Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Cigarette smoke-induced RANKL expression enhances MMP-9 production by alveolar macrophages
Authors Zhou L, Le Y, Tian J, Yang X ![]() , Jin R, Gai X, Sun Y
, Jin R, Gai X, Sun Y
Received 7 October 2018
Accepted for publication 29 November 2018
Published 20 December 2018 Volume 2019:14 Pages 81—91
DOI https://doi.org/10.2147/COPD.S190023
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chunxue Bai
Lu Zhou,1 Yanqing Le,1 Jieyu Tian,2 Xia Yang,2 Rong Jin,3 Xiaoyan Gai,1 Yongchang Sun1
1Department of Respiratory Medicine, Peking University Third Hospital, Beijing, China; 2Department of Respiratory Medicine, Beijing Tongren Hospital, Capital Medical University, Beijing, China; 3Department of Immunology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China
Background and purpose: Cigarette smoke (CS) induces alveolar destruction through overproduction of proteinases including matrix metalloproteinase (MMP)-9 by alveolar macrophages (AMs). Receptor activator of nuclear factor-κB ligand (RANKL) functions in immune regulation and cytokine secretion; whether it is involved in CS-induced MMP-9 expression is unknown. The purpose of our study was to investigate the expression and functional role of RANKL pathway in MMP-9 production pertaining to the pathogenesis of COPD.
Materials and methods: We first localized RANKL and its receptor RANK in the lungs of mice exposed to long-term CS exposure. Next, we studied RANKL and RANK expression under CS extract (CSE) stimulation in vitro. Lastly, we studied the in vitro biological function of RANKL in CS-induced production of MMP-9.
Results: Both RANKL and RANK were highly expressed in AMs in CS-exposed mice, but not in the control mice. In vitro, CSE increased the expressions of RANKL and RANK in macrophages. AMs responded to CSE and RANKL stimulation by overexpressing MMP-9, and CSE-induced MMP-9 expression was partly blocked by using monoclonal anti-RANKL antibody.
Conclusion: RANKL/RANK pathway mediates CS-induced MMP-9 expression in AMs, suggesting a novel mechanism for CS-associated emphysema.
Keywords: COPD, receptor activator of nuclear factor-κB ligand, RANK, alveolar macrophages, MMP-9
Introduction
COPD is a heterogeneous disease associated with cigarette smoke (CS) exposure, which is believed to induce proteinase-mediated injury to the alveolar tissue and extracellular matrix,1–3 leading to emphysema.
Metalloproteinases (MMPs) are a family of proteolytic enzymes which play important roles in tissue remodeling and repair associated with inflammation. MMP-9, an important member of MMPs, is found to be involved in airway inflammation and development of emphysema.1–3 A major source of MMP-9 in the lung is the alveolar macrophage; however, the pathways leading to MMP-9 overproduction associated with cigarette smoking have not been fully elucidated.
In our previous studies, we found that the circulatory levels of receptor activator of nuclear factor-κB ligand (RANKL) were increased in COPD patients and associated with bone loss,4,5 suggesting a role of cigarette smoking or inflammation in the induction of this molecule. RANKL was originally described for its key roles in bone metabolism and later in immune regulation.6–9 RANKL expression has been detected in various tissues including T lymphocytes,7 macrophages,10 osteoblasts, osteocytes and bone stroma, and the lung.8,9,11,12 However, the expression of RANKL and its potential pathological function in the lung, particularly that relates to cigarette smoking, has not been studied.
In order to investigate whether cigarette smoking induces RANKL expression and its biological function relating to COPD, we examined the expression of RANKL and its receptor RANK in a well-described model of COPD by long-term CS exposure. We found, for the first time to our knowledge, that RANKL as well as RANK were expressed and localized, together with MMP-9, in the alveolar macrophages of CS-exposed mice. In an in vitro culture of alveolar macrophages, CS extract (CSE) upregulated RANKL and RANK expression, and the cells responded to CSE or RANKL stimulation by enhanced expression of MMP-9, which was inhibited by a monoclonal anti-RANKL antibody neutralizing RANKL. These results reveal a novel function of the RANKL/RANK system by which CS exposure induces MMP-9 production by alveolar macrophages, which may have implications in the pathogenesis of emphysema and serve as a potentially new target for intervention.
Materials and methods
Animals
Female C57BL/6 mice, 6–8 weeks old, supplied by Beijing Vital River Laboratory were bred in-house. Food and water were provided ad libitum. All mice were housed in a light–dark cycle of 12 hours under specific pathogen-free conditions. All in vivo manipulations were approved by the Ethics Committee of Peking University Third Hospital, and performed in accordance with the committee’s animal care and use guidelines.
CS exposure
Six wild-type mice were exposed to CS using a nose-only, directed flow inhalation and smoke exposure system (SG-300; SIBATA, Tokyo, Japan). CS exposure parameters were as follows: cigarettes (Baisha cigarettes with filter, Hunan, China; tar 11 mg, nicotine 0.9 mg, CO 12 mg), suction 20 mL smoke per 8 seconds, two times a day for 50 minutes with 20-minute smoke-free intervals, 5 days a week for 24 weeks. An optimal smoke:air ratio of 1:9 was obtained. Control mice were exposed to room air only.
Lung function
Mouse lung function was measured using Flexivent apparatus (Scireq, Montreal, QC, Canada) as previously described.13 Mice were anesthetized by intraperitoneal injection of 1% pentobarbital sodium (0.6 mg/10 g). A tracheostomy was performed and a cannula inserted into the trachea. Animals were ventilated with a tidal volume of 10 mL/kg at a rate of 500 breaths/min with a positive end-expiratory pressure of 2 cm H2O. The total lung capacity, lung compliance (C) and airway resistance (R) were then measured.
Quantification of emphysema
To evaluate pulmonary emphysema, we determined enlargement of alveolar spaces by quantifying the mean linear intercept (MLI) and destruction of alveolar walls by measuring the destructive index (DI), as described previously.14 Briefly, the measurement of MLI was performed by means of a 100×100 μm grid passing randomly through the lung. The total length of each line of the grid divided by the number of alveolar intercepts gave the average distance between alveolar surfaces. DI was measured by a grid with 42 points which were at the center of hairline crosses superimposed on the lung field. Structures lying under these points were classified as normal (N) or destroyed (D) alveolar and/or duct spaces. Points falling over other structures, such as duct walls, alveolar walls and so on, did not enter into the calculations. DI was calculated as D/(D+N)×100.
Immunohistochemistry and immunofluorescence (IF)
The lung tissues obtained from mice were fixed by immersion in formalin and embedded in paraffin. Lung tissues were cut into sections that were 5 μm thick and placed on glass slides. Briefly, after dewaxing and hydration, sections were incubated in 0.3% hydrogen peroxide to block endogenous peroxidase activity for 15 minutes, and then sections were incubated in citrate buffer 5 mM at pH 6.0 in a microwave oven for 15 minutes for antigen retrieval. Afterward, the sections were treated with goat serum (ZLI-6056; ZSGB-Bio, Beijing, China) and incubated overnight with MMP-9 antibody (Abcam, Cambridge, UK). Sections were subsequently incubated with the secondary antibody horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (PV-6002; ZSGB-Bio) for 30 minutes. Immunoreactivity was visualized with DAB Detection System kit (ZLI-9018; ZSGB-Bio), and sections were counterstained with Mayer’s hematoxylin. Negative controls for nonspecific binding were processed omitting the primary antibody.
Confocal microscopy was applied to evaluate the co-expression of RANKL (Abcam) and the mouse alveolar macrophage marker F4/80 (Abcam). Similar protocol was used to evaluate the expression of RANK (Abcam) or MMP-9 (Abcam) in alveolar macrophages. Briefly, after dewaxing and hydration, sections were incubated in citrate buffer 5 mM at pH 6.0 in a microwave oven for 15 minutes for antigen retrieval. Afterward, sections were treated with goat serum (ZLI-6056; ZSGB-Bio) and incubated overnight with the primary antibody. Sections were subsequently incubated with the secondary antibody (goat anti-rabbit IgG conjugated with AlexaFluor 488 and goat anti-mouse IgG conjugated with AlexaFluor 594; Jackson ImmunoResearch, West Grove, PA, USA) for 30 minutes at 37°C. Slides were stored at 4°C and analyzed within 24 hours. Negative controls for nonspecific binding were processed omitting the primary antibody. IF was evaluated with confocal microscopy (TCS-SP8; Leica Microsystems, Wetzlar, Germany).
Cell culture
The mouse alveolar macrophage cell line MH-S and mouse macrophage cell line RAW264.7 were purchased from Bio-Rad Biological Technology Co. Ltd (Shanghai, China), an agent of ATCC. MH-S was cultured in Roswell Park Memorial Institute medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal calf serum (FCS) (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific), and RAW264.7 was cultured in DMEM medium (Thermo Fisher Scientific) containing 10% FCS (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific). Cells were grown at 37°C in the presence of 5% CO2.
CSE preparation
CSE was prepared by bubbling smoke from five cigarettes (Baisha cigarettes with filter; tar 11 mg, nicotine 0.9 mg, CO 12 mg) through 10 mL of FCS-free cell culture medium at a constant airflow, lasting 5 minutes for each cigarette.15,16 Smoked medium was then sterile filtered through a 0.22 μm filter, and this served as the 100% CSE work solution. Cell proliferation was detected with different concentrations of CSE (0.5%, 1%, 2%, 4%) by using Cell Counting Kit-8, and 0.5% was determined as the optimum experimental concentration.
Cell stimulation
Macrophages were stimulated with CSE at 37°C in the presence of 5% CO2 for 48 hours to detect RANKL and RANK expression. Macrophages were also stimulated with CSE, recombinant RANKL (100 ng/mL; R&D systems, Minneapolis, MN, USA), monoclonal anti-RANKL (10 μg/mL; Biolegend, San Diego, CA, USA) and rat IgG2a kappa isotype control antibody (10 μg/mL; Biolegend) at 37°C in the presence of 5% CO2 for 48 hours to detect MMP-9 expression.
Flow cytometry
At the termination of cell culture, cells in the 12-well plates were harvested, followed by incubation with fluorescence-conjugated antibodies: phycoerythrin-conjugated anti-mouse RANKL antibody (eBioscience, San Diego, CA, USA), phycoerythrin-conjugated anti-mouse RANK antibody (eBioscience) and phycoerythrin-conjugated rat IgG2a κ isotype control (eBioscience). At least 10,000 cells were counted for each sample using BD FACSCalibur.
IF for RANK co-localization with endosomal vesicles by IF analysis
Untreated and CSE-treated alveolar macrophages (for 24 hours) were fixed for 15 minutes in 4% paraformaldehyde, permeabilized for 15 minutes in PBS/0.1% Triton and stained with mouse monoclonal anti-RANK antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and rabbit monoclonal anti-LAMP1 antibody (Abcam). They were incubated overnight at 4°C, followed by staining with secondary Alexa 488 anti-rabbit IgG and Alexa 694 anti-mouse IgG (Jackson ImmunoResearch) for 30 minutes at room temperature. Pictures were taken with a confocal microscope (TCS-SP8; Leica Microsystems).
Detection of MMP-9
MMP-9 expression was assessed on the lung tissue sections by immunohistochemistry, while its mRNA levels in the lung tissue and cells were determined using quantitative PCR.
Cell counts were calculated and standardized to the number of positive cells/mm2 of the area. The quantification was performed with blinded samples, and 20 fields were randomly selected under 400× microscopy and the number of positive cells per square millimeter was counted.
MMP-9 protein levels in cells were determined using Western blotting. Proteins (30 μg) were resolved in 10% SDS-PAGE and transferred to polyvinylidene difluoride membrane (Merck-Millipore, Carrigtwohill, Ireland), which were then blocked and incubated with the following antibodies: MMP-9 (Santa Cruz Biotechnology) and β-actin (Abcam). After incubation with HRP-conjugated goat anti-mouse IgG antibody, the immunoreactive bands were detected using enhanced chemiluminescence from EMD Millipore (Billerica, MA, USA). Quantitative image analysis was performed with ImageJ software (NIH, Bethesda, MD, USA). Results are expressed as relative densities.
Reverse transcriptase PCR
Total RNA was extracted from the total lung tissues of mice and cultured cells using TRIZOL (Thermo Fisher Scientific) following manufacturer’s instructions. Isolated mRNA (1 μg each) was reverse transcribed into cDNA using the reverse transcription system in the presence of oligo dT primers (Promega, Madison, WI, USA).
Real-time PCR
Real-time PCR was performed on QuantStudio 3 and 5 Real-Time PCR System(Applied Biosystems, Foster City, CA, USA) in a 20 μL reaction using SYBR Green One-Step qRT-PCR Kit (Tiangen, Beijing, China).
The primer sequences used for the amplification were as follows: GAPDH, 5′-AAATGGTGAAGGTCGGTGTGAAC-3′ (sense) and 5′-CAACAATCTCCACTTTGCCACTG-3′ (antisense); MMP-9, 5′-GCCCTGGAACTCACACGACA-3′ (sense) and 5′-TTGGAAACTCACACGCCAGAAG-3′ (antisense); MMP-12, 5′-GATGGATGAAGCGGTACCTCACTTA-3′ (sense) and 5′-GAGTCACATCACTCCAGACTTGGAA-3′ (antisense); TIMP1, 5′-GGAACGGAAATTTGCACATCAG-3′ (sense) and 5′-CTGATCCGTCCACAAACAGTGAG-3′ (antisense). The real-time PCR conditions were: 95°C for 15 minutes, followed by 40 cycles of 95°C for 10 seconds and 60°C for 32 seconds. Results were presented as fold changes relative to GAPDH reference.
Statistical analysis
Statistical analysis was performed with SPSS20.0 (IBM Corporation, Armonk, NY, USA). Group data are expressed as mean ± standard error of the mean. For data with normal distribution, comparisons among three or more groups were performed with an ANOVA test. For nonparametric data, comparisons were performed by Tamhane’s T2 signed-rank test. Two-groups comparison was performed using Student’s t-test. P-values <0.05 were considered to be significant.
Results
Expression and cellular localization of RANKL and RANK in CS-exposed mice
After CS exposure for 24 weeks, mice showed a significant increase in total lung capacity (Figure 1A) and airway resistance (R; Figure 1B) compared to those exposed to air as controls. However, no significant difference in lung compliance (C) was observed between the study groups. Lung histologic analysis showed enlargement of airway spaces (HE staining, 400×; Figure 1C and D), with significantly increased MLI (Figure 1E) and DI (Figure 1F) in CS-exposed mice, but not in air-exposed mice.
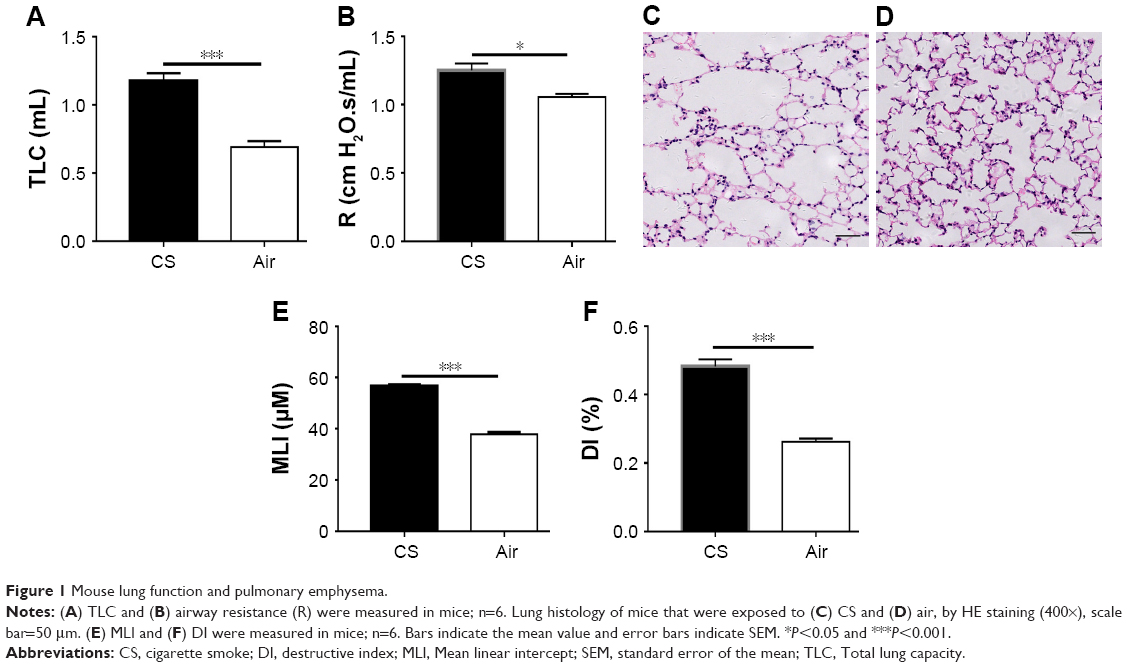 | Figure 1 Mouse lung function and pulmonary emphysema. |
We firstly examined the expression of RANKL and RANK by IF in lung tissues and found RANKL+ and RANK+ cells in the alveoli in CS-exposed mice (Figure 2A and B), but scarcely in control mice (Figure 2C and D).
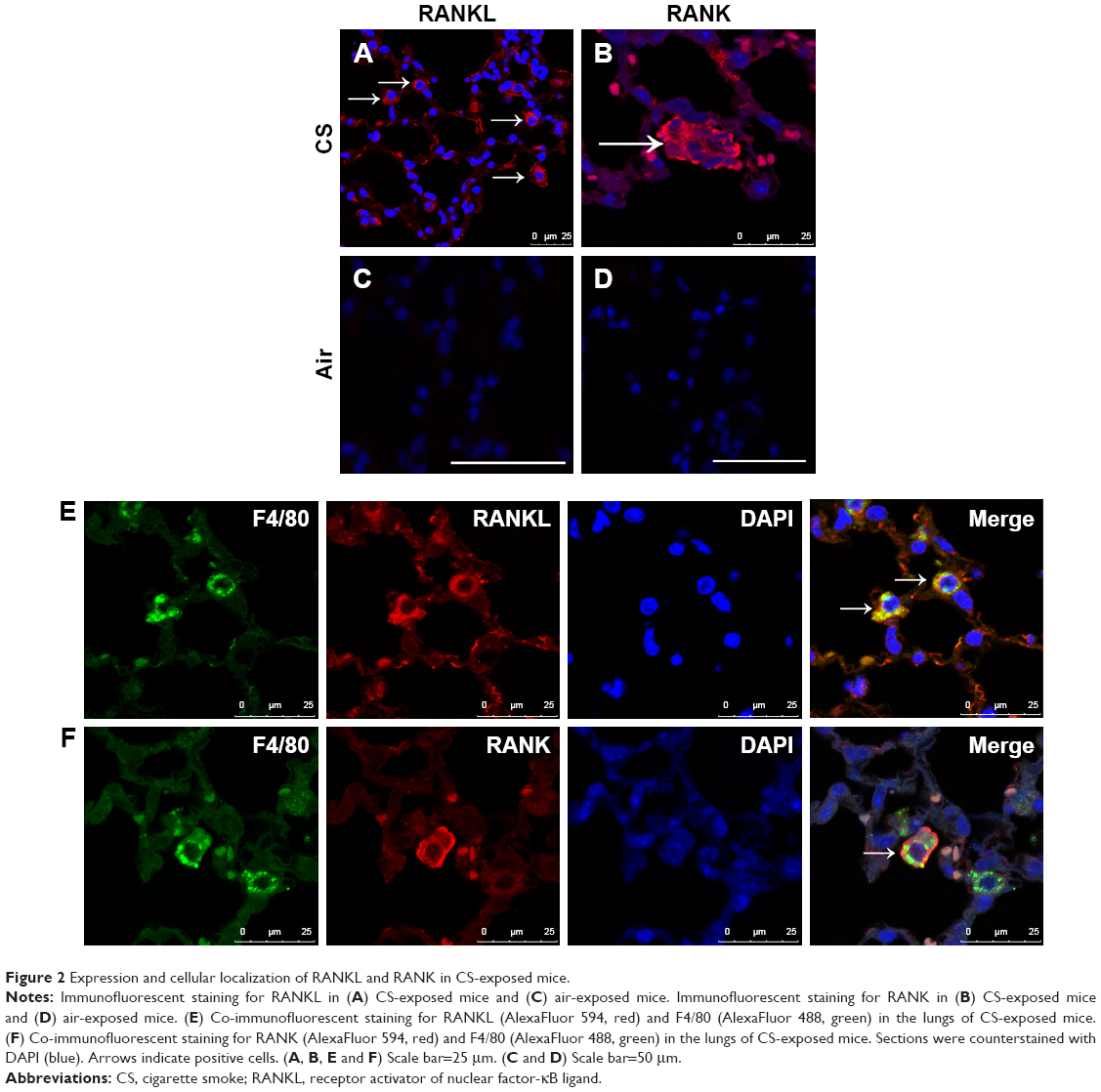 | Figure 2 Expression and cellular localization of RANKL and RANK in CS-exposed mice. |
To address the expression pattern of RANKL, we performed IF costaining for RANKL and cellular markers, focusing on alveolar macrophages. We found that RANKL+ cells in the alveoli were macrophages in CS-exposed mice (Figure 2E). Interestingly, RANK+ cells were also identified as alveolar macrophages in CS-exposed mice (Figure 2F), suggesting that CS exposure upregulated the expression of both RANKL and its receptor RANK in alveolar macrophages.
Upregulation of RANKL and RANK expression by CSE in vitro
Two macrophage cell lines, MH-S and RAW264.7, were stimulated with CSE in vitro. The result showed that the mean fluorescence intensity of RANKL measured by flow cytometry was increased significantly after CSE stimulation as compared to the control (Figure 3A, B, C and D). Similarly, RANK expression in CSE-stimulated cells was also increased significantly, and the macrophages had inherent RANK expression in the absence of stimulation (Figure 3E, F, G and H).
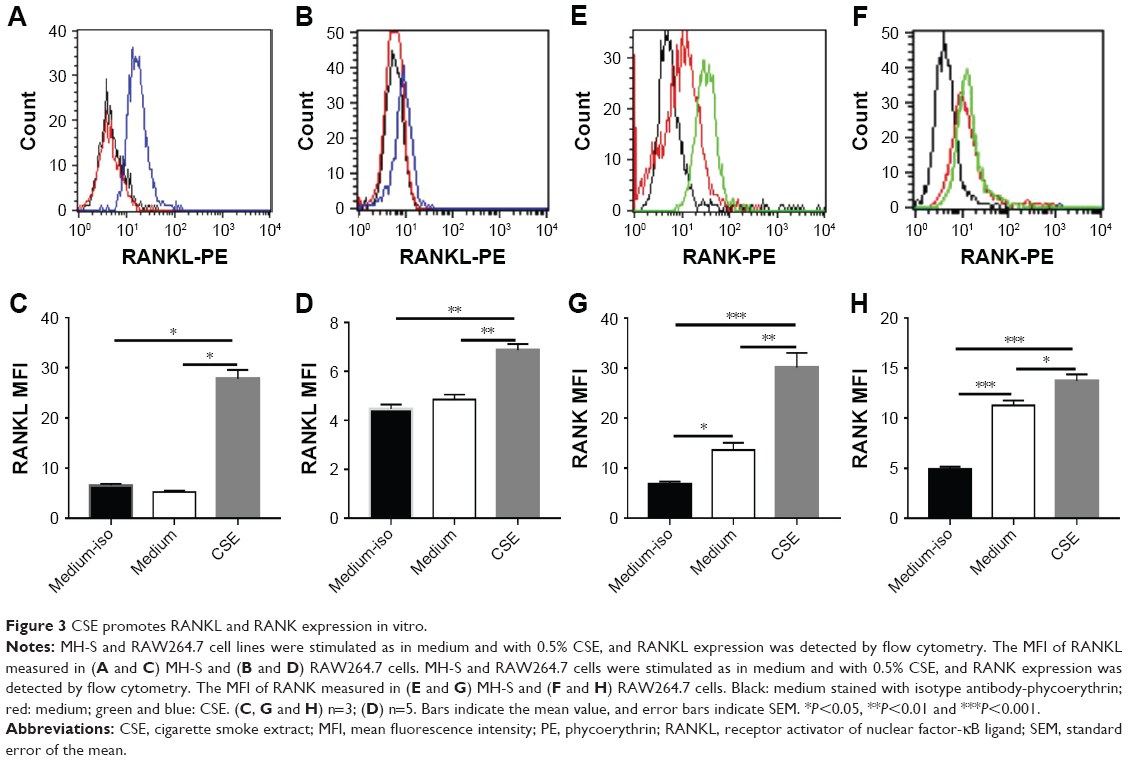 | Figure 3 CSE promotes RANKL and RANK expression in vitro. |
Intracellular mechanism for enhanced RANK expression by CSE
A recent study showed that high expression of RANK in cystic fibrosis monocytes was due to decreased receptor endocytosis,17 similar to a previous work on the lipopolysaccharide receptor.18 To test whether the high expression of RANK in alveolar macrophages exposed to CS was due to decreased RANK degradation via reduced translocation to the lysosomal compartment, RANK co-localization with lysosome-associated membrane protein 1 (LAMP1) was performed by IF. Interestingly, unstimulated macrophages had robust RANK co-localization in the lysosomal compartment (Figure 4A), while in CSE-stimulated cells, there was minimal RANK associated with LAMP1-positive vesicles (Figure 4B). LAMP1-positive vesicles were clustered in the nuclear area in unstimulated cells, while the signal was present diffusely throughout the cytoplasm in CSE-stimulated cells (Figure 4A and B). Rab7, a small GTPase, is essential for the transport from early to late endosomes19 and is a key regulator for proper aggregation and fusion of late endocytic structures in the perinuclear region and, consequently, for the biogenesis and maintenance of functional lysosomal compartment.20 We found that Rab7 failed to increase in CSE-stimulated cells compared to unstimulated cells (Figure 4C and D). Taken together, these data suggest that CSE stimulation leads to aberrant location of lysosomes and transportation of vesicles in the endosomal–lysosomal axis, resulting in reduced RANK endocytosis.
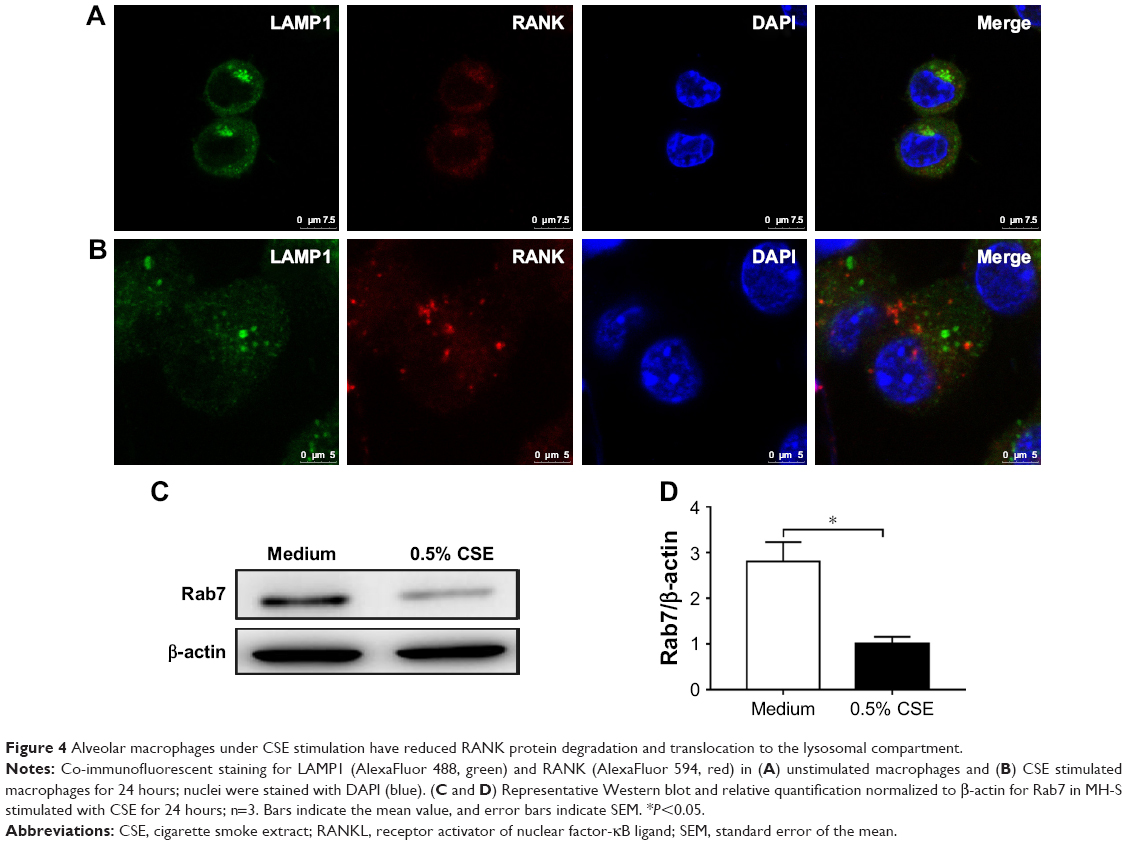 | Figure 4 Alveolar macrophages under CSE stimulation have reduced RANK protein degradation and translocation to the lysosomal compartment. |
Upregulation of MMP-9 expression by CS in vivo and in vitro
As the alveolar macrophages are a major source of MMPs, MMP-9 is increased in COPD and plays a critical role in airway inflammation and development of emphysema.1–3 We firstly examined MMP-9 expression by immunohistochemistry in the lung tissues and found MMP-9+ cells in the alveoli in CS-exposed mice (Figure 5A), but they were scarcely found in control mice (Figure 5B). The number of MMP-9+ cells was increased and significantly higher in CS-exposed mice compared to the control ones (Figure 5C). The MMP-9 mRNA level of the total lung tissue was also increased in CS-exposed mice (Figure 5D). We further found IF costaining of MMP-9 in the alveolar macrophages of CS-exposed mice (Figure 5E). In vitro culture of mouse alveolar macrophages with CSE led to enhanced MMP-9 expression at the protein level in alveolar macrophages (Figure 5F and G).
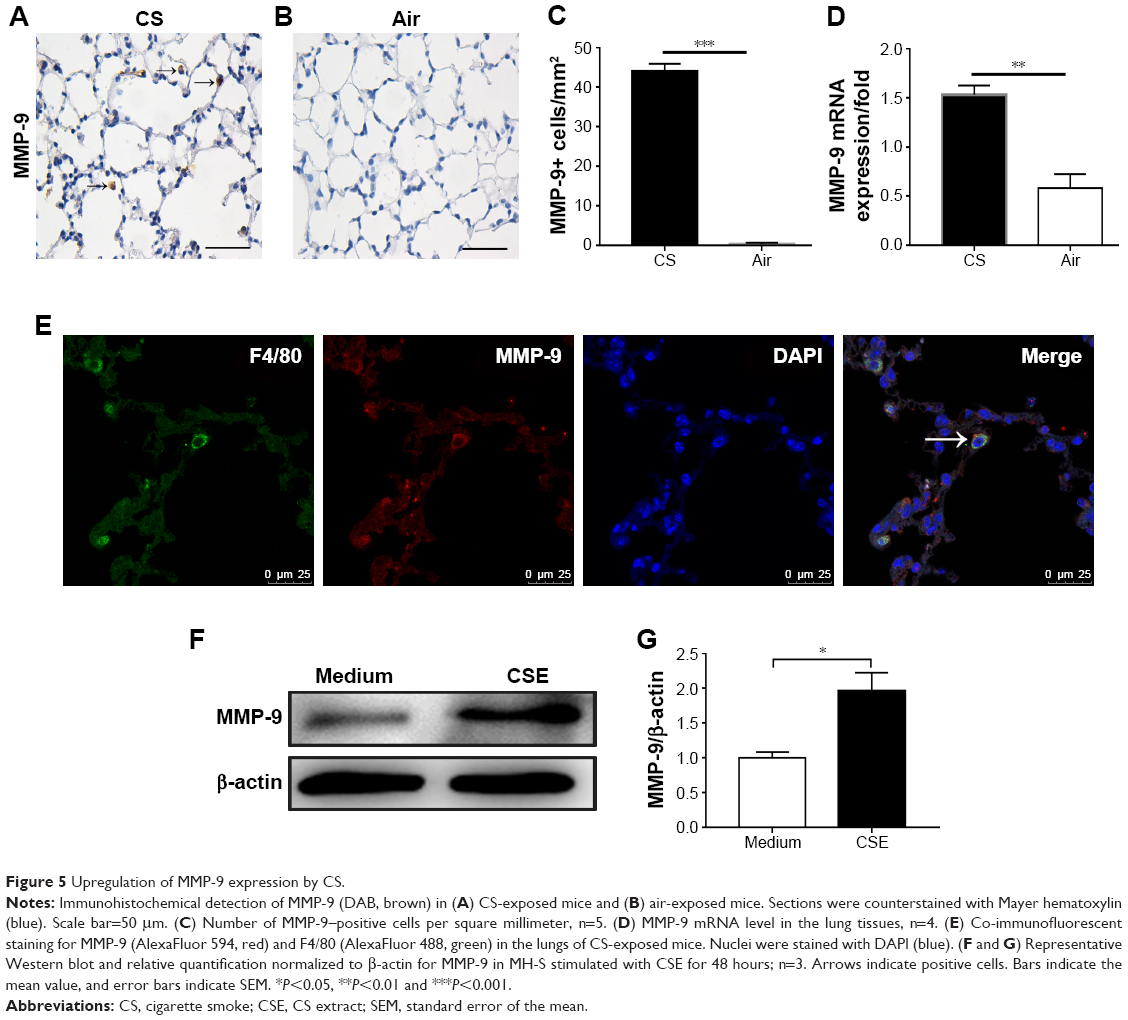 | Figure 5 Upregulation of MMP-9 expression by CS. |
MMP-9 expression was regulated by RANKL
RANKL functions in bone metabolism via osteoclast differentiation,8,9 and can also promote monocyte and dendritic cell survival by upregulation of BCL-XL protein.7,21 However, the function of RANKL in alveolar macrophages has not been investigated. In order to understand the functions of RANKL in the context of CS-induced lung destruction, we firstly used RANKL to stimulate alveolar macrophages in vitro, which showed upregulation of MMP-9 mRNA (Figure 6A), while the MMP-12 and TIMP1 mRNA showed no significant change (Figure 6B and C). Similarly, the ratio of MMP-9 mRNA to TIMP1 mRNA was increased (Figure 6D), but that of MMP-12 mRNA to TIMP1 mRNA showed no significant difference (Figure 6E) under RANKL stimulation. RANKL also upregulated MMP-9 protein expression in alveolar macrophages in vitro (Figure 6F and G).
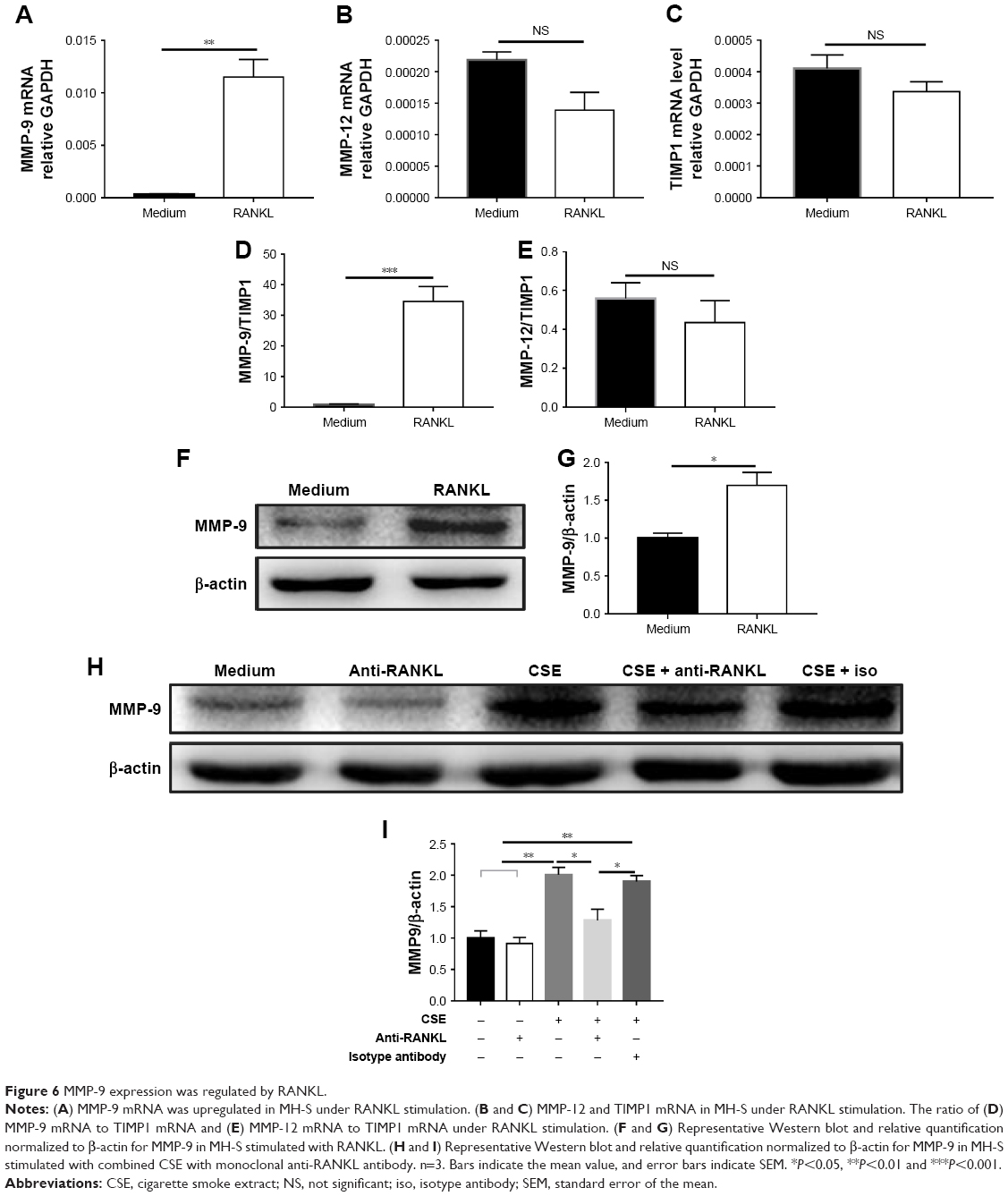 | Figure 6 MMP-9 expression was regulated by RANKL. |
Because both CSE and RANKL promoted MMP-9 expression and CSE upregulated RANKL expression, we asked whether CSE induction of MMP-9 was dependent or partly through the RANKL pathway. In the culture of alveolar macrophages with CSE stimulation, the addition of anti-RANKL antibody partly blocked the increase of MMP-9 protein (Figure 6H and I). Taken together, these results suggest that CS exposure induces MMP-9 expression partly via upregulation of RANKL and RANK, creating an autocrine mode of action for the RANKL pathway in alveolar macrophages.
Discussion
By using a well-established mouse model of COPD, we demonstrated that long-term CS exposure induced the expression of RANKL and its receptor RANK, together with MMP-9 in the alveolar macrophages in the lung tissues. We further found that CSE markedly enhanced the expression of RANKL and RANK in alveolar macrophages in vitro. Interestingly, both CSE and RANKL upregulated MMP-9 expression in the alveolar macrophages and CSE-induced MMP-9 overproduction was partly blocked by anti-RANKL antibody. Thus, our data support a novel mechanism for CS-induced MMP-9 expression relevant to the pathogenesis of COPD.
The RANKL–RANK pathway was found to play key roles in bone metabolism8,9,22–24 and immune regulation.6,7 Recent studies have also shown that RANKL functions in thermoregulation and plays a protective role in ischemic stroke in the central nervous system.25,26 RANKL expression has been detected in various tissues and cells,7–12 including the lung;11 however, the expression of RANKL and its potential pathological function in the lung, particularly that which relates to cigarette smoking, has not been described. Here, in a mouse model of COPD induced by long-term CS exposure, we found marked expression of RANKL and its receptor RANK in alveolar macrophages. To our knowledge, for the first time, we have shown that RANKL was localized on alveolar macrophages. To understand the biological effect of RANKL, we further localized its receptor RANK in alveolar macrophages. Previous studies found that RANK expression was detectable on mature dendritic cells6 and macrophages.26 Interestingly, RANK expression was also localized on alveolar macrophages in the lung tissues from the mouse model. Taken together, these findings point to a new function of RANKL–RANK pathway via alveolar macrophages in the development of COPD.
The upregulation of RANKL–RANK expression in the alveolar macrophages by CS exposure had not been described, although our previous work had revealed higher levels of RANKL in peripheral blood of COPD patients.4,5 CS was capable of promoting alveolar macrophage activation.27,28 Our in vitro study demonstrated that CSE stimulated RANKL–RANK expression in alveolar macrophages, confirming the in vivo findings. Macrophages have inherent RANK expression,29 and it is intriguing to note that the high expression of RANK induced by CSE in alveolar macrophages was due to decreased receptor endocytosis. CS inhibits phagocytosis of alveolar macrophages,30 and endocytosis is a form of phagocytosis. Recent works have shown that decreased receptor endocytosis leads to high expression of RANK and the lipopolysaccharide receptor.17,18 Our result was consistent with these findings and further demonstrated that CSE-induced downregulation of Rab7 might account for reduced trafficking of RANK to the degradation pathway in alveolar macrophages.
RANKL has a variety of biological functions, including activation of monocytes to secrete cytokines and chemokines,21 protection of monocytes from apoptosis21 and induction of osteoclast differentiation by inducing MMP-9 expression.31–33 Our in vivo model showing CS induction of co-expression of RANKL and MMP-9 in alveolar macrophages suggests an interaction of these two molecules in the pathogenesis of COPD. Indeed, our in vitro study confirmed that RANKL stimulated MMP-9 expression in alveolar macrophages. Interestingly, CSE was also shown to be able to promote MMP-9 expression in alveolar macrophages. Therefore, we sought to find whether CSE induction of MMP-9 was dependent, at least partly, on the RANKL pathway. The result that CSE-induced MMP-9 expression in the alveolar macrophages was partly blocked by an anti-RANKL antibody confirmed our hypothesis. Because MMP-12 was also found to be implicated in the pathogenesis of COPD by previous studies,1–3 we concurrently examined its expression under RANKL stimulation, which showed that the expression of MMP-12 was unresponsive to RANKL.
Smokers with COPD had increased expression of MMP-9 in the lung, compared with smokers without COPD and nonsmokers.34–36 MMP-9 functions in degradation of elastin,37 leading to alveolar destruction. There is also supportive evidence that overexpression of MMP-9 in the lungs of transgenic mice contributes to the development of emphysema.38,39 CS exposure has been found to stimulate alveolar macrophages to produce MMPs.40,41 Experimental data indicated that CS exposure induced MMP expression through different signaling pathways, for example, in the alveolar macrophages through the NF-κB pathway42 and in epithelial cells via Wingless/integrase-1 (WNT) signaling.43 Our data added new knowledge to the understanding of the molecular mechanisms underlying MMP overproduction induced by CS exposure relevant to the development and progression of COPD.
Conclusion
In conclusion, the present study showed that CS exposure induced the expression of RANKL and its receptor RANK, together with increased MMP-9, in alveolar macrophages, both in vivo and in vitro. Induction of MMP-9 by CSE in the alveolar macrophages was partly through the RANKL pathway by an autocrine mechanism of action. These data reveal a novel mechanism of interaction between RANKL and MMP-9 in the pathogenesis of COPD and a potential new target for intervention.
Acknowledgment
The authors acknowledge financial support from the National Natural Science Foundation of China (81470239 and 81770040).
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Owen CA. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3(2):253–268. | ||
Ishii T, Abboud RT, Wallace AM, et al. Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. Eur Respir J. 2014;43(1):82–91. | ||
Mocchegiani E, Giacconi R, Costarelli L. Metalloproteases/anti-metalloproteases imbalance in chronic obstructive pulmonary disease: genetic factors and treatment implications. Curr Opin Pulm Med. 2011;17(Suppl 1):S11–S19. | ||
Bai P, Sun Y, Jin J, et al. Disturbance of the OPG/RANK/RANKL pathway and systemic inflammation in COPD patients with emphysema and osteoporosis. Respir Res. 2011;12:157–164. | ||
Hu X, Sun Y, Xu W, Lin T, Zeng H. Expression of RANKL by peripheral neutrophils and its association with bone mineral density in COPD. Respirology. 2017;22(1):126–132. | ||
Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390(6656):175–179. | ||
Wong BR, Josien R, Lee SY, et al. TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med. 1997;186(12):2075–2080. | ||
Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–176. | ||
Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A. 1998;95(7):3597–3602. | ||
Crotti TN, Smith MD, Weedon H, et al. Receptor activator NF-kappaB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Ann Rheum Dis. 2002;61(12):1047–1054. | ||
Kartsogiannis V, Zhou H, Horwood NJ, et al. Localization of RANKL (receptor activator of NF kappa B ligand) mRNA and protein in skeletal and extraskeletal tissues. Bone. 1999;25(5):525–534. | ||
Zhao S, Zhang YK, Harris S, Ahuja SS, Bonewald LF. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J Bone Miner Res. 2002;17(11):2068–2079. | ||
Li JJ, Wang W, Baines KJ, et al. IL-27/IFN-γ induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J Immunol. 2010;185(7):4401–4409. | ||
Bracke KR, D’Hulst AI, Maes T, et al. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J Immunol. 2006;177(7):4350–4359. | ||
Wirtz HR, Schmidt M. Acute influence of cigarette smoke on secretion of pulmonary surfactant in rat alveolar type II cells in culture. Eur Respir J. 1996;9(1):24–32. | ||
Braber S, Thio M, Blokhuis BR, et al. An association between neutrophils and immunoglobulin free light chains in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(8):817–824. | ||
Velard F, Jourdain ML, Abdallah D. Overexpression of RANK and M-CSFR receptors in monocytes of G551D-bearing cystic fibrosis patients. Am J Respir Crit Care Med. 2018;198(7):968–970. | ||
Bruscia EM, Zhang PX, Satoh A, et al. Abnormal trafficking and degradation of TLR4 underlie the elevated inflammatory response in cystic fibrosis. J Immunol. 2011;186(12):6990–6998. | ||
Press B, Feng Y, Hoflack B, Wandinger-Ness A. Mutant Rab7 causes the accumulation of cathepsin D and cation-independent mannose 6-phosphate receptor in an early endocytic compartment. J Cell Biol. 1998;140(5):1075–1089. | ||
Bucci C, Thomsen P, Nicoziani P, Mccarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11(2):467–480. | ||
Seshasayee D, Wang H, Lee WP, et al. A novel in vivo role for osteoprotegerin ligand in activation of monocyte effector function and inflammatory response. J Biol Chem. 2004;279(29):30202–30209. | ||
Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–319. | ||
Yasuda H, Shima N, Nakagawa N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139(3):1329–1337. | ||
Nakagawa N, Kinosaki M, Yamaguchi K, et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun. 1998;253(2):395–400. | ||
Hanada R, Leibbrandt A, Hanada T, et al. Central control of fever and female body temperature by RANKL/RANK. Nature. 2009;462(7272):505–509. | ||
Shimamura M, Nakagami H, Osako MK, et al. OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc Natl Acad Sci U S A. 2014;111(22):8191–8196. | ||
Metcalfe HJ, Lea S, Hughes D, Khalaf R, Abbott-Banner K, Singh D. Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages. Clin Exp Immunol. 2014;176(3):461–472. | ||
Woodruff PG, Ellwanger A, Solon M, Cambier CJ, Pinkerton KE, Koth LL. Alveolar macrophage recruitment and activation by chronic second hand smoke exposure in mice. COPD. 2009;6(2):86–94. | ||
Arai F, Miyamoto T, Ohneda O, et al. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med. 1999;190(12):1741–1754. | ||
Bozinovski S, Vlahos R, Zhang Y, et al. Carbonylation caused by cigarette smoke extract is associated with defective macrophage immunity. Am J Respir Cell Mol Biol. 2011;45(2):229–236. | ||
Lu SH, Chen TH, Chou TC. Magnolol inhibits RANKL-induced osteoclast differentiation of raw 264.7 macrophages through heme oxygenase-1-dependent inhibition of NFATc1 expression. J Nat Prod. 2015;78(1):61–68. | ||
Fujisaki K, Tanabe N, Suzuki N, et al. Receptor activator of NF-kappaB ligand induces the expression of carbonic anhydrase II, cathepsin K, and matrix metalloproteinase-9 in osteoclast precursor RAW264.7 cells. Life Sci. 2007;80(14):1311–1318. | ||
Yoon WJ, Kim KN, Heo SJ, et al. Sargachromanol G inhibits osteoclastogenesis by suppressing the activation NF-κB and MAPKs in RANKL-induced RAW 264.7 cells. Biochem Biophys Res Commun. 2013;434(4):892–897. | ||
Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117(3):684–694. | ||
Gosselink JV, Hayashi S, Elliott WM, et al. Differential expression of tissue repair genes in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(12):1329–1335. | ||
Churg A, Zhou S, Wright JL. Series “matrix metalloproteinases in lung health and disease”: matrix metalloproteinases in COPD. Eur Respir J. 2012;39(1):197–209. | ||
Parks WC, Shapiro SD. Matrix metalloproteinases in lung biology. Respir Res. 2001;2(1):10–19. | ||
Xu B, Chen H, Xu W, et al. Molecular mechanisms of MMP9 overexpression and its role in emphysema pathogenesis of Smad3-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2012;303(2):L89–L96. | ||
Foronjy R, Nkyimbeng T, Wallace A, et al. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1149–L1157. | ||
Russell RE, Culpitt SV, Dematos C, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26(5):602–609. | ||
Bracke K, Cataldo D, Maes T, et al. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int Arch Allergy Immunol. 2005;138(2):169–179. | ||
Li L, Wang Y, Gao W, et al. Klotho reduction in alveolar macrophages contributes to cigarette smoke extract-induced inflammation in chronic obstructive pulmonary disease. J Biol Chem. 2015;290(46):27890–27900. | ||
Heijink IH, de Bruin HG, Dennebos R, et al. Cigarette smoke-induced epithelial expression of WNT-5B: implications for COPD. Eur Respir J. 2016;48(2):504–515. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.