Back to Journals » Lung Cancer: Targets and Therapy » Volume 8
Cis-oriented solvent-front EGFR G796S mutation in tissue and ctDNA in a patient progressing on osimertinib: a case report and review of the literature
Authors Klempner SJ ![]() , Mehta P, Schrock AB, Ali SM, Ou SI
, Mehta P, Schrock AB, Ali SM, Ou SI ![]()
Received 30 July 2017
Accepted for publication 10 November 2017
Published 6 December 2017 Volume 2017:8 Pages 241—247
DOI https://doi.org/10.2147/LCTT.S147129
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Joseph Locker
Samuel J Klempner,1,2 Pareen Mehta,3 Alexa B Schrock,4 Siraj M Ali,4 Sai-Hong Ignatius Ou5
1The Angeles Clinic and Research Institute, Los Angeles, CA, USA; 2Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA; 3Department of Radiology, The Angeles Clinic and Research Institute, Los Angeles, CA, USA; 4Clinical Development, Foundation Medicine, Inc., Cambridge, MA, USA; 5Department of Medicine-Hematology/Oncology, Chao Family Comprehensive Cancer Center, University of California Irvine School of Medicine, Orange, CA, USA
Abstract: Acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI) is a universal event and limits clinical efficacy. The third-generation EGFR inhibitor osimertinib is active in EGFR-mutant/T790M positive non-small-cell lung cancer. Mechanisms of acquired resistance are emerging, and here we describe a cis-oriented solvent-front EGFR G796S mutation as the resistance mechanism observed in a progression biopsy and circulating tumor DNA (ctDNA) from a patient with initial response followed by progression on osimertinib. This is one of the earliest reports of a sole solvent-front tertiary EGFR mutation as a resistance mechanism to osimertinib. Our case suggests a monoclonal resistance mechanism. We review the importance of the solvent-front residues across TKIs and describe known osimertinib resistance mechanisms. We observe that nearly all clinical osimertinib-resistant tertiary EGFR mutations are oriented in cis with EGFR T790M. This case highlights the importance of mutations affecting EGFR kinase domains and supports the feasibility of broad panel ctDNA assays for detection of novel acquired resistance and tumor heterogeneity in routine clinical care.
Keywords: EGFR G796, lung cancer, ctDNA, resistance, osimertinib, T790M
Introduction
Acquired resistance to epidermal growth factor receptor (EGFR)-directed therapies is universal and can be partly predicted by tyrosine kinase inhibitor (TKI) structure and EGFR binding features. The third-generation irreversible EGFR inhibitor osimertinib is effective in EGFR T790M positive non-small-cell lung cancer (NSCLC).1,2 The landscape of osimertinib resistance is evolving and highlighted by the acquired EGFR C797S mutation, which limits covalent binding.3 Allelic orientation of osimertinib resistance mutations with respect to EGFR T790M may dictate the spectrum of resistant mutation treatment strategies, and here we report an EGFR solvent-front G796S mutation oriented in cis with T790M in an EGFR L858R-mutant NSCLC patient at progression on osimertinib.4,5
Case presentation
A 69-year-old Asian female never smoker presented in July 2014 with back and hip pain and was found to have a 4.4-cm left lower lobe mass, a right ischium bone metastasis and malignant adenopathy (Figure 1). CT-guided left lower lobe biopsy confirmed a poorly differentiated adenocarcinoma, and hotspot polymerase chain reaction (PCR) testing identified an activating EGFR L858R mutation but no EGFR T790M. The patient was begun on erlotinib 150 mg orally once daily. She achieved a good partial response and symptomatic improvement lasting 13 months, at which time a new left lower lobe lesion 1.8 × 1.3 cm as well as increased activity in her known bony disease was seen (Figure 1). A biopsy of the right pelvic mass was performed and was consistent with poorly differentiated adenocarcinoma. Additional hotspot molecular testing post-erlotinib confirmed the EGFR L858R mutation and was negative for EGFR T790M. However, a single-gene droplet-digital PCR test from circulating tumor DNA (ctDNA) performed after progression on erlotinib identified EGFR T790M, and the patient was transitioned to osimertinib in October 2015. She had symptomatic improvement and partial response lasting 12 months, at which point a new left pelvic mass with bony involvement was identified (Figure 1). A left pelvic mass core biopsy from the soft tissue component was subjected to comprehensive genomic profiling (FoundationOne®, Foundation Medicine, Cambridge, MA, USA), which identified the original EGFR L858R at a mutant allele frequency (MAF) of 50.52%, an EGFR T790M at 37.14%, an EGFR G796S at 38.69%, EGFR amplification at 16 predicted copies, and a low mutational burden (four mutations per DNA megabase). Overlapping sequencing reads spanning the T790M and G796S confirmed cis orientation (Figure 2A). A concurrent ctDNA assay (FoundationACT™, Foundation Medicine) detected the EGFR L858R (MAF 12.8%), EGFR T790M (MAF 11.2%), and the G796S (MAF 11.6%) without other putative resistance alterations (Table S1). Immunohistochemistry confirmed high PD-L1 expression at 70% by tumor proportion score (Dako 22C3 pharmDx, Agilent Technologies, Santa Clara, CA, USA). Additional genomic alterations are shown in Table S1, and no other putative drivers were detected. In the absence of available trials, she was transitioned to carboplatin plus pemetrexed and achieved stable disease after three cycles (Figure 1) followed by disease progression. Based on lack of standard therapies and high PD-L1 expression that patient was then enrolled in a clinical trial of pembrolizumab in combination with the oral IDO-1 inhibitor epacadostat (NCT02178722). She has achieved a radiographic partial response and remains on therapy, now 5 months in duration. The patient has provided written informed consent to have the case details and any accompanying images published.
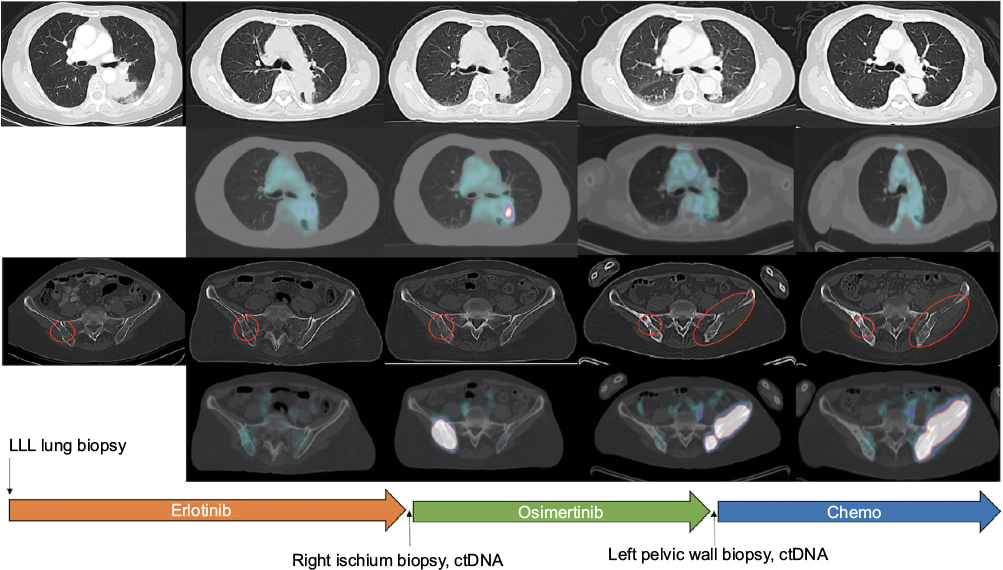 | Figure 1 Radiographic response followed by progression in a EGFR L858R NSCLC with response to first-line erlotinib and second-line osimertinib. Notes: Arrows depict treatment timeline events, and red circles denote bony lesions. Abbreviations: ctDNA, circulating tumor DNA; EGFR, epidermal growth factor receptor; LLL, left lower lobe; NSCLC, non-small-cell lung cancer. |
 | Figure 2 (A) Integrated genomics viewer highlighting the presence of a C>T at codon 790 (EGFR T790M) mutation (red) oriented in cis with a G>A at codon 796 (EGFR G796S) mutation (green). Overlapping reads spanning the T790M and G796S indicate cis orientation on the same allele. (B) The RTK sequence alignments across relevant TKIs with the gatekeeper residue highlighted in yellow and the relevant solvent-front residue in teal. Abbreviations: EGFR, epidermal growth factor receptor; RTK, receptor tyrosine kinase; TKI, tyrosine kinase inhibitor. |
Discussion
The median progression-free survival (PFS) for first-line erlotinib in EGFR-mutant NSCLC is roughly 10 months from the EURTAC (Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer) trial.6 Among patients who develop EGFR T790M-mediated resistance, the median PFS for osimertinib is 10.1 months in the AURA3 trial, with the largest proportion developing EGFR C797S-mediated resistance based on limited study.1,3 The importance of the covalent binding residue (EGFR C797) was highlighted by ibrutinib resistance mutations at the analogous cysteine residue C481 (C481S) in Bruton’s tyrosine kinase (BTK).7 With increasing clinical use of osimertinib, several additional resistance alterations have been described, the majority related to acquired changes affecting inhibitor binding (Table 1). Tertiary mutations affecting the EGFR solvent-front (G796S/R), hinge region (L792H/F), osimertinib covalent binding residue (C797S/G), and residues predicted to decrease affinity through steric interaction (L718Q/V, L844V) are described, and triple mutants (EGFR activating/T790M/tertiary EGFR) appear resistant to all third-generation EGFR TKIs.3–5,8–11 It is anticipated that the majority of clinically observed osimertinib-resistant tertiary mutations will occur in cis with the EGFR T790M. However, a recent report suggested trans-oriented T790M and C797S exists, and in fact may be sensitive to combination erlotinib and osimertinib, as would be predicted preclinically.4,12 Non-target-modifying resistance alterations, broadly defined as bypass track activation (MET, ERBB2, EGFR amplification), histologic transformation to small-cell lung cancer (SCLC), and clonal divergence of T790M-cells, have been observed, and ongoing experience will clarify relative frequencies (Table 1).
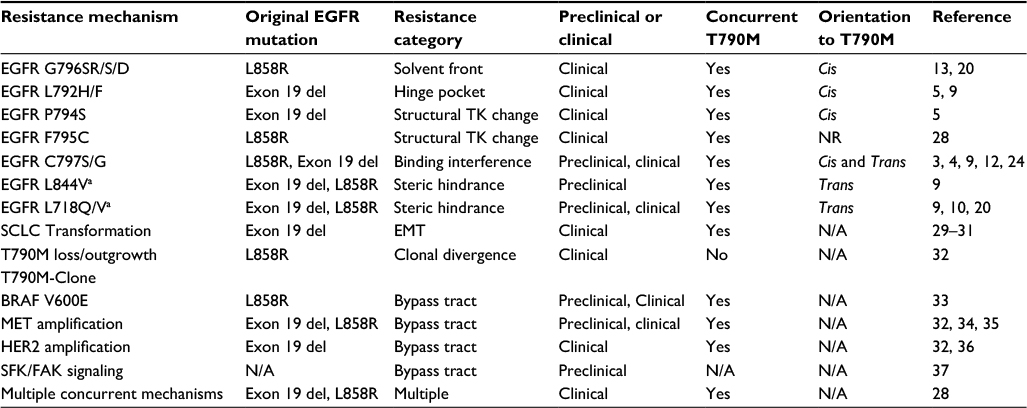 | Table 1 Currently described resistance mechanisms to the third-generation EGFR tyrosine kinase inhibitor osimertinib Notes: Representative references are included. aDenotes variable sensitivity to osimertinib in preclinical models.8 Abbreviations: del, deletion; EGFR, epidermal growth factor receptor; EMT, epithelial-mesenchymal transition; N/A, not applicable; NR, not reported; SCLC, small-cell lung cancer; TK, tyrosine kinase. |
Our case adds to the emerging resistance literature, and the lack of a detected concurrent C797S adds strong support to the suggestion that G796S is mediating osimertinib resistance in our patient. Additional support is observed in an osimertinib-resistant case with both EGFR G796S and G796R mutations.13 Orthogonal support from other oncogenic receptor tyrosine kinases (RTKs) is suggested, and we note that the EGFR G796S is analogous to the confirmed solvent-front resistance mutations ALK G1202R, ROS1 G2032R, TRKA G595R, and TRKC G623R (Figure 2B).14–17 Broadly, solvent-front resistance mutations often cause direct steric hindrance to inhibitor binding and/or destabilize the favorable electrostatic interactions between the inhibitor and its respective binding site.18 Critically, there is a need to functionally validate clinically observed solvent-front EGFR mutations, and we acknowledge a paucity of structural and functional data surrounding EGFR codon 796.
The emergence of ct DNA assays offers potential for monitoring and serial noninvasive tumor assessment, which may overcome issues with inter- and intratumoral heterogeneity.19 The tissue and ctDNA concordance observed in our case supports a likely monoclonal resistance in which the cis-oriented T790M/G796S containing clones drive the clinical progression. As would be expected, the mutant allele frequencies for the T790M and G796R are nearly identical by both tissue and ctDNA assays, and this patient would not be expected to respond to any currently available EGFR inhibitors. Although more data is needed, the MAF % is an important difference between our case and a recently reported EGFR G796D mutation mediating resistance to osimertinib.20 In the case reported by Zheng et al,20 the EGFR L858R MAF at pre-gefitinib sample (4.28%) also contained a G796D at MAF 0.61% but no EGFR T790M. At progression on gefitinib, the T790M was detected at MAF of 1.85%, but at progression on osimertinib, the T790M and L858R mutation were undetectable, and the EGFR G796D MAF had increased to 1.9%. This pattern could also be explained by more polyclonal resistance in which a preexisting population of EGFR G796D+ (without concurrent EGFR L858R or T790M) increased during osimertinib therapy. The overlapping reads observed in our case offer a more convincing argument that an acquired G796S developed in cells harboring the original L858R and acquired T790M alterations and is the driver of resistance. A larger experience with resistance to 3rd generation TKIs and ctDNA data will be required to understand the clinical implications of these differences.
Triple mutants (EGFR activating/T790M/tertiary EGFR) represent a major clinical challenge, and an iterative process of resistance assessment at each progression (ctDNA, tissue, or both) is critical.21,22 It is known that a proportion of TKI-naïve EGFR-mutant NSCLC harbor concurrent T790M, and investigation is ongoing into how first-line osimertinib affects the resistance patterns in these patients (AZENT Trial, NCT02841579).23 Ongoing combinatorial approaches combining osimertinib and bevacizumab (BOOSTER phase II, NCT03133546), dasatinib, and gefitinib are geared to delay/prevent resistance and will be eagerly awaited. The fourth-generation EGFR mutant-selective allosteric non-ATP competitive inhibitor EAI045 binds in the inactive EGFR kinase conformation (type II) and has demonstrated activity in the L858R/T790M/C797S models, partly because the C797 residue is distant from the allosteric binding pocket.24 Importantly, Jia et al24 demonstrated synergistic activity and marked tumor reduction of EAI045 with the EGFR-dimer disrupting antibody cetuximab in L858R/T790M/C797S mice.25 Structural studies with compound Go6976, a potent protein kinase C (PKC) inhibitor that also binds preferentially to EGFR T790M, bound to EGFR T790M/C797S support the idea that non-ATP competitive inhibitors may be preferential to overcome the presence of C797S.26 Whether a similar strategy is applicable to the G796S solvent-front alteration observed in our patient is unstudied. Overall, the optimal approach to EGFR tertiary mutation positive (EGFRm3+) tumors remains an unknown, and we anticipate combination approaches will be needed.
Although limited by a single patient observation, it is worth noting our patient responded to the combination of pembrolizumab and epacadostat. Data has been mixed, but it has been suggested that EGFR-mutant NSCLC are less responsive to immune checkpoint inhibitor therapies despite the 14% rate of >50% PD-L1 positivity seen in post-EGFR-TKI treated specimens.27 Whether or not PD-L1 expression continues to increase after serial lines of TKI therapy (our patient was tested after osimertinib progression) and how this impacts on immunotherapy sensitivity is unknown. Further, our patient was treated with a novel immunotherapy combination, and whether or not this can overcome the lower response rates seen in EGFR-mutant cases is unknown.
Conclusion
Here we confirm that the EGFR solvent-front G796S mutation confers clinical resistance to osimertinib without the occurrence of C79S or L792 mutations, and is oriented in cis with T790M, consistent with observations of other tertiary EGFR mutations. As endorsed by the National Comprehensive Cancer Network (NCCN) guidelines for NSCLC, comprehensive genomic profiling is critical to detect novel mutations with therapeutic implications, and serial testing is supported by our case.
Acknowledgments
The authors would like to recognize the important work of other researchers whose studies could not be cited due to space constraints.
This work was partly supported by the National Institutes of Health [5P30CA062203-20, 2017].
Disclosure
Samuel J Klempner has received honoraria from Foundation Medicine, Inc. through participation in speaker bureau activities. Pareen Mehta has no disclosures. Alexa B Schrock and Siraj M Ali are employees of Foundation Medicine, Inc. and own stocks in Foundation Medicine, Inc. Sai-Hong Ignatius Ou has received speaking and adviser honorarium from Astra Zeneca. The authors report no other conflicts of interest in this work.
References
Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376:629–640. | ||
Goss G, Tsai CM, Shepherd FA, et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016;17:1643–1652. | ||
Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. | ||
Niederst MJ, Hu H, Mulvey HE, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res. 2015;21:3924–3933. | ||
Chen K, Zhou F, Shen W, et al. Novel mutations on EGFR Leu792 potentially correlate to acquired resistance to osimertinib in advanced NSCLC. J Thorac Oncol. 2017;12(6):e65–e68. | ||
Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. | ||
Woyach JA, Furman RR, Liu TM, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–2294. | ||
Wang S, Tsui ST, Liu C, Song Y, Liu D. EGFR C797S mutation mediates resistance to third-generation inhibitors in T790M-positive non-small cell lung cancer. J Hematol Oncol. 2016;9:59. | ||
Kobayashi Y, Azuma K, Nagai H, et al. Characterization of EGFR T790M, L792F, and C797S mutations as mechanisms of acquired resistance to afatinib in lung cancer. Mol Cancer Ther. 2017;16:357–364. | ||
Ercan D, Choi HG, Yun CH, et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015;21:3913–3923. | ||
Bersanelli M, Minari R, Bordi P, et al. L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR-mutated NSCLC. J Thorac Oncol. 2016;11:e121–e123. | ||
Wang Z, Yang JJ, Huang J, et al. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J Thorac Oncol. 2017;12:1723–1727. | ||
Ou SI, Cui J, Schrock AB, et al. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer. 2017;108:228–231. | ||
Ignatius Ou SH, Azada M, Hsiang DJ, et al. Next-generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014;9:549–553. | ||
Awad MM, Katayama R, McTigue M, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368:2395–2401. | ||
Russo M, Misale S, Wei G, et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016;6:36–44. | ||
Drilon A, Li G, Dogan S, et al. What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann Oncol. 2016;27:920–926. | ||
Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17. | ||
Ahronian LG, Corcoran RB. Strategies for monitoring and combating resistance to combination kinase inhibitors for cancer therapy. Genome Med. 2017;9:37. | ||
Zheng D, Hu M, Bai Y, et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget. 2017;8:49671–49679. | ||
Wang S, Song Y, Yan F, Liu D. Mechanisms of resistance to third-generation EGFR tyrosine kinase inhibitors. Front Med. 2016;10:383–388. | ||
Wang S, Cang S, Liu D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J Hematol Oncol. 2016;9:34. | ||
Yu HA, Arcila ME, Hellmann MD, et al. Poor response to erlotinib in patients with tumors containing baseline EGFR T790M mutations found by routine clinical molecular testing. Ann Oncol. 2014;25:423–428. | ||
Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–132. | ||
Wang S, Song Y, Liu D. EAI045: the fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017;385:51–54. | ||
Kong LL, Ma R, Yao MY, et al. Structural pharmacological studies on EGFR T790M/C797S. Biochem Biophys Res Commun. 2017; 488:266–272. | ||
Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585–4593. | ||
Lin JJ, Fairclough S, Nagy R, et al. Identification of on-target mechanisms or resistance to EGFR inhibitors usinf cfDNA next-generation sequencing. J Thoracic Oncol. 2017;12(Suppl 1):S1255–S1256. | ||
Li L, Wang H, Li C, et al. Transformation to small-cell carcinoma as an acquired resistance mechanism to AZD9291: a case report. Oncotarget. 2017;8:18609–18614. | ||
Kim TM, Song A, Kim DW, et al. Mechanisms of acquired resistance to AZD9291: a mutation-selective, irreversible EGFR inhibitor. J Thorac Oncol. 2015;10:1736–1744. | ||
Ham JS, Kim S, Kim HK, et al. Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J Thorac Oncol. 2016;11:e1–e4. | ||
Planchard D, Loriot Y, Andre F, et al. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann Oncol. 2015;26:2073–2078. | ||
Ho CC, Liao WY, Lin CA, et al. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J Thorac Oncol. 2017;12:567–572. | ||
Ou SH, Agarwal N, Ali SM. High MET amplification level as a resistance mechanism to osimertinib (AZD9291) in a patient that symptomatically responded to crizotinib treatment post-osimertinib progression. Lung Cancer. 2016;98:59–61. | ||
Shi P, Oh YT, Zhang G, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016;380:494–504. | ||
Ortiz-Cuaran S, Scheffler M, Plenker D, et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin Cancer Res. 2016;22:4837–4847. | ||
Ichihara E, Westover D, Meador CB, et al. SFK/FAK signaling attenuates osimertinib efficacy in both drug-sensitive and drug-resistant models of EGFR-mutant lung cancer. Cancer Res. 2017;77:2990–3000. |
Supplementary material
 | Table S1 Complete list of genomic alterations detected in concurrent tissue and blood analyses at the time of progression on osimertinib in a patient with EGFR L858R NSCLC Notes: Note that MAF for tissue and ctDNA cannot be directly compared. For complete gene panels for tissue and ctDNA testing, please refer to Foundation Medicine technical specifications. Abbreviations: ctDNA, circulating tumor DNA; del, deletion; EGFR, epidermal growth factor receptor; MAF, mutant allele frequency; N/A, not applicable; ND, not detected; NSCLC, non-small-cell lung cancer. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.