnostic methods for CAD involve risk to the patient and are costly, so better diagnostic tools are needed. We defined four classifiers based on gene expression profiles in peripheral blood mononuclear cells and determined their potential for CAD detection.
Methods: We downloaded a CAD-related data set (GSE113079) from the Gene Expression Omnibus (GEO) database. We identified differentially expressed genes (DEGs) in peripheral blood mononuclear cells between CAD samples and healthy controls. DEGs were analyzed for functional enrichment. To create a robust CAD classifier, DEGs were identified by feature selection using the principal component analysis. Then, least absolute shrinkage and selection operator (LASSO) logistic regression, random forest, and support vector machine (SVM) models were created. Gene set variation analysis (GSVA) score and gene set enrichment analysis (GSEA) were also conducted. The performance of the models was evaluated in terms of the area under receiver operating characteristic curves (AUC).
Results: In the training set, we found 135 up-regulated genes and 104 down-regulated genes in CAD patients compared with controls. The DEGs were involved in some pathways associated with CAD, such as pathways involving calcium and interleukin-17 signaling. Twenty genes were identified as optimal features and used to generate the logistic classifier based on LASSO. The AUC for the classifier was 1.00 in the training set and 0.997 in the test set. Using the 20 DEGs, SVM and random forest classifiers were also generated and showed high diagnostic efficacy, with respective AUCs of 0.997 and 1.00 against the training set. A GSVA score was also established using the top 20 significant DEGs, which showed an AUC of 0.971 in the training set and 0.989 in the test set. Furthermore, GSEA showed autophagy and the proteasome to be major pathways involving the DEGs.
Conclusion: We identified a set of genes specific for CAD whose expression can be measured non-invasively. Using these genes, we defined four diagnostic classifiers using multiple methods.
Keywords: coronary artery disease, diagnosis, gene expression, classifier
Introduction
Coronary artery disease (CAD) is a complex pathology associated with behavioral and environmental factors.1–3 CAD shows high prevalence and is associated with a high fatality rate among cardiovascular diseases. The main manifestations of CAD are stable or unstable angina pectoris and identifiable or unrecognized myocardial infarction.4 The main risk factors for this disease are diabetes, hypertension, smoking, hyperlipidemia, and obesity,5 and its most common complications are myocardial infarction, heart failure, stroke, and death.6
Coronary angiography has become a standard diagnostic method for CAD that has improved early detection of subclinical disease.7 Furthermore, new biological mechanisms and biomarkers of CAD have been reported that can be identified by histological techniques.8 However, coronary angiography is costly and invasive, and therefore better diagnostic methods are needed.9 High-precision circulating biomarkers have emerged as a valid alternative for the diagnosis of several diseases. Combinations of biomarkers have been integrated using various methods in order to create diagnostic or prognostic tools in CAD. These methods include the least absolute shrinkage and selection operator (LASSO),10 random forest (RF) classifier,11 support vector machine (SVM),12 and the gene set variation analysis (GSVA) score.13
LASSO is a commonly used penalty regression method, which can be applied for selection of variables in high-dimensional data.10 LASSO performs via a continuous shrinking operation, minimizing regression coefficients in order to reduce the likelihood of overfitting.14 RF is a model that can deal with unbalanced sample distribution, generating less biased classifiers,11 but it often fails to be robust and is vulnerable to overfitting. However, when used as an ensemble classifier, RF builds a forest of decision trees, where each tree is based on a different subset of the features and observations of the data, thereby reducing the variance and increasing robustness. SVM is a supervised learning algorithm that analyzes data for patterns15 in order to find a max-margin separator hyperplane to classify data.16 It can quantify tumor markers as well as classify or diagnose diseases.12 GSVA is an open source R package that robustly estimates path activity changes in the sample population in an unsupervised way. However, in contrast to the other methods, GSVA calculates first an expression statistic with the kernel estimation of the empirical cumulative distribution function over the samples, which should help in protecting the method against systematic gene specific effects, such as probe effects, and thereby increase sensitivity.13
Previous studies used LASSO regression17 to predict prognosis of patients with CAD, while another study combined the k-nearest neighbor, RF and SVM into a novel heterogeneous ensemble method to diagnose CAD.18 GSVA, in contrast, has rarely been used for diagnosis or prognosis in CAD. Previous work has attempted to develop a non-invasive method to diagnose CAD based on the degree of vascular stenosis.19 Another CAD diagnostic model was established based on an active pulse wave velocity index and an artificial neural network.20 Despite these advances, better diagnostic methods are still needed to detect CAD detection.9
The purpose of the present study was to develop novel classifiers based on circulating biomarkers that may serve as diagnostic tools for CAD. We conducted a bioinformatic analysis of differentially expressed genes (DEGs) between CAD patients and healthy controls using data from the Gene Expression Omnibus (GEO) database. We explored the potential underlying molecular mechanisms for those DEGs. Using different techniques we established four classifiers for diagnosing CAD, and we evaluated their diagnostic utility.
Materials and Methods
Data Preprocessing
Gene expression profiles in the peripheral blood mononuclear cells were downloaded from the labeled data set GSE113079,21 based on the GPL20115 platform, in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database.22 The voom function23 in the limma package24 in R was applied to normalize gene expression profiles. GSE113079 includes 93 CAD samples and 48 healthy control samples. All 141 samples were randomly assigned to a training set (70%, 66 CAD samples and 34 healthy samples) or a test set (30%, 27 CAD samples and 14 health samples). If a gene was detected using multiple probes, the average value across all those probes was considered the expression level of the gene. The flow chart of the study is shown in Figure 1.
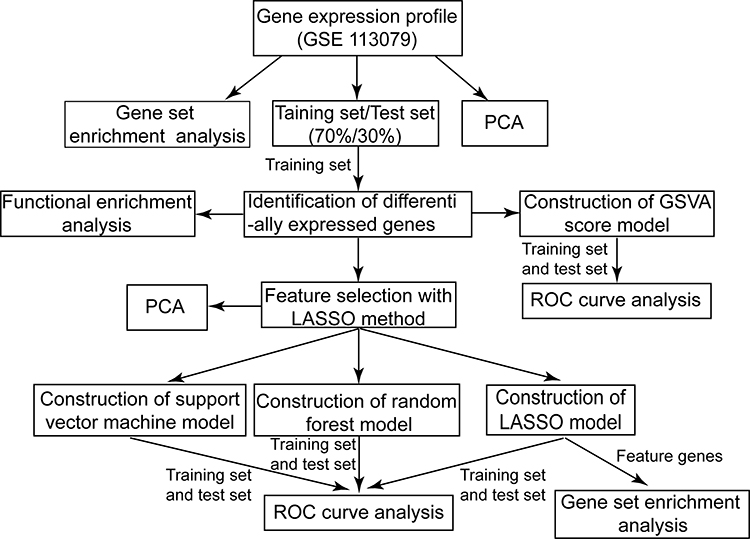 |
Figure 1 Flow chart of the present study. PCA, principal component analysis. Abbreviations: LASSO, Least absolute shrinkage and selection operator; GSVA, Gene set variation analysis; ROC, Receiver operating characteristic curve. |
Differential Expression Analysis
The limma package23 in R was used to identify DEGs between CAD and healthy samples in the training set. The genes with an expression difference of | log2 (fold change, FC) | >1 and P < 0.01 were considered DEGs. The | log2 FC | >1 was the filter standard for DEGs and P < 0.05 was considered to be statistically significant.25
Functional Enrichment Analysis and Gene Set Enrichment Analysis (GSEA)
To further explore the biological processes and pathways involving DEGs, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses using the clusterProfiler package in R.26 Pathways associated with P < 0.01 were considered significantly enriched. In addition, gene expression profiles of all training set genes were used for GSEA using GSEA software.27 The reference gene sets were immunologic signatures (c7.all.v7.0.symbols.gmt) and canonical KEGG pathways (c2.cp.kegg.v7.0.symbols.gmt), both from the MsigDB V7.0 database.28 Nominal P < 0.05 was considered significant. Moreover, biological processes were analyzed using the ClueGO plug-in29 in Cytoscape software.30
Feature Selection Using the LASSO Method and Principal Component Analysis (PCA)
Using the LASSO method, we selected the optimal features from DEGs to construct a logistic regression model. LASSO logistic regression was performed using the glmnet package (CRAN.R-project.org/package=glmnet).31 The nflods parameter for cross-validation was set to 10. The most concise LASSO model was obtained, defined as the one using the fewest characteristic genes to predict the grouping of samples well. The results of LASSO regression were output using the plotimhistory function. Five indexes were calculated to evaluate the performance of the models: sensitivity, specificity, positive predictive value, negative predictive value, and accuracy.
A subset of relevant features were selected from the original feature set.32 After feature selection, PCA was performed using the expression profiles of the optimal features. Samples were plotted in two-dimensional plots across the first two principal components. In addition, to reveal the biological functions of optimal features in CAD, we used the median expression value of each optimal feature as a threshold, and we divided the CAD samples into high- and low-expression groups to perform GSEA. A P value < 0.05 adjusted by the Benjamini & Hochberg method33 was defined as the significance threshold. GSEA was performed using the clusterProfiler package,26 and results were visualized using the enrichplot package (https://github.com/YuLab-SMU/enrichplot).
CAD Diagnostic Model Based on the SVM Method
An SVM model was constructed based on the feature genes from the LASSO model. In the tune function in the e1071 package in R,34 given a labeled training data set
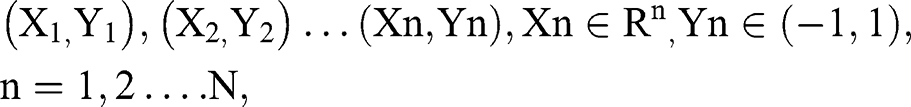
where Xn is a feature vector representation and Yn is the class label (negative or positive) of a training compound n, the optimal hyperplane can be defined as:
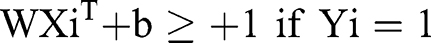
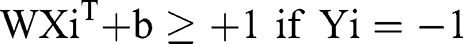
The purpose of the SVM model is to discover W and b, from which the separation hyperplane can be determined and optimal genes obtained.35 In addition, we built the SVM by filtering for the optimal gamma and performing cross-validation 10 times.
Feature Selection Using Boruta and RF Classifier Construction
Feature selection was performed using the Boruta package (http://www.jstatsoft.org/article/view) in R. In order to eliminate irrelevant variables, feature genes were searched from top to bottom. The algorithm performed 99 iterations to define the significance of variables in the data set: in the end, it determined whether characteristic genes were rejected, important or provisional.36 We used the plotimhistory function in the Boruta package to show the importance of candidate genes. Then, the expression profiles of characteristic genes were extracted, and the RF model was constructed using the randomForest function in R.
Implementation of GSVA
We extracted DEGs and defined the top 20 genes with the largest π - value as central genes, ie for which the absolute value of log2FC*-log10 (P)).37 Then we calculated the GSVA score for these genes in individual samples using the GSVA package13 in R.
Receiver Operating Characteristic (ROC) Curve Analysis
Dynamically adjusting the cut-off points to be executed, each cut-off point corresponds to a False Positive Rate and True Positive Rate, and the corresponding position of each cut-off point is drawn on the ROC diagram. The ability of each classifier model to diagnose CAD was evaluated in terms of the area under the receiver operating characteristic curve (AUC). Feature genes obtained from the LASSO method and GSVA score were extracted, and AUCs were calculated using the pROC package.38
Results
Biological Processes and Pathways of DEGs in CAD
To explore alterations in gene expression in CAD patients, we performed a differential gene expression analysis (Figure 2A), identifying 19,877 DEGs in the training set that were up or down-regulated in CAD (Figure 2B). Based on enrichment analysis, the DEGs were involved mainly in the positive regulation of interleukin (IL)-2 biosynthesis, positive regulation of vascular endothelial growth factor production, 3ʹ-UTR-mediated mRNA stabilization, polyadenylation-dependent RNA catabolism and cell-related biological processes (Figure 2C). Additionally, these genes were involved in tumor necrosis factor (TNF) and IL-17 signaling, interactions of viral proteins with cytokine and cytokine receptors, and nuclear factor (NF)-kappa B signaling (Figure 2D). ClueGO analysis also suggested the involvement of these genes in TNF and IL-17 signaling pathways, wound healing during the inflammatory response and replacement of connective tissue during wound healing (Figure 2E). GSEA results showed that CAD-associated DEGs were involved in calcium signaling, chemokine signaling, dilated cardiomyopathy, interactions between neuroactive ligands and receptors, vascular smooth muscle contraction, and interactions between cytokines and cytokine receptors (Figure 2F).
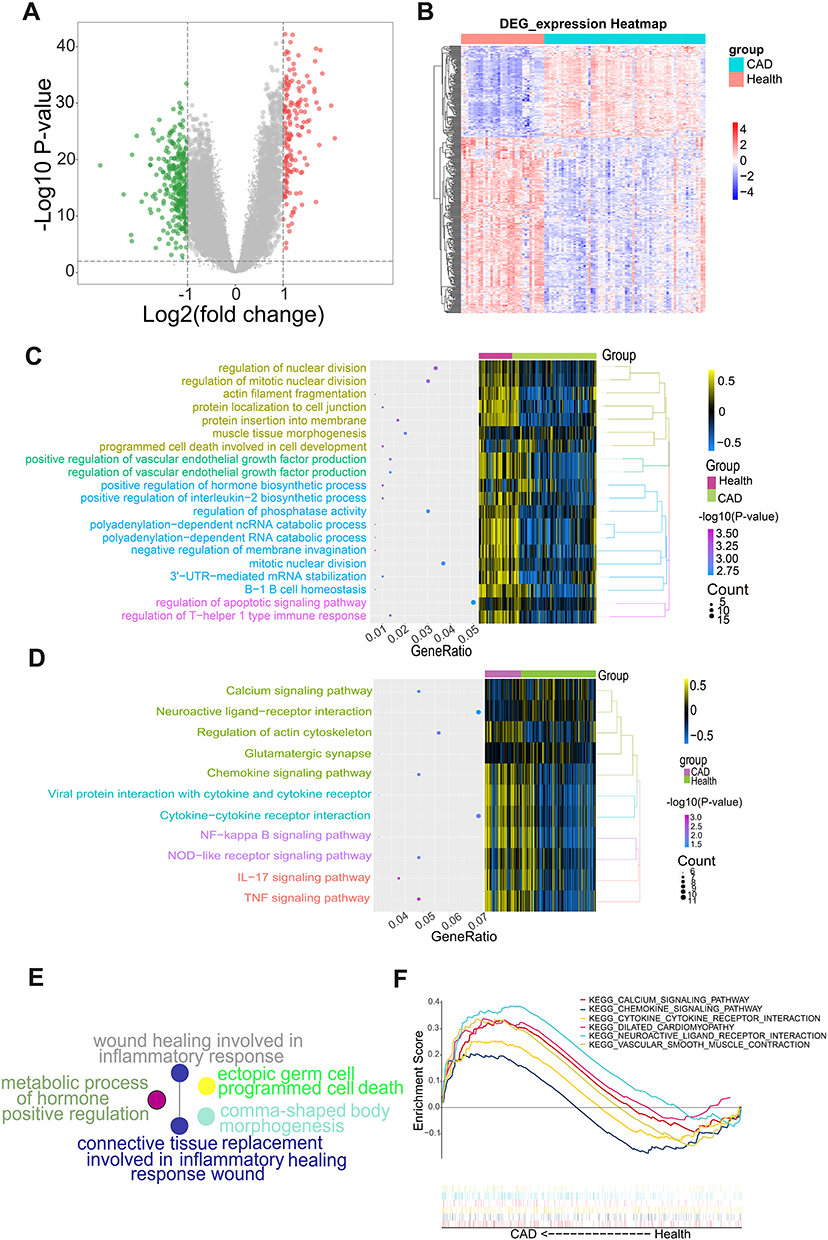 |
Figure 2 Differential gene expression analysis and functional enrichment analysis. (A) Volcano map. Red indicates genes that were up-regulated in coronary artery disease (CAD) patients compared to healthy samples, green indicates genes that were down-regulated, and gray indicates genes with similar expression between both groups. (B) Heat map of differentially expressed genes (DEGs). The depth of color reflects the level of differential expression. (C) The biological processes where DEGs may be involved according to enrichment analysis. (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in which DEGs may be involved. (E) Biological processes where DEGs may be involved according to Clue GO analysis. (F) KEGG pathways enriched in CAD samples. |
Feature Selection Using LASSO and PCA
A total of 20 DEGs were identified as the optimal genes (Figure 3A and Table 1). The results of PCA following feature selection using LASSO showed that the expression patterns of the 20 genes easily distinguished CAD from healthy samples (Figure 3B). The samples of the training set and the test set were used to verify the robustness of the model: the AUC was 1.00 in the training set (Figure 3C) and 0.997 in the test set (Figure 3D).
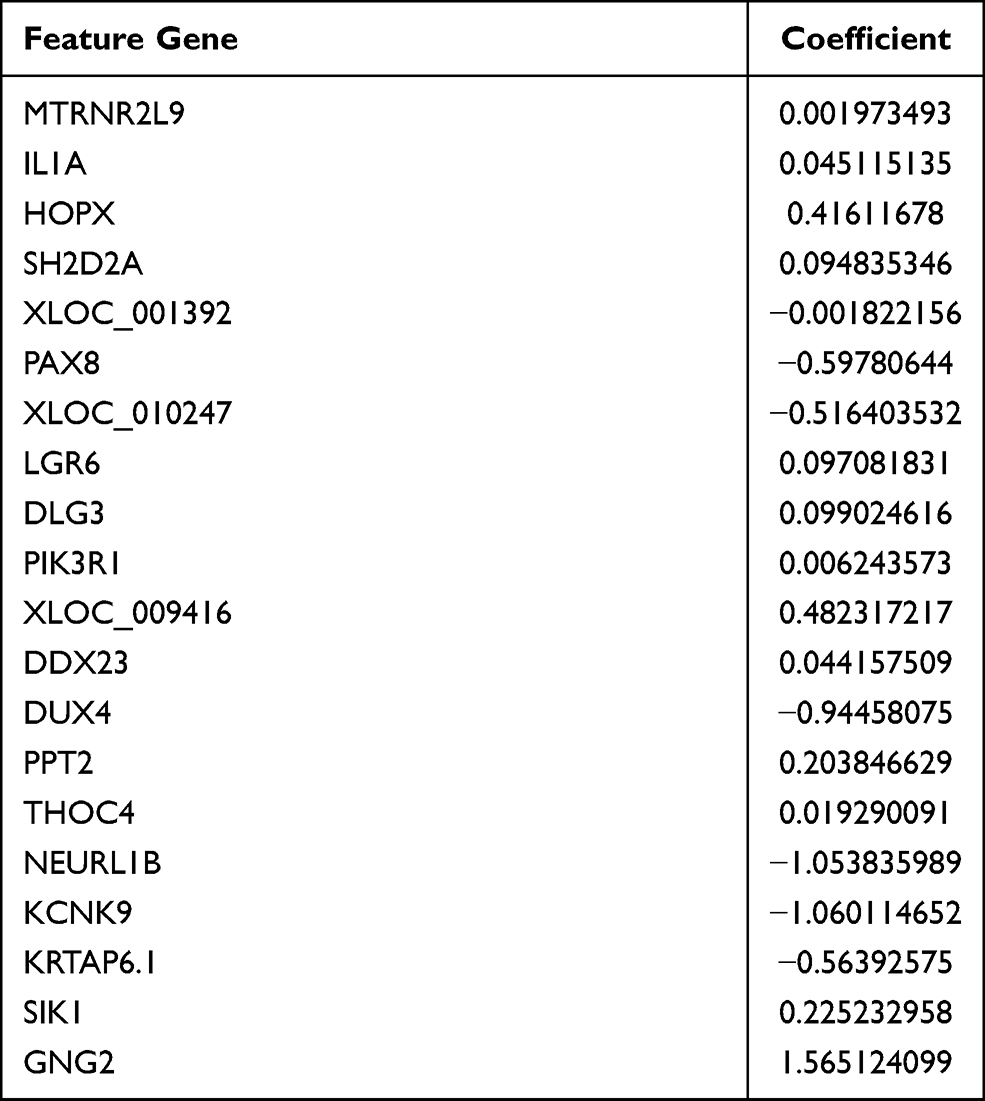 |
Table 1 The 20 Feature Genes Selected by Least Absolute Shrinkage and Selection Operator (LASSO) Method |
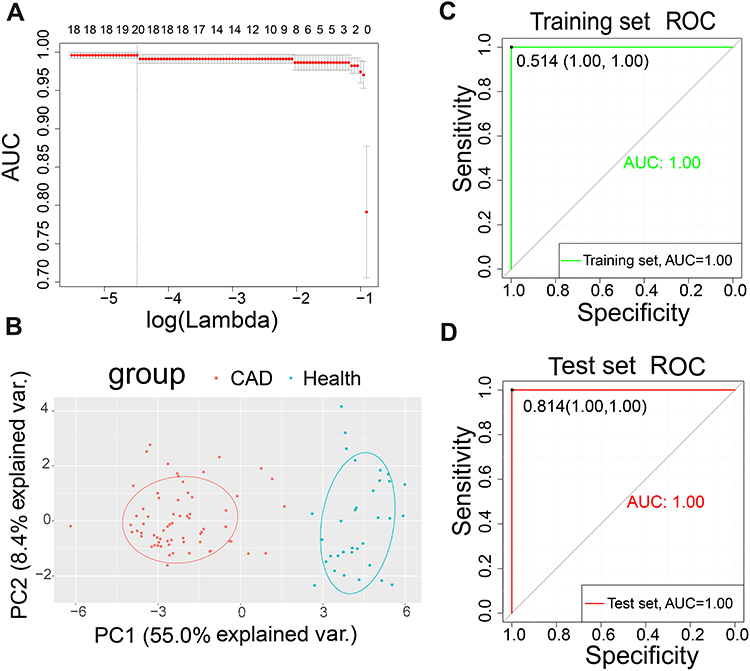 |
Figure 3 Least absolute shrinkage and selection operator (LASSO) model and principal component analysis (PCA). (A) Ten-times cross-validation of parameter selection in the LASSO model. (B) PCA after LASSO variable reduction. (C) Receiver operating characteristic (ROC) curve of the training set. (D) ROC of the test set. Abbreviations: AUC, area under the ROC curve; var., variance. |
SVM Classifier
The 20 optimal feature genes obtained from the LASSO model were 10-fold cross-validated in order to construct the SVM model. A crucial characteristic of the SVM model is that it can significantly reduce the number of false positives.39 The samples in the training and test sets were used to verify the model performance: the AUC was 1.00 in the training set (Figure 4A) and 0.997 in the test set (Figure 4B).
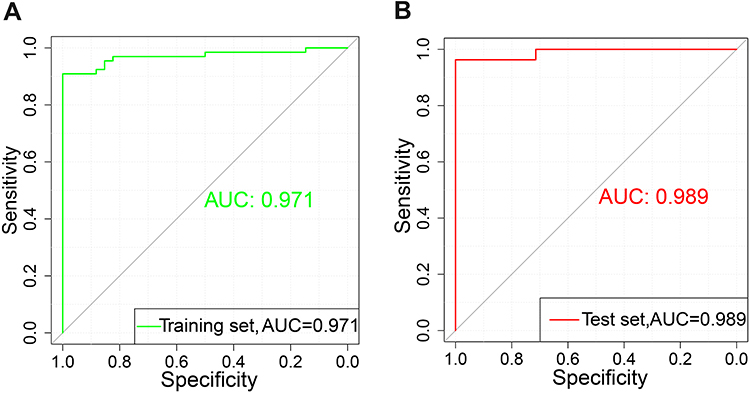 |
Figure 4 Support vector machine (SVM) model. (A) Receiver operating characteristic (ROC) curve of the training set. (B) ROC of the test set. Abbreviation: AUC, area under the ROC curve. |
Random Forest Classifier
The 20 feature genes selected by LASSO model were further used to perform feature selection (Figure 5A). Nineteen of these were identified as important genes: IL1A, HOPX, SH2D2A, XLOC_001392, PAX8, XLOC_010247, LGR6, DLG3, PIK3R1, XLOC_009416, DDX23, DUX4, PPT2, THOC4, NEURL1B, KCNK9, KRTAP6-1, SIK1, GNG2 and provisionally MTRNR2L9. The diagnostic performance of the model was evaluated: the AUC was 1.00 for both the training set (Figure 5B) and test set (Figure 5C).
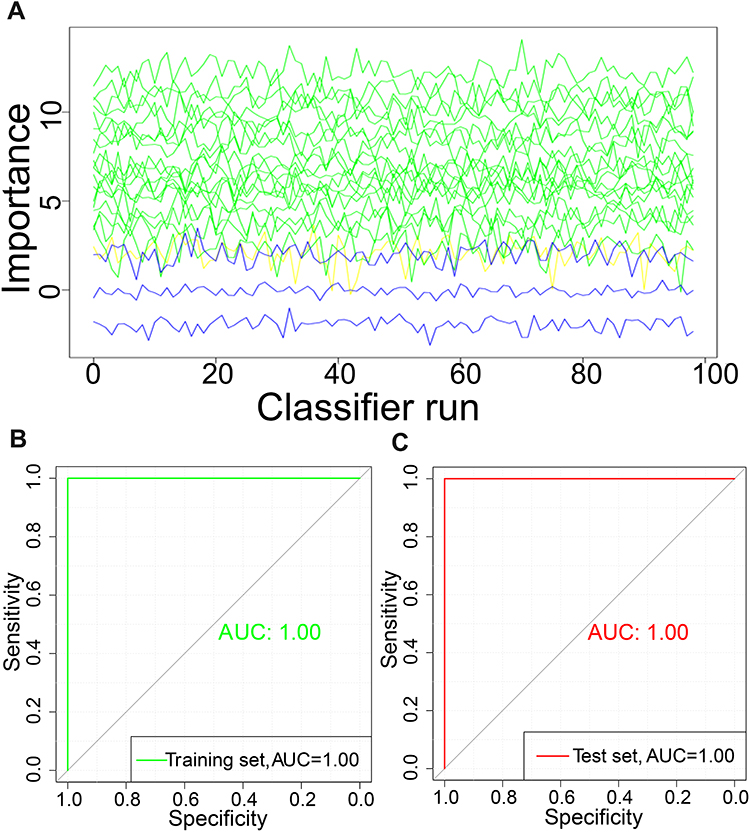 |
Figure 5 Evolution of the Z-score during Boruta operation. (A) The Boruta function was used to further select features in the 20 differentially expressed genes. (B) Receiver operating characteristic (ROC) curve of the training set. (C) ROC of the test set. Abbreviation: AUC, area under the ROC curve. |
Construction of GSVA Score Using DEGs
Among the DEGs in the training set, the top 20 significant DEGs (ranked by π value) were KIF17, SHANK1, OPN4, BIRC7, TRPM5, NUPR1, OR4C3, C16orf73, XLOC_12_013427, XLOC_001485, CSNK1A1, NMNAT2, FAM154A, GCLM, XLOC_001392, TM4SF5, AKAP5, ARHGEF33, DUX4, and FTMT. These genes were considered as the CAD-specific gene set. The CAD-specific gene set variation score for each individual in the dataset was calculated using the GSVA package. The AUC of the GSVA score was 0.971 for the training set (Figure 6A) and 0.989 for the test set (Figure 6B).
 |
Figure 6 Gene set variation analysis (GSVA) score model. (A) Receiver operating characteristic (ROC) curve of the training set. (B) ROC of the test set. Abbreviation: AUC, area under the ROC curve. |
Four diagnostic classifiers for CAD were defined based on GEO data and bioinformatics analysis. All four diagnostic classifiers showed robust performance (Table 2).
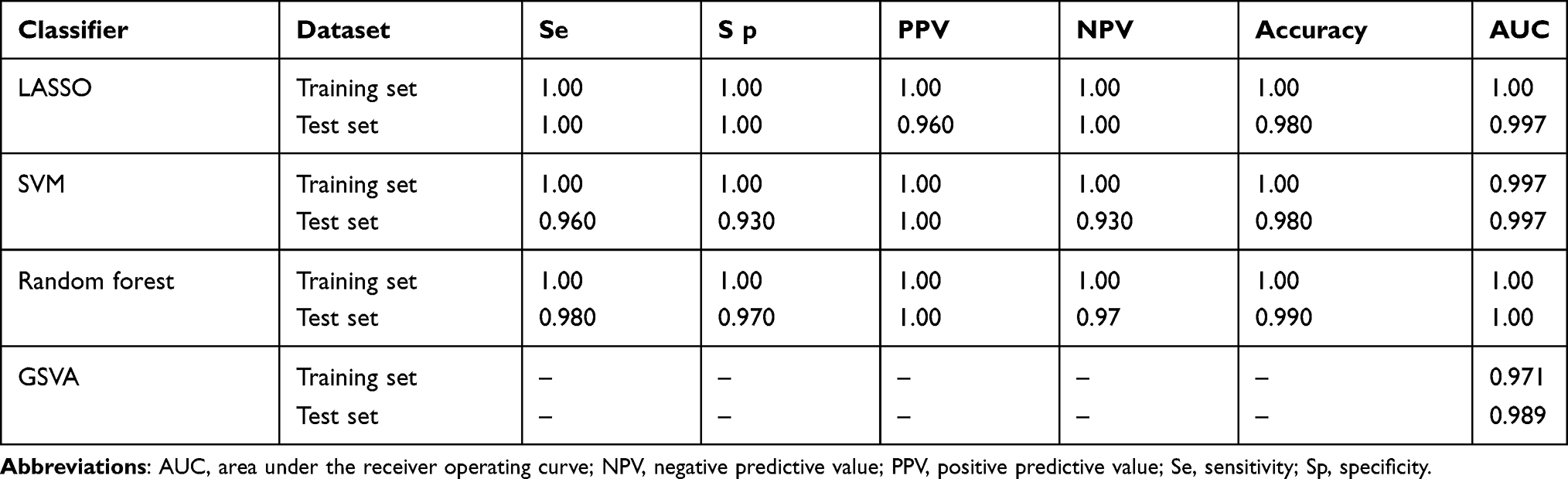 |
Table 2 Performance of the Four Classifiers for Diagnosing CAD |
Identification of Signaling Pathways
In order to reveal the biological pathways where these 20 genes may be involved, GSEA was performed (Figures 7 and 8). The genes were involved mainly in biological pathways related to immune, metabolic, and autophagy processes. In particular, the four genes DDX23, DLG3, GNG2, and THOC4 were involved in autophagy-related pathways.
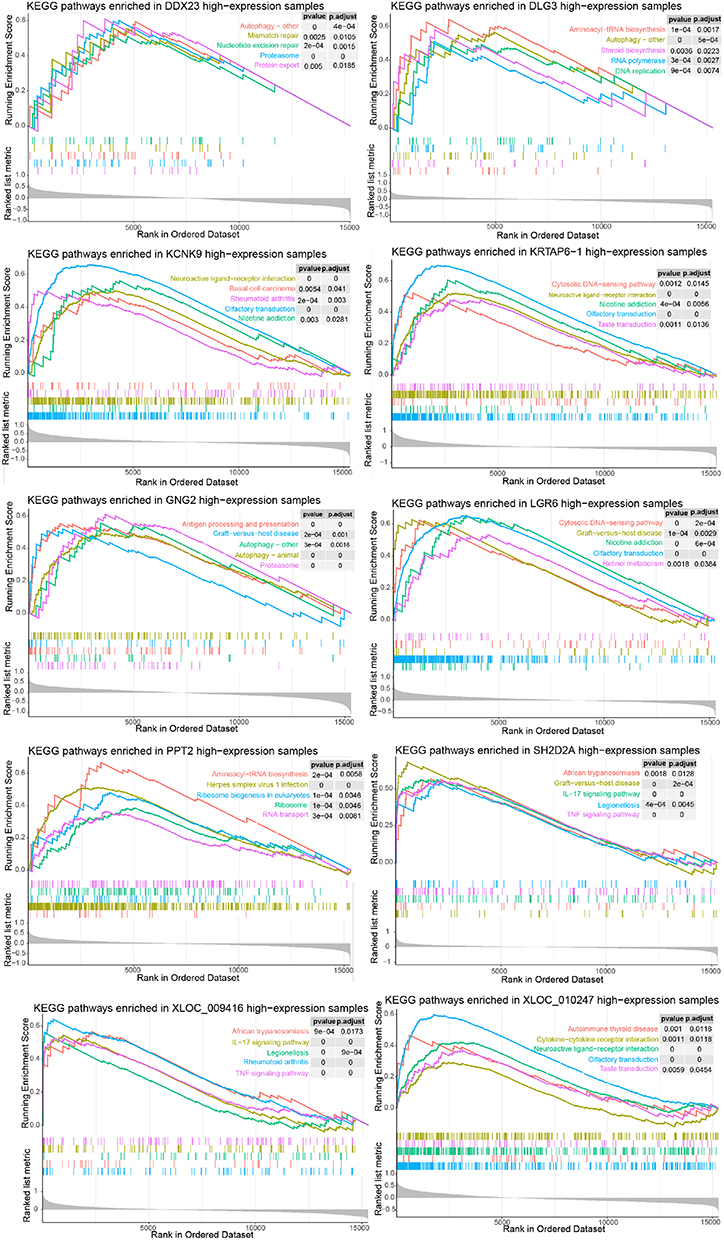 |
Figure 7 Gene set expression analysis (GSEA) of optimal genes and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched in the 10 differentially expressed genes. |
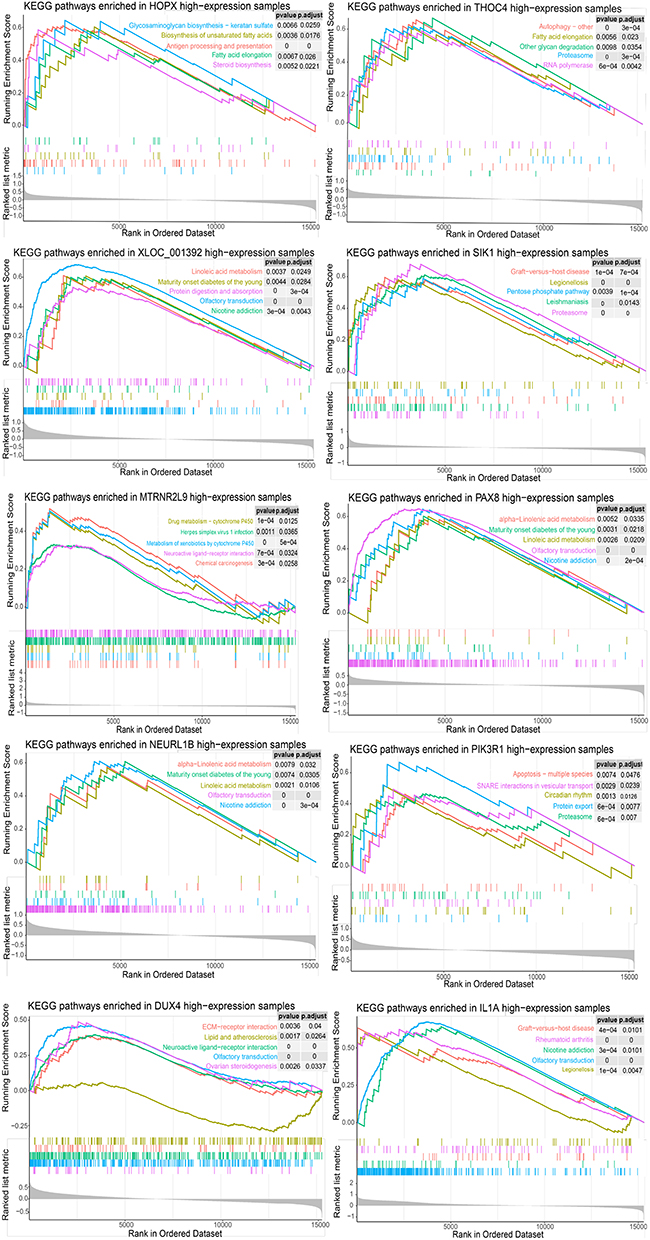 |
Figure 8 Gene set expression analysis (GSEA) of optimal genes and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched in the 10 differentially expressed genes. |
Discussion
CAD is one of the most common causes of death in the world, and its early diagnosis is essential to avoid complications and improve prognosis.40 Current CAD diagnostic methods present several limitations that can be overcome by the use of adequate combinations of circulating biomarkers. In this study, we performed functional enrichment analysis and GSEA in order to develop four diagnostic models for CAD DEGs, whose diagnostic potential we assessed in terms of AUCs.
GSEA of the gene set revealed a total of six pathways, but only two pathways were common among intersection: calcium signaling pathway and IL-17 between neuroactive ligands and receptors. Substantial clinical evidence has shown that calcium ion plays a crucial role in tumor occurrence, angiogenesis, development, and metastasis through homeostasis.41 The “neuroactive ligand receptor interaction signal pathway” has shown a cardioprotective effect.42
Previous studies showed that LASSO regression can predict the prognosis of patients with CAD. RF and SVM have been shown to help diagnose CAD in early stages.18 Nevertheless, GSVA shows higher mean and median concordance than the other methods against both training and testing data sets involving leukemia and ovarian carcinoma datasets.13 However, GSVA has rarely been applied to CAD. All four models that we assessed in this study have potential for diagnosing many diseases.
In our study, LASSO, SVM, and RF classifiers showed high diagnostic accuracy against both the training and test sets when the feature selection algorithm was used to select the 20 DEGs. The first 20 DEGs with the largest π value were extracted for GSVA scoring, which also performed well against both the training and test sets. The four classifiers were robust and therefore have good potential for future research on CAD diagnosis.
Several feature genes were extracted from the LASSO model: IL1A, HOPX, SH2D2A, XLOC_001392, PAX8, XLOC_010247, LGR6, DLG3, PIK3R1, XLOC_009416, DDX23, DUX4, PPT2, THOC4, NEURL1B, KCNK9, KRTAP6-1, SIK1, GNG2 and MTRNR2L9. Several of these genes have previously been linked to CAD. During the progression of CAD, there is a significant up regulation of IL-1A, which suggests the potential of this interleukin as a biomarker and treatment targets.43 Differential methylation of PIK3R1 and its expression have been associated with CAD pathogenesis.44 A single-nucleotide polymorphism in KCNK9 may be associated with increased risk of premature CAD and its severity.45 Other feature genes that we identified have not previously been linked to CAD. Therefore, further work needs to explore what genes may contribute to CAD and therefore serve as diagnostic biomarkers or therapeutic targets.
In our study, the models performed quite well in the training set, suggesting the possibility of overfitting. However, the models also showed good performance in the test set, confirming their robustness. Additionally, the LASSO and SVM models were cross-validated to reduce risk of overfitting. The RF model is not vulnerable to overfitting. Thus, we consider the risk of overfitting to be negligible. The main limitations of our study are that it is based entirely on bioinformatics analysis and that the sample is small. Given that dataset size and presence of noise can affect model performance,46 our results should be verified and extended with larger samples and in experimental studies.
Conclusions
Our study proposes four diagnostic classifiers for CAD based on GEO data and bioinformatics analysis. The performance of the four diagnostic classifiers was robust, so they merit further investigation for non-invasive diagnosis of CAD.
Data Sharing Statement
The dataset supporting the conclusions of this article is available in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database. The persistent identifier and hyperlink to the dataset is https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113079.
Funding
This study was supported by the Project of Nanning Scientific Research and Technology Development Plan (ZC20203010), the Project of Qingxiu District of Nanning Scientific Research and Technology Development Plan (2020059), the Scientific Research Project of Guangxi Health Commission (Z20201226) and Guangxi Medical and Health Key Discipline Construction Project (Department of Cardiology, The First People’s Hospital of Nanning).
Disclosure
The authors declare that they have no competing interests.
References
1. Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135(10):e146–e603.
2. Ades PA, Gaalema DE. Coronary heart disease as a case study in prevention: potential role of incentives. Prev Med. 2012;55(Suppl):S75–79.
3. Mallika V, Goswami B, Rajappa M. Atherosclerosis pathophysiology and the role of novel risk factors: a clinicobiochemical perspective. Angiology. 2007;58(5):513–522.
4. Murabito JM, Evans JC, Larson MG, Levy D. Prognosis after the onset of coronary heart disease. An investigation of differences in outcome between the sexes according to initial coronary disease presentation. Circulation. 1993;88(6):2548–2555.
5. Malakar AK, Choudhury D, Halder B, Paul P, Uddin A, Chakraborty S. A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol. 2019;234(10):16812–16823.
6. Trujillo TC, Dobesh PP. Traditional management of chronic stable angina. Pharmacotherapy. 2007;27(12):1677–1692.
7. McCullough PA. Coronary artery disease. Clin J Am Soc Nephrol. 2007;2(3):611–616.
8. Vernon ST, Hansen T, Kott KA, Yang JY, O’Sullivan JF, Figtree GA. Utilizing state-of-The-art “omics” technology and bioinformatics to identify new biological mechanisms and biomarkers for coronary artery disease. Microcirculation. 2019;26(2):e12488.
9. Guerreiro S, Ferreira AM, Abecasis J, et al. Additional cardiac investigation prior to the introduction of the CAD-RADS classification in coronary computed tomography angiography reports. Rev Port Cardiol. 2019;38(1):45–50.
10. Nghiem L, Potgieter C. Simulation-selection-extrapolation: estimation in high-dimensional errors-in-variables models. Biometrics. 2019;75(4):1133–1144.
11. Rigatti SJ. Random Forest. J Insur Med. 2017;47(1):31–39.
12. Sun Z, Fu X, Zhang L, Yang X, Liu F, Hu G. A protein chip system for parallel analysis of multi-tumor markers and its application in cancer detection. Anticancer Res. 2004;24(2C):1159–1165.
13. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7.
14. McEligot AJ, Poynor V, Sharma R, Panangadan A. Logistic LASSO regression for dietary intakes and breast cancer. Nutrients. 2020;12:9.
15. Nedaie A, Najafi AA. Support vector machine with Dirichlet feature mapping. Neural Netw. 2018;98:87–101.
16. Noble WS. What is a support vector machine?. Nat Biotechnol. 2006;24(12):1565–1567.
17. Mehta A, Liu C, Nayak A, et al. Untargeted high-resolution plasma metabolomic profiling predicts outcomes in patients with coronary artery disease. PLoS One. 2020;15(8):e0237579.
18. Velusamy D, Ramasamy K. Ensemble of heterogeneous classifiers for diagnosis and prediction of coronary artery disease with reduced feature subset. Comput Methods Programs Biomed. 2021;198:105770.
19. Jang JJ, Bhapkar M, Coles A, et al. Predictive model for high-risk coronary artery disease. Circ Cardiovasc Imaging. 2019;12(2):e007940.
20. Vallee A, Cinaud A, Blachier V, Lelong H, Safar ME, Blacher J. Coronary heart disease diagnosis by artificial neural networks including aortic pulse wave velocity index and clinical parameters. J Hypertens. 2019;37(8):1682–1688.
21. Li L, Wang L, Li H, et al. Characterization of LncRNA expression profile and identification of novel LncRNA biomarkers to diagnose coronary artery disease. Atherosclerosis. 2018;275:359–367.
22. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue):D991–995.
23. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29.
24. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
25. Ho AM, Cabello-Arreola A, Markota M, et al. Label-free proteomics differences in the dorsolateral prefrontal cortex between bipolar disorder patients with and without psychosis. J Affect Disord. 2020;270:165–173.
26. Yu G, Wang LG, Han Y, He QY. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287.
27. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics. 2007;23(23):3251–3253.
28. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425.
29. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504.
30. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093.
31. Engebretsen S, Bohlin J. Statistical predictions with glmnet. Clin Epigenetics. 2019;11(1):123.
32. Yong S, Jianyu M, Zhengyu W, Peng Z, Lingfeng N. Feature selection with $\ell_{2,1-2}$ regularization. IEEE Trans Neural Netw Learn Syst. 2018;29(10):4967–4982.
33. Ferreira JA. The Benjamini-Hochberg method in the case of discrete test statistics. Int J Biostat. 2007;3(1):Article 11.
34. Jiang H, Gu J, Du J, Qi X, Qian C, Fei B. A 21-gene support vector machine classifier and a 10-gene risk score system constructed for patients with gastric cancer. Mol Med Rep. 2020;21(1):347–359.
35. Huang S, Cai N, Pacheco PP, Narrandes S, Wang Y, Xu W. Applications of Support Vector Machine (SVM) learning in cancer genomics. Cancer Genomics Proteomics. 2018;15(1):41–51.
36. Su X, Xu Y, Tan Z, et al. Prediction for cardiovascular diseases based on laboratory data: an analysis of random forest model. J Clin Lab Anal. 2020;34(9):e23421.
37. Xiao Y, Hsiao TH, Suresh U, et al. A novel significance score for gene selection and ranking. Bioinformatics. 2014;30(6):801–807.
38. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77.
39. Corona RI, Sudarshan S, Aluru S, Guo JT. An SVM-based method for assessment of transcription factor-DNA complex models. BMC Bioinform. 2018;19(Suppl 20):506.
40. Mack M, Gopal A. Epidemiology, traditional and novel risk factors in coronary artery disease. Heart Fail Clin. 2016;12(1):1–10.
41. Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7(1):3–17.
42. Lauss M, Kriegner A, Vierlinger K, Noehammer C. Characterization of the drugged human genome. Pharmacogenomics. 2007;8(8):1063–1073.
43. Wang C, Song C, Liu Q, et al. Gene expression analysis suggests immunological changes of peripheral blood monocytes in the progression of patients with coronary artery disease. Front Genet. 2021;12:641117.
44. Miao L, Yin RX, Zhang QH, et al. Integrated DNA methylation and gene expression analysis in the pathogenesis of coronary artery disease. Aging. 2019;11(5):1486–1500.
45. Chen B, Xie F, Tang C, Ma G, Wei L, Chen Z. Study of five pubertal transition-related gene polymorphisms as risk factors for premature coronary artery disease in a Chinese Han population. PLoS One. 2015;10(8):e0136496.
46. Yao J, Zhao Q, Yuan Y, et al. Identification of common prognostic gene expression signatures with biological meanings from microarray gene expression datasets. PLoS One. 2012;7(9):e45894.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.