Back to Journals » Drug Design, Development and Therapy » Volume 20
Circulating Butyrate Attenuates Cetuximab Efficacy in Colorectal Cancer Through EGFR and AMPK–Wip1 Signaling
Authors Zhang J, Zhang M ![]() , Wu X, Jing H, Li P, Wang W, Guo X, Zhao Z, Zhu S, Wang Y
, Wu X, Jing H, Li P, Wang W, Guo X, Zhao Z, Zhu S, Wang Y ![]()
Received 14 October 2025
Accepted for publication 15 January 2026
Published 22 January 2026 Volume 2026:20 574116
DOI https://doi.org/10.2147/DDDT.S574116
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leonidas Panos
Jiayao Zhang,1– 3 Mingqing Zhang,1– 3 Xiaojing Wu,1,2 Haoren Jing,1– 3 Peiran Li,4 Wei Wang,5 Xi Guo,5 Zhenying Zhao,6 Siwei Zhu,1,2 Yijia Wang2
1School of Medicine, Nankai University, Tianjin, People’s Republic of China; 2Tianjin Institute of Coloproctology, Tianjin Union Medical Center, The First Affiliated Hospital of Nankai University, Tianjin, People’s Republic of China; 3Department of Colorectal Surgery, Tianjin Union Medical Center, The First Affiliated Hospital of Nankai University, Tianjin, People’s Republic of China; 4Human Biology and Society, University of California, Los Angeles, CA, USA; 5TEDA Institute of Biological Sciences and Biotechnology, Nankai University, Tianjin, People’s Republic of China; 6Department of Pharmacy, Tianjin Union Medical Center, The First Affiliated Hospital of Nankai University, Tianjin, People’s Republic of China
Correspondence: Siwei Zhu, School of Medicine, Nankai University, Tianjin, China; Tianjin Institute of Coloproctology, Tianjin Union Medical Center, The First Affiliated Hospital of Nankai University, Tianjin, People’s Republic of China, Email [email protected] Yijia Wang, Tianjin Institute of Coloproctology, Tianjin Union Medical Center, The First Affiliated Hospital of Nankai University, Tianjin, People’s Republic of China, Email [email protected]
Background: Cetuximab is an approved therapy for metastatic colorectal cancer (CRC) with wild-type RAS and BRAF; however, additional resistance mechanisms beyond genetic mutations remain poorly understood. Butyrate, a key metabolite produced by the gut microbiome and present in the circulatory system, has been reported to supply cellular energy and modulate the epidermal growth factor receptor (EGFR) downstream signaling pathway. However, whether butyrate affects the resistance to cetuximab is still unknown.
Methods: In this work, Cell Counting Kit-8 (CCK-8) and colony formation assays were used to evaluate the efficacy of cetuximab. Glycolysis/oxidative phosphorylation (OXPHOS) Assay Kit was applied to assess metabolic activity. Human Phospho-Kinase Array and RNA sequencing were employed to screen targets of butyrate. Overexpression plasmids and short hairpin RNAs (shRNAs) targeting these molecules were transfected into cells for further validation. Subcutaneous tumor and pulmonary metastasis models were used for in vivo studies.
Results: The findings showed that physiological concentrations of butyrate increased cetuximab resistance in KRAS wild-type cells only. Further investigation found that butyrate upregulated EGFR signaling through facilitating the binding reaction between epidermal growth factor (EGF) and EGFR. In parallel, butyrate activated AMP-activated protein kinase (AMPK)–wild-type p53-induced phosphatase 1 (Wip1) signaling, leading to suppression of p53 and p38 mitogen-activated protein kinase (p38 MAPK)-mediated pro-apoptotic signaling. These two mechanisms are the reason that butyrate attenuates the efficacy of cetuximab. Results of subcutaneous tumor and pulmonary metastasis models exhibited a similar conclusion to in vitro experiments.
Conclusion: Butyrate reduces cetuximab efficacy in KRAS wild-type colorectal cancer through EGFR and AMPK–Wip1 signaling, and may represent a candidate predictive biomarker for treatment response.
Keywords: cetuximab resistance, butyrate, colorectal cancer, EGFR signaling, AMPK/wip1 pathway
Introduction
Cetuximab (CTX) is a human/mouse chimeric IgG1 monoclonal antibody that targets the extracellular domain of the epidermal growth factor receptor (EGFR), competitively blocking ligand binding.1,2 It is approved for the treatment of colorectal cancer (CRC) with wild-type RAS. However, resistance frequently emerges even in patients who meet this genotypic criterion.3 Therefore, it is not accurate to use genotype alone to predict the therapeutic effect of CTX; other factors need to be referenced to realize precision medicine for individuals. For example, tumors with higher levels of p38 mitogen-activated protein kinase (p38) were more sensitive to CTX.4 In addition to markers expressed in tumor cells themselves, the tumor microenvironment is another important factor related to CTX resistance.
Gut microbiota composition and microbiota-derived metabolites are important components of the CRC microenvironment. Substantial inter-individual variation in gut microbiota composition has been linked to colorectal carcinogenesis.5,6 Drug resistance is also affected by gut microbiota. For example, Fusobacterium nucleatum was reported to promote chemoresistance to CRC by modulating autophagy.7 Metabolites are the main substances involved in the interaction between the bacterium and the host. Short-chain fatty acids (SCFAs) are key gut microbiota–derived metabolites produced via dietary fiber fermentation8 and are highly concentrated in the colon (~70–140 mM proximally; ~20–70 mM distally).
As a major component of SCFAs, butyrate not only affects the intestinal environment but also provides energy for cells via absorption by colonocytes to enter the tricarboxylic acid (TCA) cycle. The fraction of butyrate that is not metabolized by colonocytes is transported into the portal circulation and subsequently metabolized by the liver. About 2% (0–20 μM) butyrate enters peripheral circulation.9–12 Although only a small amount of butyrate enters the circulation, it may still exert effects beyond the gut, including at metastatic sites. In CRC mouse models, butyrate has been reported to reduce liver metastasis, potentially through immune modulation and HDAC inhibition.13,14 Importantly, sodium butyrate (NaBu), commonly used as a butyrate donor and histone deacetylase (HDAC) inhibitor,15 has been reported to modulate EGFR downstream signaling in multiple experimental settings.16–18 Given that CTX exerts its antitumor activity by blocking EGFR, these findings raise the possibility that butyrate may modulate response to CTX. However, these observations were largely obtained at supraphysiological, millimolar concentrations, which are far higher than physiological circulating levels and may introduce cytotoxicity or off-target effects.19 Therefore, whether physiological circulating butyrate modulates CTX efficacy remains unknown.
In this study, we integrated in vitro experiments, mouse models, and exploratory clinical analyses to test whether circulating-level butyrate modulates CTX efficacy in CRC and to elucidate the underlying EGFR-related mechanisms.
Materials and Methods
Cell Culture
Human colorectal cancer cell lines (Caco2, Sw48, HCT116, and LoVo) were obtained from the Shanghai Institutes for Biological Sciences (Chinese Academy of Sciences, Shanghai, China). Cells were cultured in their recommended media (minimum essential medium (MEM) with 20% fetal bovine serum (FBS) for Caco2; Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS for Sw48; RPMI-1640 medium with 10% FBS for HCT116 and LoVo; all from Gibco, Thermo Fisher Scientific) supplemented with 1% penicillin–streptomycin. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cell culture conditions were selected according to the supplier’s recommendations and commonly used protocols for these colorectal cancer cell lines.20–22 To exclude potential confounding factors, we verified that baseline butyrate concentrations in the culture medium were approximately two orders of magnitude lower than reported circulating plasma levels in patients 9–12 (Figure S1A), indicating negligible background interference. Furthermore, NaBu supplementation did not significantly alter the pH of the culture medium (Figure S1B).
Cell Proliferation Assays
Cell viability was measured using the Cell Counting Kit-8 (CCK-8; Yeasen, Shanghai, China). Colony formation assays were performed in 6-well plates, with colonies fixed in methanol and stained with crystal violet after 2 weeks.
Plasmid Transfection
Cells were transfected with short hairpin RNA (shRNA) or overexpression plasmids using Lipofectamine™ 3000 (Invitrogen, USA) following the manufacturer’s instructions. Cells were collected 48 h post-transfection for subsequent assays.
Western Blotting
Proteins were extracted with radio-immunoprecipitation assay (RIPA) buffer containing protease inhibitors, quantified by bicinchoninic acid (BCA) assay, and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were blocked with 5% milk and incubated with primary antibodies overnight at 4 °C, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Signals were visualized with enhanced chemiluminescence (ECL) (Solarbio, Beijing, China). Antibodies are listed in Supplementary Table 1.
Human Phospho-Kinase Array
Protein phosphorylation profiling was performed using the Proteome Profiler™ Array Kit (ARY003B, R&D Systems) according to the manufacturer’s protocol.
Microscale Thermophoresis (MST)
MST measurements were performed using a Monolith NT.115 instrument (NanoTemper Technologies, Germany). Recombinant human EGFR protein (TargetMol) was fluorescently labeled with Alexa Fluor™ 647 NHS Ester (A37573, Invitrogen) according to the manufacturer’s instructions. Excess unbound dye was removed by buffer exchange prior to measurements. Binding of labeled EGFR to EGF was assessed in the presence or absence of 0.02 mM sodium butyrate (NaBu). Control measurements without EGF were included to exclude non-specific binding. Each condition was measured in at least three independent experiments. Dissociation constants (Kd) were calculated using MO. Affinity Analysis software (NanoTemper Technologies).
RNA Sequencing and Bioinformatics
RNA was extracted using TRIzol (Invitrogen). Libraries were prepared with the NEBNext® Ultra™ II Kit and sequenced on the Illumina NovaSeq 6000 platform. Reads were processed with fastp, aligned to GRCh38 with HISAT2, and quantified with featureCounts. Differential expression analysis was performed using DESeq2. Genes with |log2 fold change (log2FC)| ≥ 0.5 and false discovery rate (FDR) < 0.05 were considered significant. Functional enrichment was analyzed using clusterProfiler.
Animal Studies
Male BALB/c nude mice (4–6 weeks old, specific pathogen-free) were purchased from SPF Biotechnology (Beijing, China) and housed under specific pathogen-free conditions. After acclimatization, mice were allocated to either the subcutaneous xenograft model or the pulmonary metastasis model according to the experimental design. Within each model, mice were then randomly assigned to four treatment groups (n = 5 per group): control, sodium butyrate (NaBu), cetuximab (CTX), and NaBu + CTX, using a random number–based randomization approach prior to any treatment administration. Following group assignment, sodium butyrate was administered via drinking water, with concentrations gradually increased from 1 to 10 mmol/L, initiated prior to tumor inoculation and maintained throughout the experiment. Cetuximab (CTX; 0.5 mg/mouse) was administered intraperitoneally every 3 days, starting at the end of week 2 after tumor inoculation and continuing through the end of week 6. Investigators were blinded to group allocation during tumor measurement. Group sizes were determined based on feasibility and commonly used designs for exploratory mechanistic studies in colorectal cancer models, rather than on a priori power calculations.
For subcutaneous xenograft models, 1 × 107 Caco2 or Sw48 cells were injected into the flank of each mouse. Tumor volume of subcutaneous xenografts was measured every 3 days. Mice were euthanized at the end of week 6 after tumor implantation, and tumors were harvested for histological and immunohistochemical analyses.
For pulmonary metastasis models, 2 × 105 cells were injected via the tail vein. Mice were monitored for survival for up to 60 days after tumor inoculation. Animals that reached predefined humane endpoints were euthanized and recorded as events for survival analysis. Lung metastatic burden was assessed based on visible nodules and histological evaluation at the time of death or at the end of follow-up.
Clinical Samples and Study Design
This study included an exploratory, observational analysis of patients with colorectal cancer treated at a single center. Clinical samples were collected according to availability. Among these patients, those who received cetuximab were analyzed for treatment response in relation to circulating butyrate levels, while the overall cohort was used for immunohistochemical evaluation of molecular markers. Given the exploratory nature of the clinical component, the sample size was determined by patient availability rather than by formal power calculation.
Histology and Immunohistochemistry (IHC)
Paraffin-embedded tissues were subjected to hematoxylin and eosin (H&E) staining. IHC was performed after antigen retrieval, overnight incubation with primary antibodies, and HRP/3,3′-diaminobenzidine (DAB) detection. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays were carried out using the CF488 kit (Servicebio, Wuhan, China).
Butyrate Measurement
Butyric acid concentrations in human plasma, mouse serum, and cell culture media were quantified using a commercial ELISA kit (Elk Biotechnology, ELK8174), according to the manufacturer’s instructions.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 8.0. Data normality was assessed using the Shapiro–Wilk test. Data are expressed as mean ± standard deviation (SD) unless otherwise indicated. Comparisons between two groups were performed with two-tailed Student’s t-test or Mann–Whitney U-test; multiple-group comparisons were performed with one-way analysis of variance (ANOVA) (Tukey’s post hoc test) or Kruskal–Wallis (Dunn’s post hoc test). Two-way ANOVA was used for dose–response analyses. A two-sided p < 0.05 was considered statistically significant. For longitudinal tumor volume data, mixed-effects models were used to account for repeated measurements within individual mice.
Results
Butyrate Preferentially Promotes KRAS Wild-Type Colorectal Cancer Cell Proliferation and Reduces Cetuximab Efficacy
Circulating butyrate concentrations did not differ significantly among healthy controls, untreated CRC patients, and CTX-treated CRC patients (Figure S1C), and overall levels fell within previously reported ranges.9–12 In an exploratory cohort of 22 patients receiving cetuximab, circulating butyrate levels were significantly higher in those with progressive disease than stable disease (Figure S1D, p=0.01). When dichotomized by the median, a greater proportion of patients with progressive disease was observed in the high-butyrate group (Fisher’s exact p=0.03, Figure S1E). To model patient-relevant exposure, NaBu was applied to KRAS wild-type (Caco2, Sw48) and KRAS-mutant (HCT116, LoVo) colorectal cancer cell lines. At low concentrations, NaBu promoted the proliferation of KRAS wild-type cells in a dose-dependent manner, with minimal effects on KRAS-mutant cells (Figure 1A–C). At higher concentrations (>0.32 mM), NaBu exerted cytotoxic effects on both KRAS wild-type and KRAS-mutant cells.
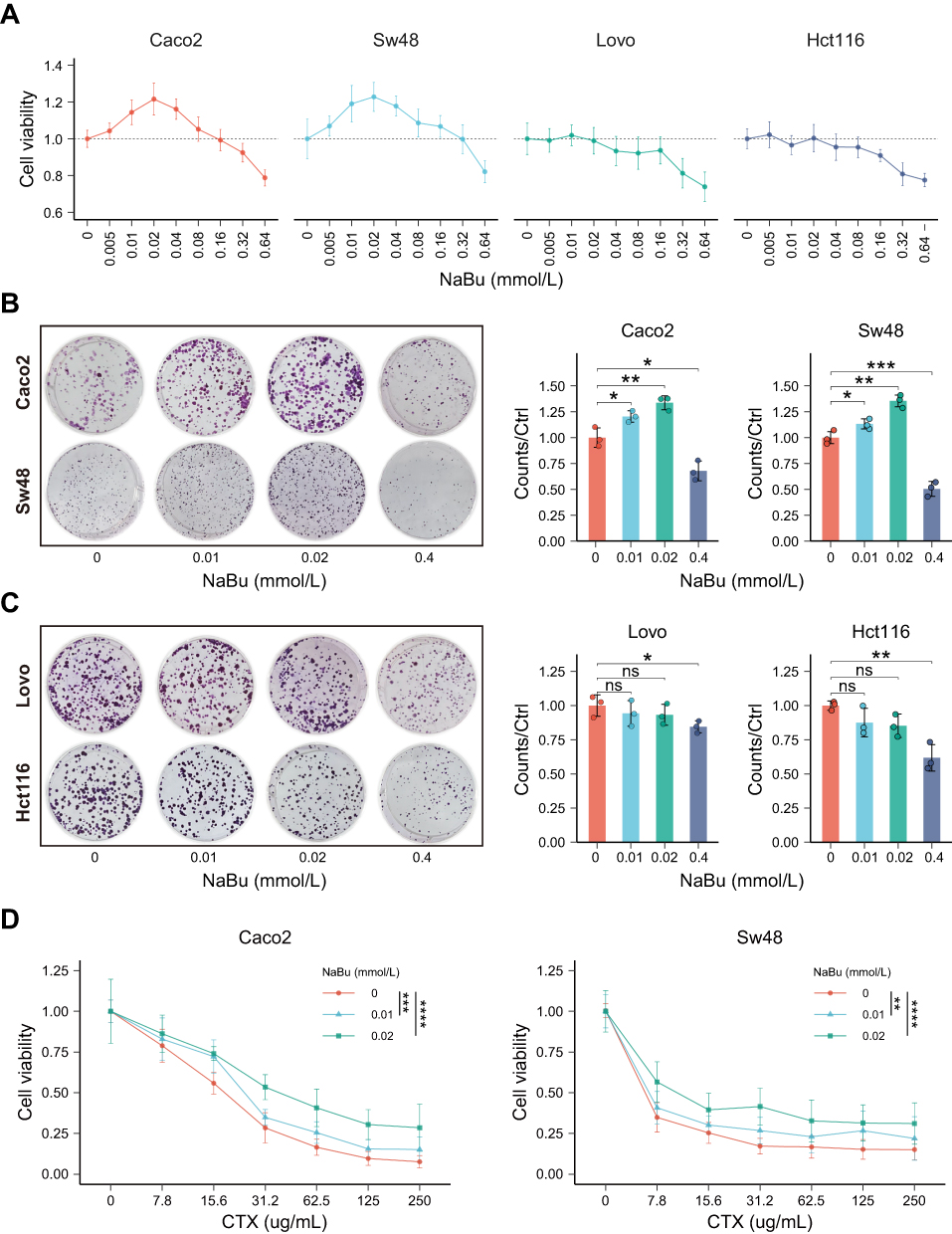 |
Figure 1 NaBu promotes proliferation of KRAS-WT CRC cells and reduces CTX sensitivity. (A) CCK-8 assay of four CRC cell lines (Caco2, Sw48, HCT116, LoVo) treated with increasing concentrations of NaBu. (B and C) Colony formation assays showing enhanced growth of KRAS-WT cells (B: Caco2, Sw48) but minimal effects on KRAS-mutant cells (C: HCT116, LoVo). (D) Combination treatment of NaBu (0.02 mM) and CTX in KRAS-WT cells showing reduced CTX sensitivity. Statistical significance: ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. |
We next compared glycolytic activity between KRAS–wild-type and KRAS-mutant CRC cells. Under baseline conditions, KRAS–wild-type cells exhibited significantly lower glycolysis than KRAS-mutant cells, as indicated by the proportion of glycolytic ATP (Figure S1F).
To assess the combined effect of NaBu and CTX, KRAS wild-type cells were co-treated with 0.02 mM NaBu and increasing CTX doses. NaBu consistently shifted the CTX dose–response curve to the right across the tested range (0–250 μg/mL), resulting in higher viability at matched CTX doses (Figure 1D).
Butyrate Upregulates the EGFR/AKT/ERK Signaling Pathway
To examine the effects of NaBu on EGFR signaling, a Human Phospho-Kinase Array was used to screen phosphorylation changes in downstream effectors. Increased phosphorylation of EGFR, AKT, and ERK1/2 was observed in NaBu-treated Sw48 cells (Figure 2A and B).
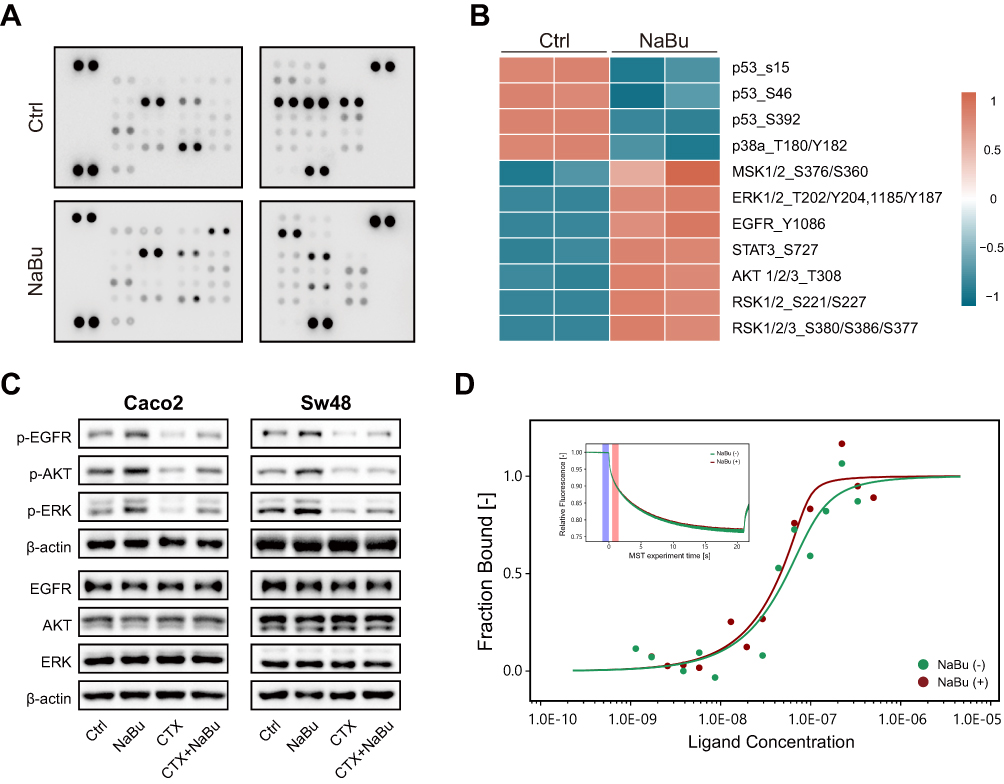 |
Figure 2 NaBu enhances EGFR signaling in KRAS-WT CRC cells. (A) Human phospho-kinase array of Sw48 cells treated with NaBu, showing increased phosphorylation of EGFR, AKT, and ERK1/2, and decreased phosphorylation of p53 and p38. (B) Heatmap representation of differential phosphorylation. (C) Western blot validation of p-EGFR, p-AKT, and p-ERK1/2 in KRAS-WT cells (Caco2, Sw48) treated with NaBu ± CTX. (D) Microscale thermophoresis (MST) analysis of EGF–EGFR binding in the presence or absence of NaBu. Enhanced binding in the presence of NaBu is reflected by a leftward shift of the binding curve and a reduced apparent dissociation constant. The inset shows representative MST time traces used for curve fitting and binding analysis. NaBu (+), presence of sodium butyrate; NaBu (−), absence of sodium butyrate. |
Western blotting further validated that NaBu enhanced phosphorylation of EGFR, AKT, and ERK1/2 in KRAS wild-type cell lines (Caco2 and Sw48), irrespective of CTX treatment (Figure 2C).
Using microscale thermophoresis, we found that Kd of the EGF–EGFR interaction was reduced in the presence of NaBu, indicating strengthened ligand binding (Figure 2D).
Butyrate Disturbed the Efficacy of CTX, Partially Dependent on the MEK/ERK Signaling Pathway
MEK1/2 knockdown was achieved in Caco2 and Sw48 cells using shRNA (Figure 3A). In cell proliferation assays (CCK-8 and colony formation), NaBu did not promote cell growth following MEK/ERK silencing. In contrast, NaBu continued to attenuate the cytotoxic effects of CTX, although to a lesser extent (Figure 3B and C).
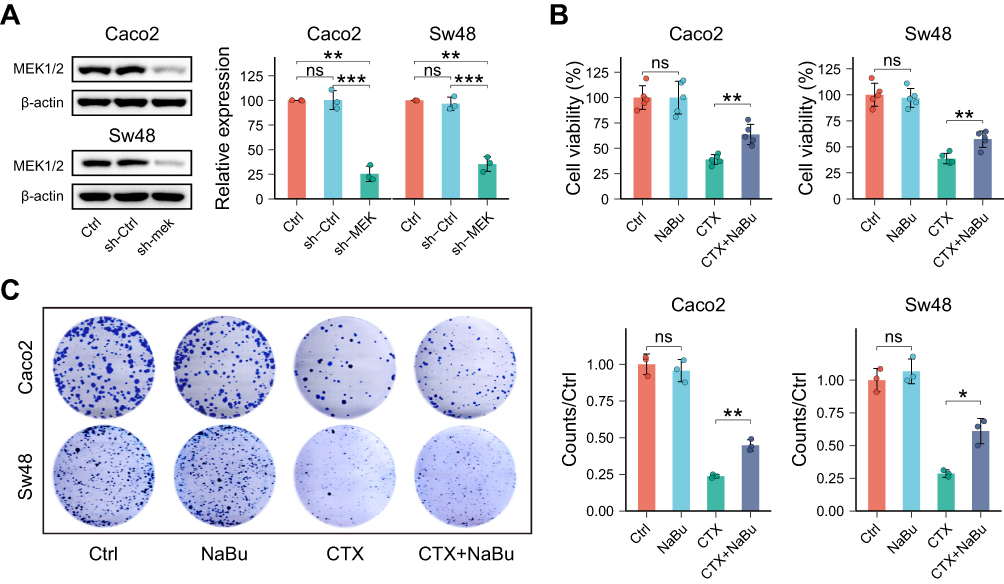 |
Figure 3 NaBu’s proliferative effect requires MEK/ERK signaling, but partial CTX resistance persists. (A) Validation of MEK1/2 knockdown by shRNA in Caco2 and Sw48 cells. (B) CCK-8 assay showing that NaBu failed to promote cell growth after MEK1/2 silencing, whereas its ability to attenuate CTX cytotoxicity was only partially reduced. (C) Colony formation assay confirming that MEK1/2 knockdown abolished NaBu-induced proliferation, but NaBu still impaired CTX efficacy to some extent. Statistical significance: ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001. |
Butyrate Upregulates Glycolysis and Activates the AMPK–Wip1 Axis
RNA sequencing comparing CTX+NaBu with CTX alone revealed enrichment of pathways related to glycolysis and oxidative phosphorylation in both Sw48 and Caco2 cells (Figures 4A, B and S2A–B). Functional assays showed a dose-dependent shift toward glycolytic ATP production, with a corresponding relative decrease in OXPHOS contribution (Figure 4C). Western blot analysis showed increased phosphorylation of AMPK after NaBu exposure (Figure 4D). NaBu treatment also increased Wip1 protein levels and reduced phosphorylation of p53 and p38 (Figure 4D).
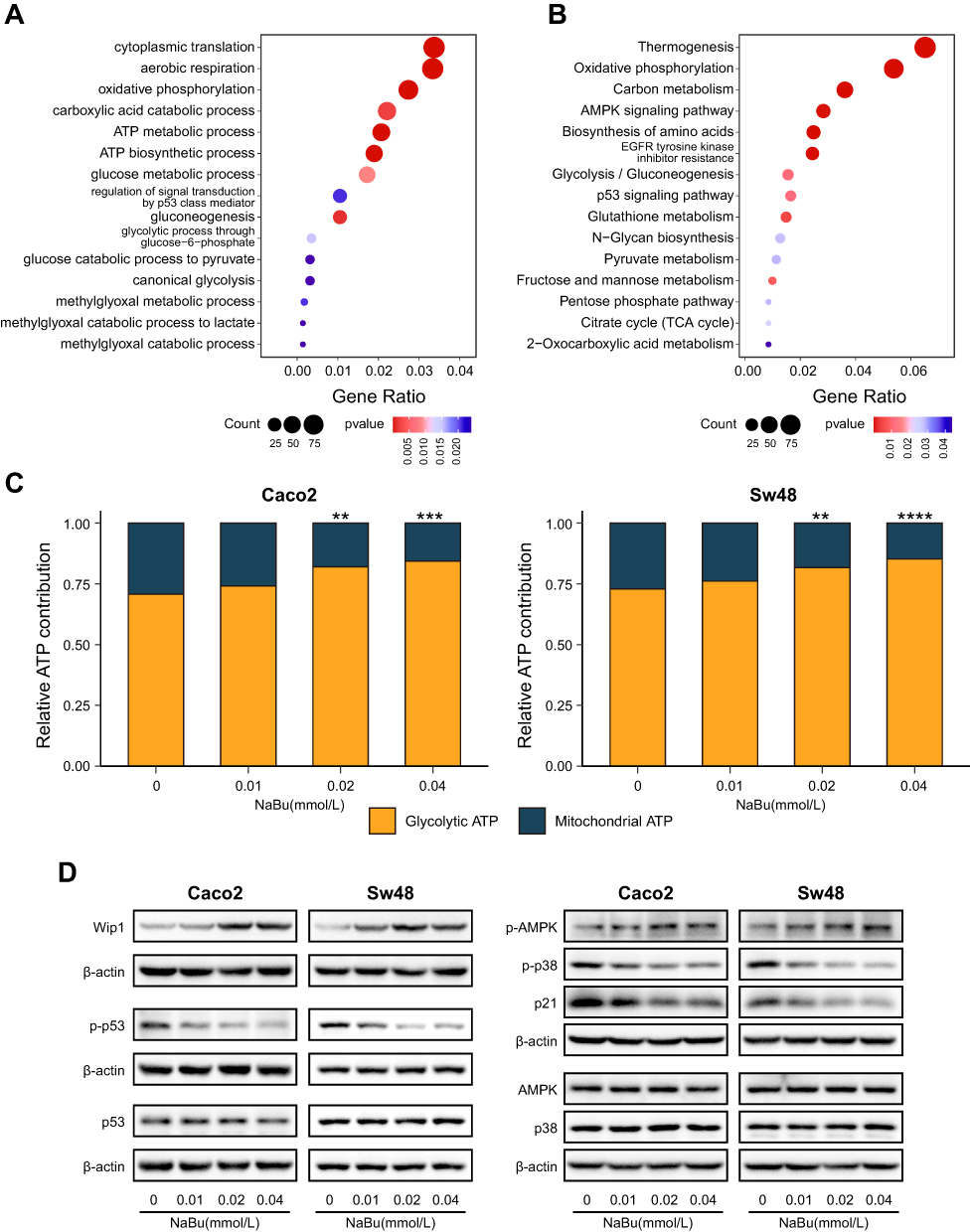 |
Figure 4 NaBu enhances glycolysis and activates the AMPK–Wip1 signaling. (A and B) GO biological process (A) and KEGG pathway (B) enrichment analyses of RNA sequencing (CTX+NaBu vs CTX, Sw48) showing significant enrichment of pathways related to energy metabolism, AMPK signaling, and p53 regulation. (C) Functional ATP assay in Caco2 and Sw48 cells under CTX treatment, showing a dose-dependent shift of ATP production from oxidative phosphorylation toward glycolysis after NaBu exposure. (D) Western blot analysis showing that, under CTX treatment, additional NaBu exposure increased phosphorylation of AMPK and elevated Wip1 protein, while reducing phosphorylation of p53 and p38 in Caco2 and Sw48 cells. Statistical significance: ns, not significant; **p < 0.01; ***p < 0.001; ****p < 0.0001. |
Wip1 knockdown enhanced CTX sensitivity, whereas Wip1 overexpression conferred resistance (Figure 5A–C). In parallel, Wip1 overexpression markedly reduced p-p53 and p-p38, while Wip1 silencing abolished NaBu’s suppressive effect on these proteins (Figure 5D and E).
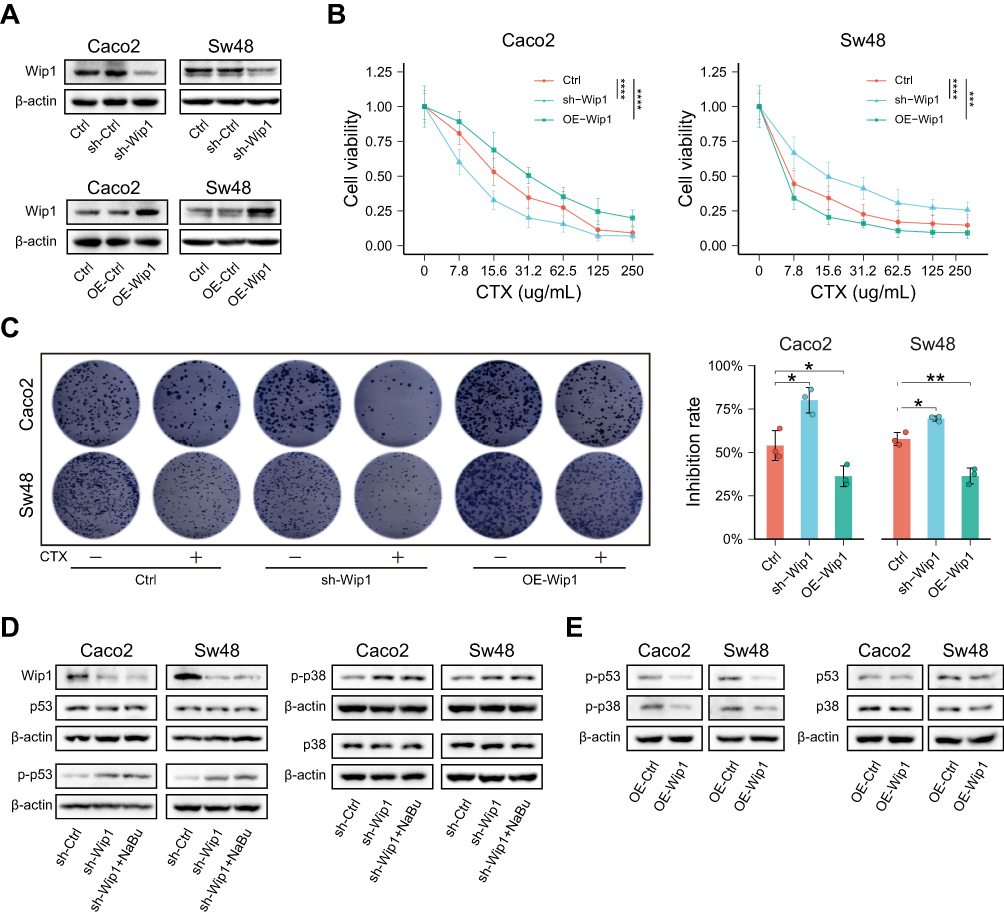 |
Figure 5 Wip1 modulates CTX sensitivity by suppressing p53 and p38 signaling. (A) Validation of Wip1 knockdown (shRNA) and overexpression (OE) in Caco2 and Sw48 cells (Western blot). (B and C) Functional assays showing that Wip1 knockdown enhanced CTX sensitivity, whereas Wip1 overexpression conferred resistance, as measured by CCK-8 (B) and colony formation (C) assays. (D) Western blot showing that NaBu failed to suppress p53 and p38 phosphorylation in Wip1-silenced cells. (E) Wip1 overexpression reduced p-p53 and p-p38 even without NaBu treatment. Statistical significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. +, CTX present; −, CTX absent. |
Butyrate Increased the Resistance of CRC Cell Lines to CTX in vivo
Two KRAS wild-type CRC cell lines, Caco2 and Sw48, were used to establish subcutaneous xenograft and pulmonary metastasis models in BALB/c nude mice. When NaBu was administered via drinking water, serum butyrate levels were significantly elevated compared with controls and were not affected by tumor model type or CTX treatment (Figure 6A).
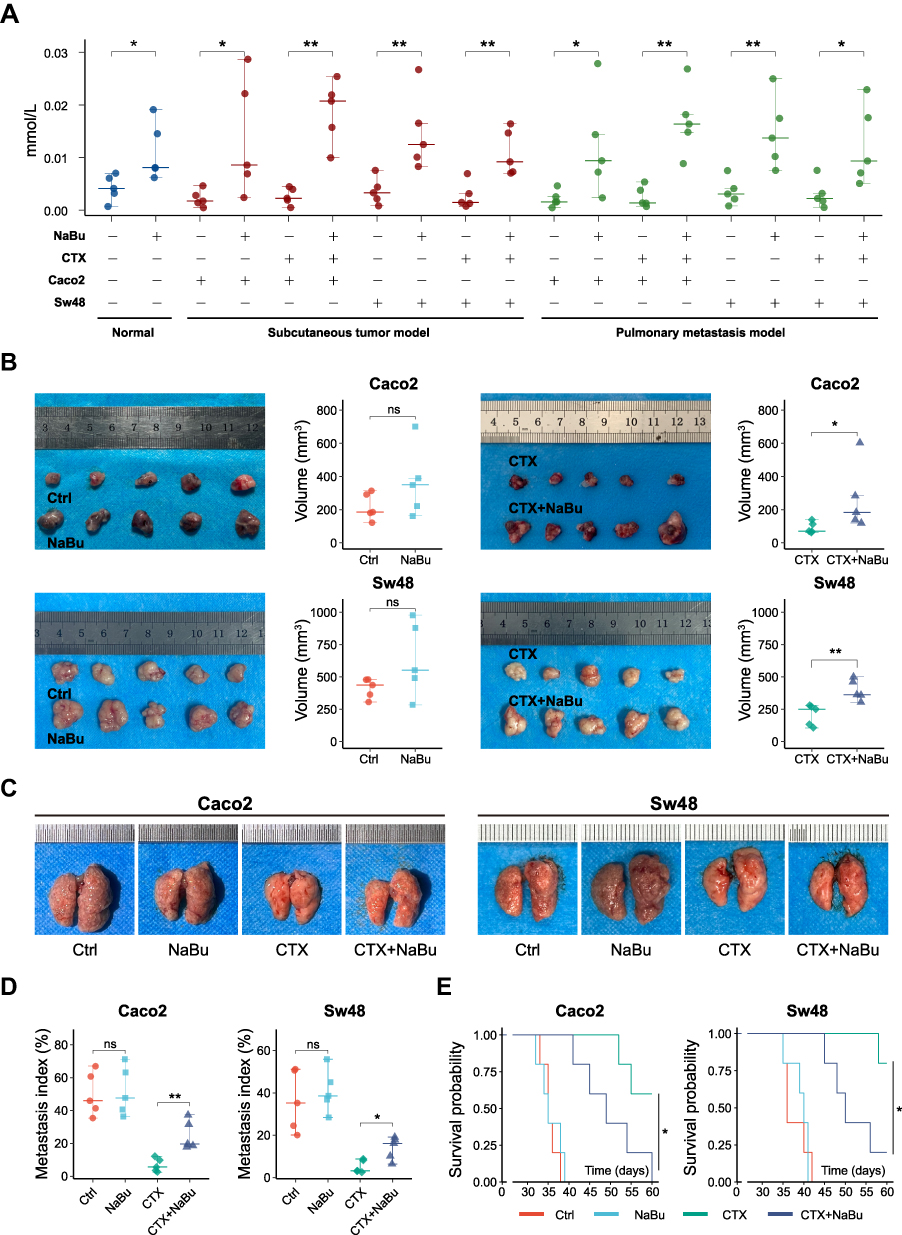 |
Figure 6 NaBu reduces the antitumor efficacy of CTX in vivo. (A) Serum butyrate levels measured by ELISA in normal mice and in Caco2 and Sw48 xenograft models (subcutaneous and pulmonary metastasis) under the indicated treatments. (B) Subcutaneous xenograft growth of Caco2 and Sw48 cells. (C) Representative lung images from pulmonary metastasis models. (D) Quantification of lung metastatic burden (metastasis index based on nodule counts). (E) Kaplan–Meier survival curves of pulmonary metastasis models. Statistical significance: ns, not significant; *p < 0.05; **p < 0.01. |
In both models, NaBu supplementation attenuated the antitumor efficacy of CTX. Tumor volume in the subcutaneous model (Figures 6B and S3A) and the pulmonary metastasis index, calculated based on visible nodules (Figures 6C, 6D and S3B), were higher in NaBu-supplemented mice compared with controls under CTX treatment. Longitudinal tumor growth curves are provided in Supplementary Figure S3A and were analyzed using a mixed-effects model to account for repeated measurements. In the absence of CTX, NaBu slightly promoted tumor growth and metastasis, although these differences did not reach statistical significance. CTX treatment significantly suppressed tumor growth regardless of NaBu administration.
Survival analysis demonstrated that CTX markedly prolonged survival compared with the control group, whereas NaBu shortened survival only in the context of CTX treatment (Figure 6E). TUNEL and Ki67 staining indicated that CTX induced apoptosis and inhibited proliferation, while co-treatment with NaBu reduced these effects (Figures 7A and S4A–B). H&E staining of lung metastases further confirmed that CTX preserved alveolar architecture more effectively than the NaBu+CTX combination group (Figures 7B and S5A).
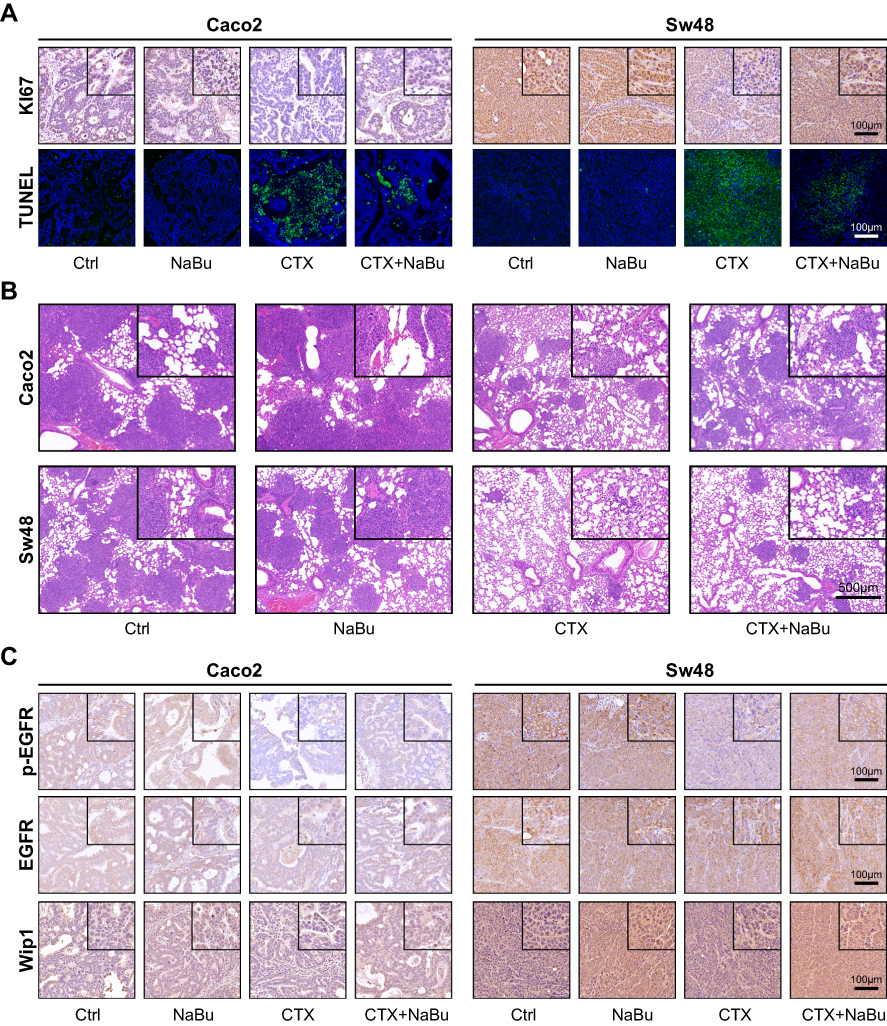 |
Figure 7 Histological and immunohistochemical analyses of in vivo models. (A) TUNEL and Ki67 staining of subcutaneous tumor sections from Caco2 and Sw48 xenografts. CTX markedly induced apoptosis (TUNEL) and reduced proliferation (Ki67), whereas NaBu co-treatment attenuated these effects. Scale bars = 100 μm. (B) H&E staining of lung sections from pulmonary metastasis models. CTX preserved alveolar architecture and reduced tumor burden, while NaBu co-treatment diminished these protective effects. Scale bars = 500 μm. (C) Immunohistochemical staining of subcutaneous tumors showing that NaBu increased p-EGFR and Wip1 expression, consistent with in vitro findings, whereas total EGFR levels were relatively unchanged. Scale bars = 100 μm. The square boxes in the upper-right corners indicate the regions shown at higher magnification in the insets. |
Further immunohistochemical analysis indicated that NaBu increased the expression of phosphorylated EGFR (Figures 7C and S6A–B) and Wip1 (Figures 7C and S7A), regardless of CTX treatment.
Human Validation of p-EGFR and Wip1 Expression
To assess whether the experimental findings extend to the clinical setting, we analyzed tumor samples from 50 CRC patients. Immunohistochemical staining revealed stronger p-EGFR and Wip1 signals in tumors from patients with higher circulating butyrate (Figure 8A). Quantitative analysis further demonstrated that circulating butyrate levels were positively correlated with H-scores of p-EGFR (R = 0.52, p < 0.05) and Wip1 (R = 0.60, p < 0.05) (Figure 8B).
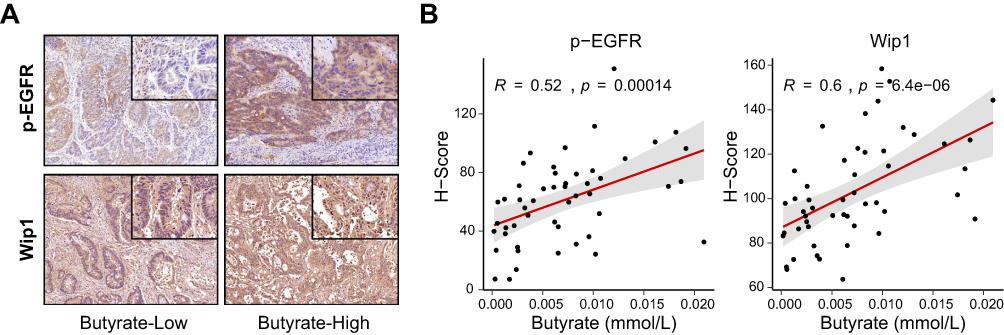 |
Figure 8 Human validation of p-EGFR and Wip1 expression in colorectal cancer. (A) Representative immunohistochemical staining of p-EGFR and Wip1 in CRC patient samples stratified by circulating butyrate levels (low vs high). Insets show higher magnification of tumor regions. The square boxes in the upper-right corners indicate the regions shown at higher magnification in the insets. (B) Correlation analysis of circulating butyrate concentration with H-scores of p-EGFR (left) and Wip1 (right) in 50 CRC patients, showing significant positive associations (Pearson’s correlation). |
Discussion
Cetuximab remains an established targeted therapy for patients with KRAS wild-type colorectal cancer; however, substantial inter-individual variability in therapeutic response persists despite appropriate molecular selection. In this study, we combined patient-derived clinical serum analyses with in vitro and in vivo models and found that circulating butyrate attenuates cetuximab-induced growth inhibition and apoptosis in KRAS wild-type colorectal cancer. Our data indicate the involvement of sustained EGFR–MEK/ERK activity together with activation of the AMPK–Wip1–p53/p38 axis in shaping treatment responsiveness.
Butyrate can be oxidized as a carbon source through acetyl-CoA entry into the TCA cycle in intestinal epithelial cells.23 In tumor cells, however, butyrate utilization is closely associated with the Warburg effect, such that glycolysis-dominant states favor intracellular accumulation and altered functional consequences.24 In our study, colorectal cancer cells displayed distinct proliferative responses to butyrate depending on KRAS status: KRAS wild-type cells exhibited increased proliferation under physiologic butyrate concentrations, whereas KRAS-mutant cells did not show a significant proliferative response under the same conditions. This divergence is consistent with differences in cellular energy metabolism. Mutant KRAS is known to enhance aerobic glycolysis,25 which limits mitochondrial oxidative metabolism and favors intracellular accumulation of butyrate, more likely to act through HDAC–related mechanisms.24,26 In KRAS wild-type cells, efficient entry of butyrate into the TCA cycle may limit its intracellular accumulation and maintain it at relatively low levels. Notably, when sodium butyrate concentrations exceeded 0.32 mM, growth inhibition was observed in both KRAS-mutant and KRAS wild-type cells, suggesting saturation of metabolic utilization. We speculate that this low intracellular butyrate state may be permissive for the proliferative response observed under physiologic butyrate concentrations, although additional mechanisms are likely involved. Importantly, proliferation and resistance to CTX are not always coupled.27 Therefore, additional mechanisms involving butyrate may affect CTX resistance. To investigate the relation between butyrate and CTX efficacy, we first assessed whether CTX treatment or the presence of CRC alters blood butyrate concentration. Although CRC development28 or drug treatment29 can alter gut microbial composition, our results showed that blood butyrate levels were not affected by either CRC or CTX treatment. Prior studies have similarly reported that blood butyrate concentrations remain relatively unaffected across different disease states, such as multiple sclerosis.10 More recent work indicates that inter-individual variability, shaped by genetic and microbiome backgrounds, is the predominant determinant of SCFA levels in blood.30,31 In fecal analyses, butyrate concentrations also show substantial variation among individuals.32 Taken together, these findings suggest that circulating butyrate levels are largely independent of cancer or treatment status, but instead reflect individual differences. This may explain why patients with higher butyrate levels constitute a subgroup more prone to poor CTX responses. Indeed, in our exploratory clinical cohort, higher circulating butyrate was significantly associated with progressive disease, and high-butyrate patients exhibited stronger p-EGFR and Wip1 expression by immunohistochemistry, in line with our experimental results.
Mechanistically, our study uncovered two major pathways through which butyrate attenuates CTX efficacy. First, butyrate enhanced EGF–EGFR binding, increased EGFR phosphorylation, and sustained MEK/ERK signaling despite CTX treatment. Because CTX exerts its antitumor activity by competitively blocking ligand binding to EGFR and suppressing downstream MEK/ERK signaling,2 the NaBu-induced increase in EGF–EGFR affinity provides a plausible explanation for the persistence of EGFR activation and compromised pathway suppression observed under CTX treatment.
Second, RNA sequencing and functional assays indicated that butyrate activated AMPK, which in turn upregulated Wip1. Multiple studies have shown that butyrate can activate AMPK across different systems, including hepatocytes and renal tissue.33,34 Given that AMPK promotes glycolysis,35 our results suggest that NaBu enhances glycolytic flux through AMPK activation. Beyond metabolic regulation, AMPK activation has also been linked to stress-response regulatory networks and DNA damage response, in which Wip1 (PPM1D), a protein phosphatase 2C (PP2C)-family phosphatase, functions as a critical phosphatase dampening p53 and p38 signaling.36–38 Although Wip1 is transcriptionally induced by p53, its expression does not strictly correlate with p53 mutation status.39 While p53 upregulates Wip1, Wip1 in turn inhibits p53 signaling by dephosphorylating Ser15 on p53. Wild-type p53 increases the expression of its downstream target p21 and promotes apoptosis by regulating BCL-2 (B-cell lymphoma 2) and Bax (BCL-2–associated X protein). The level of BCL-2 family members plays a key role in apoptosis and influences the efficacy of EGFR TKIs.40 Clinical studies also showed that metastatic CRC with wild-type RAS/BRAF exhibits high resistance to CTX when p53 function is lost.41 Wip1 additionally inhibits p38, another positive regulator for CTX efficacy.4 We therefore hypothesized that the upregulation of AMPK/Wip1 by butyrate may represent an additional mechanism contributing to CTX resistance. Our own data support this model: NaBu increased phosphorylated AMPK and Wip1 protein levels, accompanied by reduced phosphorylation of p53 and p38. Functionally, Wip1 knockdown enhanced CTX sensitivity, whereas its overexpression conferred resistance. Moreover, clinical immunohistochemistry revealed that tumors from high-butyrate patients displayed higher Wip1 expression, consistent with its role in mediating CTX resistance. This dual role of AMPK—regulating both metabolic adaptation and apoptotic signaling via Wip1—may explain why butyrate reduces CTX efficacy beyond its proliferative effect. Taken together, these findings highlight AMPK–Wip1 signaling as an important secondary mechanism by which butyrate attenuates CTX efficacy, complementing the EGFR/MEK/ERK pathway.
Our in vivo models further corroborated these findings: NaBu reduced the therapeutic benefit of CTX in both subcutaneous xenografts and pulmonary metastasis, with diminished apoptosis and sustained proliferation observed histologically. Notably, NaBu alone did not markedly increase tumor burden, which may reflect immune suppression of tumor growth and the influence of hypoxic glycolysis in vivo.
In summary, our study demonstrates that butyrate, an important blood metabolite derived from the gut microbiome, attenuates the efficacy of CTX in CRC through dual mechanisms: enhancing EGFR activation and activating AMPK–Wip1 signaling to suppress p53/p38-mediated apoptosis. Complementary clinical observations in two patient cohorts—circulating butyrate associated with CTX resistance, and tumor immunohistochemistry showing higher p-EGFR and Wip1 expression in high-butyrate patients—support the translational relevance of our findings. A schematic summary of the proposed integrative mechanism is shown in Figure 9.
 |
Figure 9 Proposed model of butyrate-induced resistance to cetuximab in colorectal cancer. Arrows indicate proposed activation or positive regulation, whereas blunt-ended lines indicate inhibition. Upward arrows indicate increased activity. |
Nevertheless, this study has several limitations. First, the clinical association between circulating butyrate and cetuximab outcome was observed in a small, exploratory cohort and should be validated prospectively in larger, independent populations. Second, we did not collect information on gut microbiota composition, dietary intake, or antibiotic exposure, which may influence circulating butyrate levels. Third, our in vivo experiments were performed in immunodeficient mice, which may not fully capture immune-mediated components of cetuximab activity. In addition, systemic NaBu administration may not completely recapitulate physiological butyrate exposure, and HDAC-related off-target effects cannot be excluded. Finally, the clinical analyses focused mainly on KRAS/BRAF status; other genomic alterations relevant to EGFR signaling were not comprehensively assessed. Taken together, these limitations preclude recommending routine pre-treatment serum butyrate testing for all KRAS wild-type CRC patients at present. Future studies should standardize butyrate quantification, establish clinically actionable thresholds, and evaluate whether controlled modulation of systemic butyrate exposure (eg, dietary or microbiota-targeted interventions) can improve cetuximab efficacy while ensuring safety.
Conclusion
Our findings show that physiologically relevant concentrations of butyrate can weaken the therapeutic effect of cetuximab in KRAS wild-type colorectal cancer. This occurs through two interconnected mechanisms: maintaining EGFR–MEK/ERK signaling activity and activating the AMPK–Wip1 pathway, which together suppress p53/p38-mediated apoptosis. Consistent results from cell, animal, and patient studies suggest that circulating butyrate may influence individual treatment responses. These observations highlight the importance of considering metabolic and microbial factors when evaluating cetuximab efficacy and may provide a basis for developing new strategies to improve targeted therapy outcomes in colorectal cancer.
Abbreviations
AMPK, adenosine monophosphate-activated protein kinase; ANOVA, analysis of variance; BCL-2, B-cell lymphoma 2; Bax, BCL-2–associated X protein; CCK-8, Cell Counting Kit-8; CRC, colorectal cancer; CTX, cetuximab; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ELISA, enzyme-linked immunosorbent assay; ERK, extracellular signal-regulated kinase; H&E, hematoxylin and eosin; HDAC, histone deacetylase; HRP, horseradish peroxidase; IHC, immunohistochemistry; Kd, dissociation constant; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; MST, microscale thermophoresis; NaBu, sodium butyrate; OXPHOS, oxidative phosphorylation; p-EGFR, phosphorylated epidermal growth factor receptor; SCFAs, short-chain fatty acids; shRNA, short hairpin RNA; TCA, tricarboxylic acid; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; Wip1, wild-type p53-induced phosphatase 1.
Data Sharing Statement
All data are provided in the manuscript and Supplementary files or are available from the corresponding author, Yijia Wang, upon reasonable request.
Ethics Approval and Informed Consent
All protocols for animal experiments involving the use of mice were approved by the Animal Care and Use Committees of Nankai University, Tianjin, China (No.2025-SYDWLL-000265), and were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the National Institutes of Health. Human samples were collected with written informed consent from all participants. The use of human specimens was approved by the Ethics Committee of Tianjin Union Medical Center, Tianjin, China (No. GZR-2024-011), and conducted in accordance with the Declaration of Helsinki.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by the foundation of Tianjin Municipal Education Commission Research Plan Project Grant number 2024ZXZD015, and the foundation of Hebei Province Administration of Traditional Chinese Medicine Grant number T2025021, and Tianjin Key Medical Discipline Construction Project (Grant No. TJYXZDXK-3-019C), and the foundation of the committee on science and technology of Tianjin under Grant number 22JCYBJC00570.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11(9):777–16. doi:10.4161/cbt.11.9.15050
2. Liu YF, Feng ZQ, Chu TH, et al. Andrographolide sensitizes KRAS-mutant colorectal cancer cells to cetuximab by inhibiting the EGFR/AKT and PDGFRbeta/AKT signaling pathways. Phytomedicine. 2024;126:155462. doi:10.1016/j.phymed.2024.155462
3. Bridgewater JA, Pugh SA, Maishman T, et al. Systemic chemotherapy with or without cetuximab in patients with resectable colorectal liver metastasis (New EPOC): long-term results of a multicentre, randomised, controlled, phase 3 trial. Lancet Oncol. 2020;21(3):398–411. doi:10.1016/S1470-2045(19)30798-3
4. Yang J, Mo J, Dai J, et al. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021;12(11):1079. doi:10.1038/s41419-021-04367-3
5. Sarhadi V, Lahti L, Saberi F, et al. Gut microbiota and host gene mutations in colorectal cancer patients and controls of iranian and finnish origin. Anticancer Res. 2020;40(3):1325–1334. doi:10.21873/anticanres.14074
6. Wang Y, Wan X, Wu X, Zhang C, Liu J, Hou S. Eubacterium rectale contributes to colorectal cancer initiation via promoting colitis. Gut Pathog. 2021;13(1):2. doi:10.1186/s13099-020-00396-z
7. Yu T, Guo F, Yu Y, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170(3):548–563e16. doi:10.1016/j.cell.2017.07.008
8. Parada Venegas D, la Fuente MK D, Landskron G, et al. Short Chain Fatty Acids (SCFAs)-Mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277. doi:10.3389/fimmu.2019.00277
9. Majchrzak-Celinska A, Kleszcz R, Stasilowicz-Krzemien A, Cielecka-Piontek J. Sodium butyrate enhances curcuminoids permeability through the blood-brain barrier, restores wnt/beta-catenin pathway antagonists gene expression and reduces the viability of glioblastoma cells. Int J Mol Sci. 2021;22(20). doi:10.3390/ijms222011285
10. Trend S, Leffler J, Jones AP, et al. Associations of serum short-chain fatty acids with circulating immune cells and serum biomarkers in patients with multiple sclerosis. Sci Rep. 2021;11(1):5244. doi:10.1038/s41598-021-84881-8
11. Saresella M, Marventano I, Barone M, et al. Alterations in circulating fatty acid are associated with gut microbiota dysbiosis and inflammation in multiple sclerosis. Front Immunol. 2020;11:1390. doi:10.3389/fimmu.2020.01390
12. Jakobsdottir G, Bjerregaard JH, Skovbjerg H, Nyman M. Fasting serum concentration of short-chain fatty acids in subjects with microscopic colitis and celiac disease: no difference compared with controls, but between genders. Scand J Gastroenterol. 2013;48(6):696–701. doi:10.3109/00365521.2013.786128
13. Ma X, Zhou Z, Zhang X, et al. Sodium butyrate modulates gut microbiota and immune response in colorectal cancer liver metastatic mice. Cell Biol Toxicol. 2020;36(5):509–515. doi:10.1007/s10565-020-09518-4
14. Xu Z, Zhou Z, Zhang J, et al. Targeting BMI-1-mediated epithelial-mesenchymal transition to inhibit colorectal cancer liver metastasis. Acta Pharm Sin B. 2021;11(5):1274–1285. doi:10.1016/j.apsb.2020.11.018
15. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–691. doi:10.1038/nrd4360
16. Huang W, Zeng C, Liu J, et al. Sodium butyrate induces autophagic apoptosis of nasopharyngeal carcinoma cells by inhibiting AKT/mTOR signaling. Biochem Biophys Res Commun. 2019;514(1):64–70. doi:10.1016/j.bbrc.2019.04.111
17. Kochetkova EY, Blinova GI, Bystrova OA, Martynova MG, Pospelov VA, Pospelova TV. Targeted elimination of senescent Ras-transformed cells by suppression of MEK/ERK pathway. Aging. 2017;9(11):2352–2375. doi:10.18632/aging.101325
18. Dou X, Gao N, Lan J, Han J, Yang Y, Shan A. TLR2/EGFR are two sensors for pBD3 and pEP2C induction by sodium butyrate independent of HDAC inhibition. J Agric Food Chem. 2020;68(2):512–522. doi:10.1021/acs.jafc.9b06569
19. Wang F, Wu H, Fan M, et al. Sodium butyrate inhibits migration and induces AMPK-mTOR pathway-dependent autophagy and ROS-mediated apoptosis via the miR-139-5p/Bmi-1 axis in human bladder cancer cells. FASEB J. 2020;34(3):4266–4282. doi:10.1096/fj.201902626R
20. Kim H, Park C, Wei X, et al. Golgi condensation causes intestinal lipid accumulation through HIF-1alpha-mediated GM130 ubiquitination by NEDD4. Exp Mol Med. 2025;57(2):349–363. doi:10.1038/s12276-025-01396-2
21. Mi Y, Mu L, Huang K, et al. Hypoxic colorectal cancer cells promote metastasis of normoxic cancer cells depending on IL-8/p65 signaling pathway. Cell Death Dis. 2020;11(7):610. doi:10.1038/s41419-020-02797-z
22. Liu X, Cooper DE, Cluntun AA, et al. Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell. 2018;175(2):502–513e13. doi:10.1016/j.cell.2018.08.040
23. Donohoe DR, Garge N, Zhang X, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13(5):517–526. doi:10.1016/j.cmet.2011.02.018
24. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48(4):612–626. doi:10.1016/j.molcel.2012.08.033
25. Serna-Blasco R, Sanz-Alvarez M, Aguilera O, Garcia-Foncillas J. Targeting the RAS-dependent chemoresistance: the Warburg connection. Semin Cancer Biol. 2019;54:80–90. doi:10.1016/j.semcancer.2018.01.016
26. Liang C, Qin Y, Zhang B, et al. ARF6, induced by mutant Kras, promotes proliferation and Warburg effect in pancreatic cancer. Cancer Lett. 2017;388:303–311. doi:10.1016/j.canlet.2016.12.014
27. Liu K, Jin M, Ye S, Yan S. CHI3L1 promotes proliferation and improves sensitivity to cetuximab in colon cancer cells by down-regulating p53. J Clin Lab Anal. 2020;34(1):e23026. doi:10.1002/jcla.23026
28. Zhang M, Lv Y, Hou S, Liu Y, Wang Y, Wan X. Differential mucosal microbiome profiles across stages of human colorectal cancer. Life. 2021;11(8). doi:10.3390/life11080831
29. Xu J, Lu L, Jiang S, et al. Paeoniflorin ameliorates oxaliplatin-induced peripheral neuropathy via inhibiting neuroinflammation through influence on gut microbiota. Eur J Pharmacol. 2024;971:176516. doi:10.1016/j.ejphar.2024.176516
30. Nogal A, Asnicar F, Vijay A, et al. Genetic and gut microbiome determinants of SCFA circulating and fecal levels, postprandial responses and links to chronic and acute inflammation. Gut Microbes. 2023;15(1):2240050. doi:10.1080/19490976.2023.2240050
31. Holmes ZC, Villa MM, Durand HK, et al. Microbiota responses to different prebiotics are conserved within individuals and associated with habitual fiber intake. Microbiome. 2022;10(1):114. doi:10.1186/s40168-022-01307-x
32. McOrist AL, Miller RB, Bird AR, et al. Fecal butyrate levels vary widely among individuals but are usually increased by a diet high in resistant starch. J Nutr. 2011;141(5):883–889. doi:10.3945/jn.110.128504
33. Ye K, Zhao Y, Huang W, Zhu Y. Sodium butyrate improves renal injury in diabetic nephropathy through AMPK/SIRT1/PGC-1alpha signaling pathway. Sci Rep. 2024;14(1):17867. doi:10.1038/s41598-024-68227-8
34. Ding J, Liu J, Chen J, et al. Sodium butyrate alleviates free fatty acid-induced steatosis in primary chicken hepatocytes via the AMPK/PPARalpha pathway. Poult Sci. 2024;103(4):103482. doi:10.1016/j.psj.2024.103482
35. Zhang P, Fu HJ, Lv LX, et al. WSSV exploits AMPK to activate mTORC2 signaling for proliferation by enhancing aerobic glycolysis. Commun Biol. 2023;6(1):361. doi:10.1038/s42003-023-04735-z
36. Marzi L, Combes E, Vie N, et al. FOXO3a and the MAPK p38 are activated by cetuximab to induce cell death and inhibit cell proliferation and their expression predicts cetuximab efficacy in colorectal cancer. Br J Cancer. 2016;115(10):1223–1233. doi:10.1038/bjc.2016.313
37. Bouali S, Chretien AS, Ramacci C, et al. P53 and PTEN expression contribute to the inhibition of EGFR downstream signaling pathway by cetuximab. Cancer Gene Ther. 2009;16(6):498–507. doi:10.1038/cgt.2008.100
38. Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA. The Wip1 phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell. 2007;12(4):342–354. doi:10.1016/j.ccr.2007.08.033
39. Wang J, Wang G, Cheng D, et al. Her2 promotes early dissemination of breast cancer by suppressing the p38-MK2-Hsp27 pathway that is targetable by Wip1 inhibition. Oncogene. 2020;39(40):6313–6326. doi:10.1038/s41388-020-01437-2
40. Bain NT, Wang Y, Arulananda S. Minimal residual disease in EGFR-mutant non-small-cell lung cancer. Front Oncol. 2022;12:1002714. doi:10.3389/fonc.2022.1002714
41. Ziranu P, Lai E, Schirripa M, et al. The role of p53 expression in patients with RAS/BRAF wild-type metastatic colorectal cancer receiving irinotecan and cetuximab as later line treatment. Target Oncol. 2021;16(4):517–527. doi:10.1007/s11523-021-00816-3
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.