Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
Cigarette Smoke Extract–Induced Necroptosis Causes Mitochondrial DNA Release and Inflammation of Bronchial Epithelial Cells
Authors Mizumura K ![]() , Ozoe R, Nemoto Y, Furusho N, Kurosawa Y, Kozu Y, Oki T, Maruoka S
, Ozoe R, Nemoto Y, Furusho N, Kurosawa Y, Kozu Y, Oki T, Maruoka S ![]() , Gon Y
, Gon Y ![]()
Received 28 February 2025
Accepted for publication 23 July 2025
Published 1 August 2025 Volume 2025:20 Pages 2685—2695
DOI https://doi.org/10.2147/COPD.S523610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Kenji Mizumura, Ryosuke Ozoe, Yosuke Nemoto, Naho Furusho, Yusuke Kurosawa, Yutaka Kozu, Takashi Oki, Shuichiro Maruoka, Yasuhiro Gon
Division of Respiratory Medicine, Department of Internal Medicine, Nihon University School of Medicine, Tokyo, Japan
Correspondence: Kenji Mizumura, Division of Respiratory Medicine, Department of Internal Medicine, Nihon University School of Medicine, 30-1 Oyaguchi Kami-cho, Itabashi-ku, Tokyo, 173-8610, Japan, Tel +81-3-3972-8111 Ext 2402, Fax +81-3-3972-2893, Email [email protected]
Purpose: Chronic obstructive pulmonary disease (COPD) is characterized by airway inflammation and structural changes in the lungs, including emphysema. Cigarette smoke exposure induces mitochondrial damage, necroptosis-mediated pulmonary epithelial cell death, and emphysematous changes. However, the association between these events and airway inflammation remains unclear. Here, we focused on mitochondrial DNA (mtDNA) as a second messenger linking mitochondrial damage to airway inflammation, aiming to elucidate the mechanisms underlying extracellular mtDNA release and its role in airway inflammation.
Methods: Human bronchial epithelial BEAS-2B cells were exposed to cigarette smoke extract and the release of mtDNA into the cytoplasm and extracellular space was examined. Real-time polymerase chain reaction was used to measure mtDNA levels. To examine the involvement of necroptosis, a necroptosis inhibitor (Nec-1) and mitochondria-targeted antioxidant (MitoQ) were used. To evaluate the inflammatory response induced by extracellular mtDNA, we quantified the levels of specific cytokines—interleukin (IL)-6 and IL-8—in the cell culture supernatants after mtDNA transfection, as these mediators are widely accepted as key markers of inflammation in bronchial epithelial cells.
Results: Cigarette smoke extract treatment induced the translocation of mtDNA from the mitochondria to the cytoplasm in BEAS-2B cells, followed by its extracellular release. Nec-1 and MitoQ inhibited the extracellular release of mtDNA without affecting its cytoplasmic translocation. Introducing mtDNA into BEAS-2B cells markedly elevated IL-6 and IL-8 levels, indicating that mtDNA may play a pro-inflammatory role.
Conclusion: Necroptosis facilitated the release of extracellular mtDNA after cigarette smoke extract exposure, establishing a connection between mitochondrial damage and airway inflammation. mtDNA acted as a pro-inflammatory mediator by inducing cytokine production in pulmonary epithelial cells. These findings suggest that targeting necroptosis could offer a novel therapeutic strategy for COPD by addressing both airway inflammation and structural lung damage.
Keywords: oxidative stress, airway inflammation, mitochondrial damage, cytokine release, chronic obstructive pulmonary disease
Introduction
In addition to generating energy through oxidative phosphorylation, mitochondria are pivotal in the regulation of various cellular functions, including cell death and inflammatory responses.1,2 Mounting evidence indicates that mitochondrial DNA (mtDNA) functions as a damage-associated molecular pattern (DAMP), contributing to the activation of molecular pathways involved in the progression of various diseases.3 Damage to mitochondria leads to their dysfunction, resulting in the elevated production of mitochondrial reactive oxygen species within cells.4 Furthermore, mitochondrial components, such as mtDNA, can be transported beyond these organelles to locations such as the cytosol, cell surface, or extracellular spaces, where they play a role in activating immune responses.3–5 Cytosolic mtDNA activates the NLRP3 inflammasome, which promotes the maturation and secretion of pro-inflammatory cytokines such as interleukin (IL)-1β and IL-18.4 Additionally, it can induce the production of type I interferons (IFN-I) and other inflammatory cytokines through the cGAS–STING pathway, which recognizes intracellular DNA.6 Notably, the source of cytosolic mtDNA is not limited to mitochondria within the same cell; mtDNA derived from dead or dying cells or pathogens can also be taken up from the extracellular environment and exert similar effects.6 Furthermore, extracellular mtDNA can be recognized by toll-like receptor (TLR) 9 within endosomes, which leads to increased expression of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, while simultaneously reducing anti-inflammatory IL-10 levels.7 Recent studies have shown that circulating cell-free mtDNA levels increase in response to various stimuli, including chronic obstructive pulmonary disease (COPD),8 idiopathic pulmonary fibrosis,9 shock,10 and infection.11,12 However, the mechanisms by which mtDNA is released into the extracellular space and exerts its biological functions in the lung remain unclear.
COPD is a significant contributor to the global burden of disease and is among the top three causes of death worldwide.13 It is characterized by ongoing respiratory symptoms and restricted airflow. This chronic airflow limitation results from emphysema, which involves a reduction in the alveolar surface area, and bronchitis, which leads to mucus-induced airway obstruction and is typically associated with prolonged exposure to harmful particles or gases.14 Cigarette smoke exposure is a key risk factor for COPD, and individuals who smoke have a higher mortality rate from COPD than do those who do not smoke.15 Smoking-induced epithelial cell death is believed to play a key role in the development of pulmonary emphysema, leading researchers in COPD to concentrate on understanding the mechanisms underlying programmed cell death.
Apoptosis is recognized as the primary cell death process responsible for the development of emphysema.16 However, apoptosis is a form of cell death that involves minimal release of DAMPs and does not induce inflammation. Thus, while apoptosis can explain the mechanisms underlying emphysema, it is insufficient to fully account for the airway inflammation observed in COPD. Necrosis was once considered an unregulated form of cell death triggered by severe physical or chemical stress. However, recent studies have uncovered multiple types of programmed cell death, including necroptosis.17–19 Necroptosis represents a regulated form of necrotic cell death that depends on the genetic programming and activity of receptor-interacting protein kinase (RIPK) 3.20,21 RIPK3, RIPK1, and mixed lineage kinase domain-like protein (MLKL) assemble into a multiprotein structure known as the “necrosome”, which activates MLKL and triggers a rapid rupture of the plasma membrane.22,23 Unlike apoptosis, necroptosis is a programmed cell death mechanism that triggers inflammation by releasing large amounts of DAMPs such as mtDNA.24 Therefore, necroptosis is more suitable than apoptosis for understanding the pathogenesis of emphysema, which is characterized by epithelial cell death accompanied by chronic inflammation.25 In addition to apoptosis and necroptosis, pyroptosis and ferroptosis—other forms of cell death—have also been implicated in COPD.26,27 Pyroptosis, an inflammasome-dependent and gasdermin-mediated inflammatory cell death, enhances the release of IL-1β and IL-18.28 Ferroptosis, a form of iron-dependent oxidative cell death, may contribute to the pathogenesis of COPD through smoking-induced oxidative stress.29 Although these cell death modalities are relevant, necroptosis is uniquely positioned to explain both epithelial cell death and the sustained inflammatory response via DAMP release. This dual role highlights the novelty and importance of studying necroptosis in the context of COPD.
We have reported previously that cigarette smoke extract (CSE) induces mitochondrial damage in pulmonary epithelial cells, which leads to necroptosis via mitophagy.30 These findings demonstrate the involvement of necroptosis in emphysematous lesions of COPD. However, the relationship between cigarette smoke–induced necroptosis and chronic airway inflammation in patients with COPD remains unclear. Based on these findings, and those of earlier studies, we hypothesized that necroptosis governs the cigarette smoke–induced release of extracellular mtDNA from pulmonary epithelial cells, potentially contributing to bronchitis in patients with COPD. If it can be demonstrated that necroptosis regulates the two most critical pathological features of COPD—emphysema and airway inflammation—via mtDNA, necroptosis could serve as a novel therapeutic target for COPD. Thus, this study was designed to elucidate the mechanisms underlying extracellular mtDNA release and its role in airway inflammation.
Materials and Methods
Reagents
A necroptosis inhibitor (Nec-1; 30 μM, pretreated 1 h before CSE exposure) and mitochondria-targeted antioxidant (MitoQ; 500 nM, pretreated 1 h before CSE exposure) were used to evaluate their effects on mtDNA translocation and extracellular release. Nec-1 was purchased from Sigma-Aldrich (St. Louis, MO, USA) and MitoQ was purchased from BioVision (Milpitas, CA, USA).
Cell Culture
The human bronchial epithelial cell line BEAS-2B was obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal calf serum.
CSE Treatment
CSE was prepared by lighting a paper cigarette (Peace; Japan Tobacco, Tokyo, Japan; tar: 28 mg, nicotine: 2.3 mg) without a filter and drawing the smoke through a peristaltic pump. The smoke was introduced into 10 mL DMEM per cigarette for 6 min and then sterilized using a 0.45 μm Durapore polyvinylidene difluoride membrane filter (Stericup, 250 mL; EMD Millipore, Burlington, MA, USA). CSE treatment was conducted by replacing 20% of the culture medium with freshly prepared 20% CSE solution. For example, in a total volume of 1 mL, 200 μL medium was removed and replaced with 200 μL 20% CSE. The duration of exposure and timing of sample collection varied depending on the experimental context; these are detailed in the respective figure legends. As a control condition, 20% of the culture medium was replaced with an equal volume of DMEM—the solvent used to prepare the CSE—to replicate the procedural conditions without exposing the cells to cigarette smoke components. All experiments were independently performed in triplicate. The number of replicates is indicated in the respective figure legends.
Real-Time Polymerase Chain Reaction Analysis
Complementary DNA was synthesized from mRNA isolated from the cytoplasmic fraction and cell culture supernatant of the BEAS-2B cells using the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA). The mitochondria and cytosol were isolated using the Mitochondria/Cytosol Fractionation Kit according to the manufacturer’s protocol (Abcam, Cambridge, UK). Real-time quantitative polymerase chain reaction (PCR) was performed using the StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). MT-CO1 (#qHsaCED0048374; Bio-Rad Laboratories, Hercules, CA, USA) was used as a primer for mtDNA. For all experiments, mtDNA levels were quantified relative to untreated control samples (ie, cells not exposed to CSE). The fold change from baseline, CSE (-), was calculated to evaluate the relative increase in cytoplasmic or extracellular mtDNA abundance after CSE exposure. Because mtDNA abundance reflects its physical release into the cytoplasm or extracellular space rather than transcriptional activity, normalization to a housekeeping gene was not performed.
Enzyme-Linked Immunosorbent Assays
The concentrations of IL-6 and IL-8 in the supernatant of the BEAS-2B cells were determined using human IL-6 (BioLegend, San Diego, CA, USA) and IL-8 (BD Biosciences, Franklin Lakes, NJ, USA) enzyme-linked immunosorbent assay (ELISA) kits.
mtDNA Isolation and Transfection
mtDNA was isolated from BEAS-2B cells using a Mitochondrial DNA Isolation Kit (Abcam). The isolated mtDNA was transfected into BEAS-2B cells using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific). The cells were transfected with 1 μg/mL mtDNA, and culture supernatants were collected 24 h later for cytokine analysis. This condition was selected based on previous studies and preliminary experiments demonstrating reproducible cytokine induction.31,32
Statistical Analyses
Statistical analyses were performed using Prism version 8.4.3 (GraphPad Software, La Jolla, CA, USA). Results are presented as means ± standard error of the mean. The primary outcome measures included the levels of mtDNA translocated from the mitochondria to the cytoplasm, extracellular mtDNA release, and cytokine (IL-6 and IL-8) production. Between-group comparisons were performed using an unpaired t-test, whereas a one-way or two-way analysis of variance was used for experiments with multiple groups, followed by Tukey’s test for multiple comparisons to determine significant differences. A P-value < 0.05 was considered statistically significant.
Results
CSE Causes Translocation of mtDNA from Mitochondria to Cytosol in Pulmonary Epithelial Cells
To determine whether CSE causes the translocation of mtDNA from the mitochondria to the cytosol, we treated BEAS-2B cells with CSE and quantified the mtDNA copy number in the cytosol. The treatment resulted in the accumulation of mtDNA in the cytosol of the pulmonary epithelial cells (Figure 1). The cytosolic mtDNA level reached a maximum at 3 h after treatment and then gradually declined. These findings indicate that CSE promotes the movement of mtDNA from the mitochondria to the cytoplasm in pulmonary epithelial cells.
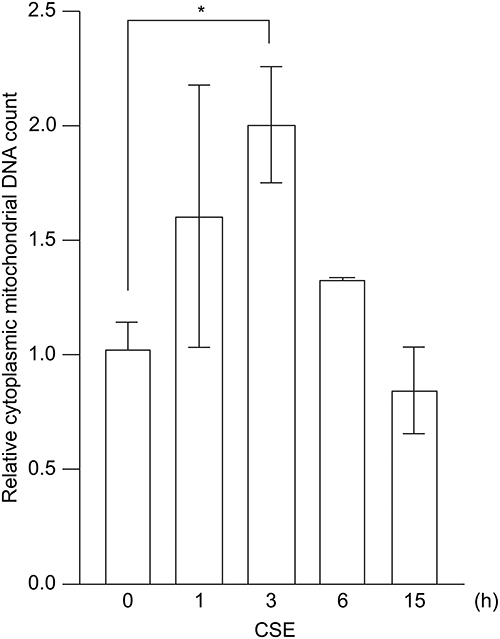 |
Figure 1 Translocation of mtDNA from the mitochondria to the cytoplasm induced by CSE. BEAS-2B cells were treated with 20% CSE, and the cytoplasmic mtDNA content was quantified using real-time PCR. Data are shown as fold change relative to untreated control cells. The results are presented as the mean ± SEM of three independent experiments. *P < 0.05 using one-way analysis of variance with Tukey’s post-hoc test. Abbreviations: CSE, cigarette smoke extract; mtDNA, mitochondrial DNA; PCR, polymerase chain reaction; SEM, standard error of the mean. |
CSE Induces Extracellular mtDNA Release in Pulmonary Epithelial Cells
We next examined whether mtDNA was released into the extracellular environment after translocation from the mitochondria to the cytoplasm in pulmonary epithelial cells following CSE treatment. CSE exposure significantly increased mtDNA levels in the cell culture supernatant compared to the control conditions (Figure 2). Extracellular mtDNA release peaked at 15 h, which was later than the 3 h peak of cytoplasmic translocation, and subsequently declined. These findings indicate that CSE triggers the movement of mtDNA from the mitochondria to the cytoplasm, followed by its release into the extracellular space of pulmonary epithelial cells.
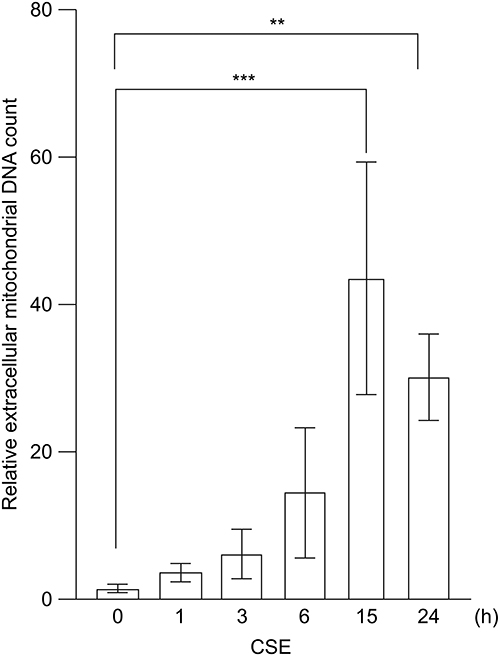 |
Figure 2 Extracellular release of mtDNA induced by CSE. BEAS-2B cells were treated with 20% CSE, and the mtDNA content in the cell culture supernatant was quantified using real-time PCR. Data are shown as fold change relative to untreated control cells. The results are presented as the mean ± SEM of three independent experiments. **P < 0.01 and ***P < 0.005 using one-way analysis of variance with Tukey’s post-hoc test. Abbreviations: CSE, cigarette smoke extract; mtDNA, mitochondrial DNA; PCR, polymerase chain reaction; SEM, standard error of the mean. |
The Mitochondria-Targeted Antioxidant, MitoQ, and Necroptosis Inhibitor, Nec-1, Do Not Inhibit CSE-Induced mtDNA Translocation to the Cytoplasm
To investigate the mechanism underlying the translocation of mtDNA from the mitochondria to the cytoplasm, we examined the effects of MitoQ and Nec-1 (Figure 3). While CSE induced the translocation of mtDNA from the mitochondria to the cytoplasm, neither MitoQ nor Nec-1 inhibited this movement (Figure 3A and B). These data indicate that mitochondrial oxidative stress and necroptosis are not involved in the translocation of mtDNA from the mitochondria to the cytoplasm.
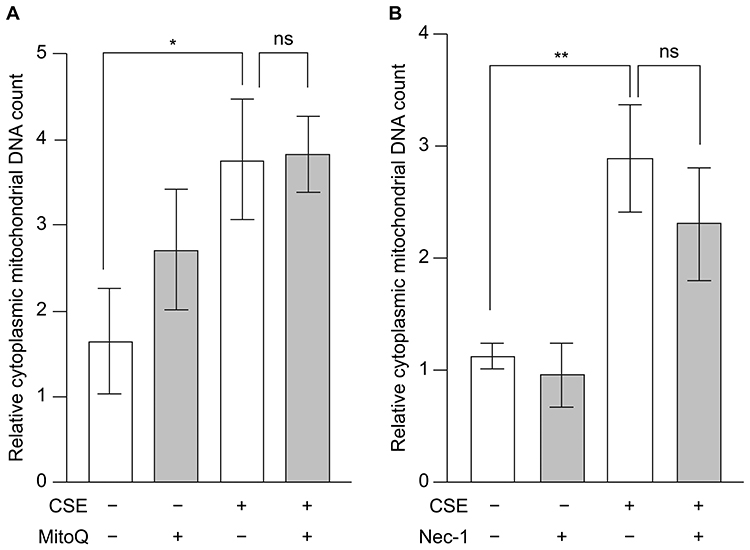 |
Figure 3 Effects of mitochondrial oxidative stress and necroptosis on CSE-induced mtDNA translocation to the cytoplasm. BEAS-2B cells were treated with 20% CSE for 3 h in the presence of the (A) mitochondria-targeted antioxidant MitoQ and (B) necroptosis inhibitor Nec-1, and the cytoplasmic mtDNA content was quantified using real-time PCR. Data are presented as fold change relative to the untreated control group (no CSE and no inhibitor). Results are presented as the mean ± SEM of three independent experiments. *P < 0.05 and **P < 0.01 using two-way analysis of variance with Tukey’s post-hoc test. Abbreviations: CSE, cigarette smoke extract; mtDNA, mitochondrial DNA; ns, not significant; PCR, polymerase chain reaction; SEM, standard error of the mean. |
The Mitochondria-Targeted Antioxidant, MitoQ, and Necroptosis Inhibitor, Nec-1, Suppress CSE-Induced Extracellular Release of mtDNA
To investigate the mechanism underlying the extracellular release of mtDNA, we examined this release in the presence of MitoQ and Nec-1 (Figure 4). CSE induced the translocation of mtDNA from the mitochondria to the cytoplasm, with both MitoQ and Nec-1 significantly suppressing the extracellular release of mtDNA (Figure 4A and B). These data suggest that mitochondrial oxidative stress and necroptosis are involved in the extracellular release of mtDNA.
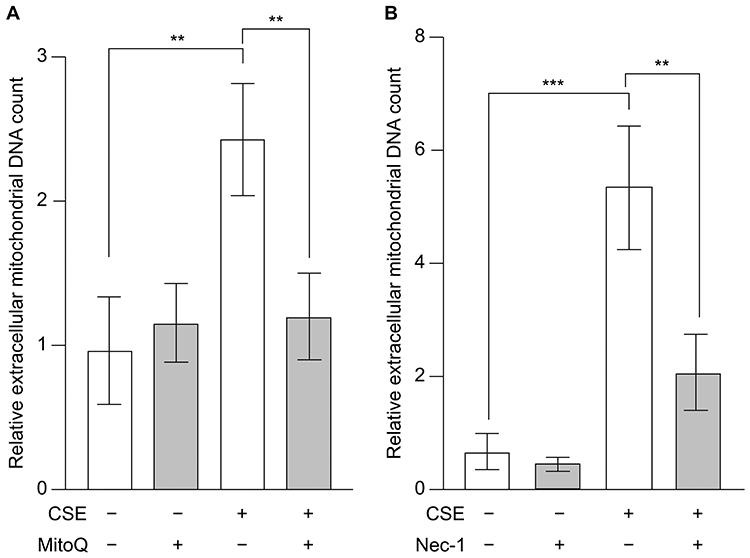 |
Figure 4 Effects of mitochondrial oxidative stress and necroptosis on CSE-induced extracellular release of mtDNA. BEAS-2B cells were treated with 20% CSE for 15 h in the presence of the (A) mitochondria-targeted antioxidant MitoQ and (B) necroptosis inhibitor Nec-1, and the mtDNA content in the cell culture supernatant was quantified using real-time PCR. Data are presented as fold change relative to the untreated control group (no CSE and no inhibitor). Results represent the mean ± SEM of three independent experiments. **P < 0.01 and ***P < 0.005 using two-way analysis of variance with Tukey’s post-hoc test. Abbreviations: CSE, cigarette smoke extract; mtDNA, mitochondrial DNA; PCR, polymerase chain reaction; SEM, standard error of the mean. |
Transfection of mtDNA Induces IL-6 and IL-8 Production in Pulmonary Epithelial Cells
Because the mechanism by which mtDNA exerts its biological functions remains unclear, we transfected BEAS-2B cells with isolated mtDNA and measured the production of IL-6 and IL-8 to evaluate the role of mtDNA. Transfection with mtDNA significantly induced IL-6 and IL-8 production in pulmonary epithelial cells (Figure 5). We also measured other cytokines associated with CSE-induced inflammation—including IL-17A, IL-25, IL-33, and thymic stromal lymphopoietin—but their concentrations were below the detection limit in our BEAS-2B cell–based system (data not shown).
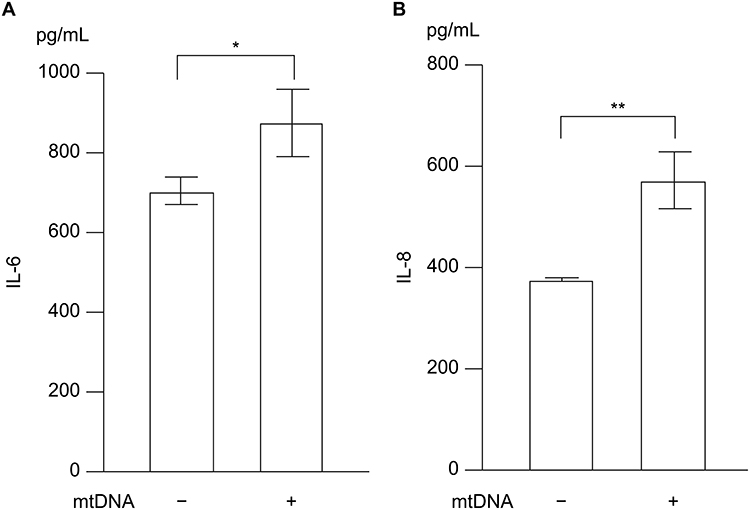 |
Figure 5 Involvement of mtDNA in inflammation. BEAS-2B cells were transfected with 1 μg/mL mtDNA, and cytokine levels in the cell culture supernatant were measured 24 h after transfection using ELISA kits: (A) IL-6 and (B) IL-8. The experiment was conducted three times independently under the same conditions. Data are presented as the mean ± SEM. *P < 0.05 and **P < 0.01 using the unpaired t-test. Abbreviations: mtDNA, mitochondrial DNA; IL, interleukin; SEM, standard error of the mean. |
Discussion
Various stimuli reportedly induce the release of mtDNA from the mitochondria to the cytoplasm and subsequently to the extracellular space.4,33 However, it remains unclear whether this extracellular release occurs as an incidental consequence of cell lysis or through a regulated mechanism involving translocation from the mitochondria to the cytoplasm and eventually to the extracellular space. In this study, we demonstrate that CSE induces the translocation of mtDNA from the mitochondria to the cytoplasm in pulmonary epithelial cells, which is followed by its extracellular release via necroptosis. Moreover, introducing mtDNA into these cells increases the secretion of the pro-inflammatory cytokines, IL-6 and IL-8. These findings suggest that necroptosis contributes not only to epithelial cell death in COPD but also to the regulation of inflammatory responses.
When necroptosis is induced, phosphorylated RIPK3 phosphorylates MLKL, which forms oligomers that create pores in the plasma membrane leading to the release of DAMPs.22,34,35 mtDNA is considered a DAMP and is known to trigger inflammation in various diseases.36 The efficient extracellular release of mtDNA requires its initial translocation from the mitochondria to the cytoplasm. However, the role of cytoplasmic mtDNA translocation in the development of COPD remains unclear. In this study, we showed that introducing mtDNA into pulmonary epithelial cells increases the production of the inflammatory cytokines IL-6 and IL-8. This suggests two potential mechanisms underlying the pathogenesis of COPD. First, mtDNA translocated from the mitochondria to the cytoplasm due to CSE may induce inflammation within the same cell. Second, mtDNA released extracellularly via necroptosis may propagate inflammation to neighboring cells. We used pulmonary epithelial cells and showed that prolonged exposure to CSE resulted in their death. Collectively, these findings suggest that CSE induces inflammation by translocating mtDNA from the mitochondria to the cytoplasm in pulmonary epithelial cells. Furthermore, during the process leading to cell death via necroptosis, mtDNA may mediate inflammation that extends to the surrounding cells. The translocation of mtDNA to the cytoplasm induces a chronic senescence-associated secretory phenotype,37 which is consistent with the involvement of cellular senescence in the pathogenesis of COPD. This aligns with the emerging understanding that COPD is a condition in which cellular senescence plays a significant role in disease progression.
COPD induces both systemic and pulmonary inflammation. One proposed mechanism for this phenomenon is the spillover hypothesis that suggests inflammation originating in the lungs extends throughout the body.38 Indeed, studies have shown that the levels of cell-free mtDNA in the blood are significantly higher in patients with than in those without COPD.8 IL-6 and IL-8 are recognized biomarkers for evaluating systemic inflammation in patients with COPD,39 and the current study provides insights into the mechanisms by which inflammation propagates from single cells in the lungs to neighboring cells, and eventually to the entire body. If mtDNA, translocated from the mitochondria to the cytoplasm and subsequently to the extracellular space, serves as a key molecule linking pulmonary and systemic inflammation in COPD, targeting necroptosis as a regulatory mechanism for the release of mtDNA could represent a novel therapeutic strategy. This approach has the potential to address both emphysematous changes and inflammation in patients with COPD.
This study has certain limitations. First, it was conducted using BEAS-2B cells, an immortalized human bronchial epithelial cell line, under in vitro conditions. While this model enables reproducible and well-controlled mechanistic investigations, it does not comprehensively capture the cellular diversity or microenvironmental complexity of the human airway. Moreover, although we used a standardized protocol to prepare and apply CSE, this artificial exposure system cannot replicate the dynamic nature of cigarette smoke inhalation in vivo, including variations in smoke composition and exposure duration. These factors may influence the biological responses observed and should be carefully considered when interpreting the results. Previous research has revealed that circulating cell-free mtDNA significantly contributes to the pathogenesis of COPD,8 and the present study offers valuable insights into the mechanisms involved. Nevertheless, future studies using primary human bronchial epithelial cells and in vivo COPD models are needed to validate our findings and to assess their physiological relevance. We demonstrated that CSE induced the translocation of mtDNA from the mitochondria to the cytoplasm and its subsequent extracellular release, with mitochondrial oxidative stress and necroptosis implicated in the extracellular release. However, these mechanisms do not appear to govern the translocation of mtDNA from the mitochondria to the cytoplasm, which leaves some questions unanswered. This translocation may involve alternative pathways, such as specific mitochondrial channels. Indeed, the authors of previous studies have reported that mtDNA can translocate through VDAC and BAX/BAK-mediated mechanisms.40,41 Further investigations are required to elucidate the precise mechanisms underlying this process. Additionally, while our study was focused on mtDNA as a DAMP, other DAMPs have also been implicated in the pathogenesis of COPD.42 These include high-mobility group box 1,43,44 S100 proteins,44 extracellular adenosine triphosphate,45 and heat shock proteins,46 all of which are known to activate various innate immune receptors, such as TLRs, the receptor for advanced glycation end-products, and purinergic receptors. Therefore, mtDNA alone cannot comprehensively explain the complex inflammatory network involved, and additional research is necessary to explore the specific roles and interactions of these DAMPs in COPD. Furthermore, although CSE exposure alone is well known to elicit inflammation in airway epithelial cells,47 we did not evaluate cytokine production under CSE-only conditions in this study because our primary objective was to clarify the specific role of mtDNA as a second messenger linking mitochondrial damage to inflammatory signaling. By focusing on cytokine responses after mtDNA transfection, we could isolate its direct effect independently of the other components present in cigarette smoke. This targeted approach provided a more mechanistic understanding of the contribution of mtDNA to COPD-associated airway inflammation.
Conclusion
In this study, we focused on mtDNA as a key factor linking emphysematous changes and inflammation in COPD. We demonstrated that CSE induces the release of mtDNA from the mitochondria to the cytoplasm and subsequently into the extracellular space, with mitochondrial damage and necroptosis playing critical roles in this process. We also showed that mtDNA promotes the production of the pro-inflammatory cytokines IL-6 and IL-8, thereby implicating necroptosis as a key regulator of the two primary pathological features of COPD: emphysema and airway inflammation. These findings suggest that targeting necroptosis may offer a novel therapeutic approach for COPD by addressing both structural lung damage and inflammatory responses. Importantly, necroptosis-mediated plasma membrane rupture, driven by RIPK3-activated MLKL oligomerization, is thought to enable the passive release of cytoplasmic DAMPs such as mtDNA into the extracellular space. This process may represent a regulated mechanism of mtDNA release that links necroptotic signaling to the propagation of inflammation.
Data Sharing Statement
The data that support the findings of this study are available on request from the corresponding author, KM.
Acknowledgments
We express our sincere gratitude to Kaori Soda-Urushima (Division of Respiratory Medicine, Department of Internal Medicine, Nihon University School of Medicine, Tokyo, Japan) for her valuable assistance and support in conducting the experiments for this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by JSPS KAKENHI (grant number JP18K08159). The funding source had no role in the study design, data collection, analysis, or interpretation, in the writing of the report, or in the decision to submit the article for publication.
Disclosure
Professor Yasuhiro Gon reports grants from GSK, personal fees from Sanofi, grants from AstraZeneca, outside the submitted work. The authors declare that they have no other competing interests.
References
1. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–632. doi:10.1038/nrm2952
2. Vringer E, Tait SWG. Mitochondria and inflammation: cell death heats up. Front Cell Dev Biol. 2019;7:100. doi:10.3389/fcell.2019.00100
3. Nakahira K, Hisata S, Choi AM. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid Redox Signal. 2015;23(17):1329–1350. doi:10.1089/ars.2015.6407
4. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi:10.1038/ni.1980
5. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
6. Kim J, Kim HS, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. 2023;55(3):510–519. doi:10.1038/s12276-023-00965-7
7. De Gaetano A, Solodka K, Zanini G, et al. Molecular mechanisms of mtDNA-mediated inflammation. Cells. 2021;10(11):2898. doi:10.3390/cells10112898
8. Giordano L, Gregory AD, Pérez Verdaguer M, et al. Extracellular release of mitochondrial DNA: triggered by cigarette smoke and detected in COPD. Cells. 2022;11(3):369. doi:10.3390/cells11030369
9. Ryu C, Sun H, Gulati M, et al. Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(12):1571–1581. doi:10.1164/rccm.201612-2480OC
10. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock. 2010;34(1):55–59. doi:10.1097/SHK.0b013e3181cd8c08
11. Tanaka A, Wakayama K, Fukuda Y, et al. Increased levels of circulating cell-free DNA in COVID-19 patients with respiratory failure. Sci Rep. 2024;14(1):17399. doi:10.1038/s41598-024-68433-4
12. Sursal T, Stearns-Kurosawa DJ, Itagaki K, et al. Plasma bacterial and mitochondrial DNA distinguish bacterial sepsis from sterile systemic inflammatory response syndrome and quantify inflammatory tissue injury in nonhuman primates. Shock. 2013;39(1):55–62. doi:10.1097/SHK.0b013e318276f4ca
13. Halpin DMG, Celli BR, Criner GJ, et al. The GOLD Summit on chronic obstructive pulmonary disease in low- and middle-income countries. Int J Tuberc Lung Dis. 2019;23(11):1131–1141. doi:10.5588/ijtld.19.0397
14. Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. doi:10.1183/09031936.03.00040703
15. Kohansal R, Martinez-Camblor P, Agustí A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med. 2009;180(1):3–10. doi:10.1164/rccm.200901-0047OC
16. Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7(1):53. doi:10.1186/1465-9921-7-53
17. Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
18. He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. doi:10.1016/j.cell.2009.05.021
19. Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi:10.1126/science.1172308
20. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–714. doi:10.1038/nrm2970
21. Mizumura K, Maruoka S, Gon Y, Choi AM, Hashimoto S. The role of necroptosis in pulmonary diseases. Respir Investig. 2016;54(6):407–412. doi:10.1016/j.resinv.2016.03.008
22. Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. doi:10.1016/j.cell.2011.11.031
23. Zhao J, Jitkaew S, Cai Z, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–5327. doi:10.1073/pnas.1200012109
24. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. doi:10.1038/nature14191
25. Mizumura K, Maruoka S, Shimizu T, Gon Y. Autophagy, selective autophagy, and necroptosis in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3165–3172. doi:10.2147/COPD.S175830
26. Shu HM, Lin CQ, He B, et al. Pyroptosis-related genes as diagnostic markers in chronic obstructive pulmonary disease and its correlation with immune infiltration. Int J Chron Obstruct Pulmon Dis. 2024;19:1491–1513. doi:10.2147/COPD.S438686
27. Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10(1):3145. doi:10.1038/s41467-019-10991-7
28. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. doi:10.1038/nrmicro2070
29. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
30. Mizumura K, Cloonan SM, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. doi:10.1172/JCI74985
31. Liu T, Hecker J, Liu S, et al. The asthma risk gene, GSDMB, promotes mitochondrial DNA-induced ISGs expression. J Respir Biol Transl Med. 2024;1(1):10005. doi:10.35534/jrbtm.2024.10005
32. Dib B, Lin H, Maidana DE, et al. Corrigendum to “Mitochondrial DNA has a pro-inflammatory role in AMD” [Biochim. Biophys. Acta 1853 (2015) 2897-2906]. Biochim Biophys Acta Mol Cell Res. 2024;1871(8):119790. doi:10.1016/j.bbamcr.2024.119790
33. Fukihara J, Sakamoto K, Ikeyama Y, et al. Mitochondrial DNA in bronchoalveolar lavage fluid is associated with the prognosis of idiopathic pulmonary fibrosis: a single cohort study. Respir Res. 2024;25(1):202. doi:10.1186/s12931-024-02828-9
34. Hildebrand JM, Tanzer MC, Lucet IS, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111(42):15072–15077. doi:10.1073/pnas.1408987111
35. Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146. doi:10.1016/j.molcel.2014.03.003
36. Bushra SI, Ahmed S, Begum MS, et al. Molecular basis of sepsis: a new insight into the role of mitochondrial DNA as a damage-associated molecular pattern. Mitochondrion. 2024;79:101967. doi:10.1016/j.mito.2024.101967
37. Victorelli S, Salmonowicz H, Chapman J, et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature. 2023;622(7983):627–636. doi:10.1038/s41586-023-06621-4
38. Barnes PJ. Chronic obstructive pulmonary disease: effects beyond the lungs. PLoS Med. 2010;7(3):e1000220. doi:10.1371/journal.pmed.1000220
39. Celli BR, Locantore N, Yates J, et al. Inflammatory biomarkers improve clinical prediction of mortality in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(10):1065–1072. doi:10.1164/rccm.201110-1792OC
40. Hu H, Guo L, Overholser J, Wang X. Mitochondrial VDAC1: a potential therapeutic target of inflammation-related diseases and clinical opportunities. Cells. 2022;11(19):3174. doi:10.3390/cells11193174
41. Riley JS, Quarato G, Cloix C, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018;37(17):e99238. doi:10.15252/embj.201899238
42. Pouwels SD, Hesse L, Faiz A, et al. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L881–L892. doi:10.1152/ajplung.00135.2016
43. Ferhani N, Letuve S, Kozhich A, et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(9):917–927. doi:10.1164/rccm.200903-0340OC
44. Pouwels SD, Heijink IH, ten Hacken NH, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014;7(2):215–226. doi:10.1038/mi.2013.77
45. Cicko S, Lucattelli M, Müller T, et al. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol. 2010;185(1):688–697. doi:10.4049/jimmunol.0904042
46. Hulina-Tomašković A, Heijink IH, Jonker MR, Somborac-Bačura A, Grdić Rajković M, Rumora L. Pro-inflammatory effects of extracellular Hsp70 and cigarette smoke in primary airway epithelial cells from COPD patients. Biochimie. 2019;156:47–58. doi:10.1016/j.biochi.2018.09.010
47. Kode A, Yang SR, Rahman I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir Res. 2006;7(1):132. doi:10.1186/1465-9921-7-132
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.