Back to Journals » Journal of Inflammation Research » Volume 18
Chronic Sleep Fragmentation Differentially Affects Alzheimer’s Disease Pathology in Male and Female APPSAA Knock-in Mice
Authors Hawkins MR ![]() , Whitlock HR
, Whitlock HR ![]() , Johnson CE, Macheda T, Cox MF
, Johnson CE, Macheda T, Cox MF ![]() , Lapid MG
, Lapid MG ![]() , Roberts KN, Hash HM, Drinkard EG
, Roberts KN, Hash HM, Drinkard EG ![]() , Sunderam S, O’Hara BF, Murphy MP, Duncan MJ, Bachstetter AD
, Sunderam S, O’Hara BF, Murphy MP, Duncan MJ, Bachstetter AD ![]()
Received 4 June 2025
Accepted for publication 26 September 2025
Published 16 October 2025 Volume 2025:18 Pages 14325—14341
DOI https://doi.org/10.2147/JIR.S544625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Margaret R Hawkins,1,* Haleigh R Whitlock,2,* Carrie E Johnson,3,4 Teresa Macheda,2 MaKayla F Cox,1 Madison G Lapid,1 Kelly N Roberts,1 Heather M Hash,1 Esther G Drinkard,1 Sridhar Sunderam,5 Bruce F O’Hara,6 Michael P Murphy,3,4 Marilyn J Duncan,2 Adam D Bachstetter1,2,4
1Spinal Cord and Brain Injury Research Center, University of Kentucky, Lexington, KY, USA; 2Department of Neuroscience, University of Kentucky, Lexington, KY, USA; 3Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY, USA; 4Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, USA; 5Department of Biomedical Engineering, University of Kentucky, Lexington, KY, USA; 6Department of Biology, University of Kentucky, Lexington, KY, USA
*These authors contributed equally to this work
Correspondence: Adam D Bachstetter, University of Kentucky, Spinal Cord and Brain Injury Research Center and Department of Neuroscience, 741 S. Limestone Street, BBSRB Room B459, Lexington, KY, 40536-0509, USA, Tel +1 859 218 4315, Email [email protected]
Introduction: Sleep fragmentation often precedes Alzheimer’s disease (AD) diagnosis and represents a potential modifiable risk factor, especially among women who have higher prevalence of both sleep disorders and AD.
Methods: This study investigated how chronic sleep fragmentation affects neuroinflammation and amyloid-beta (Aβ) accumulation in male and female APPSAA knock-in mice, a physiologically relevant AD model expressing APP at normal levels. APPSAA mice of both sexes (N=8/sex/strain, 8 months old) underwent either 5 weeks of chronic sleep fragmentation administered during the light phase using an automated sweeper system or undisturbed sleep. Sleep-wake patterns and circadian rhythms were monitored using piezoelectric sensors. Following intervention, we assessed neuroinflammatory markers via immunohistochemistry and multiplex cytokine analysis, Aβ levels in different solubility fractions, and Aβ plaque characteristics through digital pathology.
Results: Sleep fragmentation effectively disrupted sleep patterns in both sexes, reducing light-phase sleep and increasing intradaily variability. Sleep fragmentation increased GFAP immunoreactivity in both sexes, with larger effects in females than males. Surprisingly, sleep fragmentation decreased expression of the microglial activation markers MHCII and Dectin-1 in males. Pro-inflammatory cytokines (IL-1β, CCL2, CXCL2) were significantly elevated following sleep fragmentation, with distinct regional and sex-specific patterns. In females, sleep fragmentation increased PBS-soluble and formic acid-soluble Aβ in the neocortex and medium-sized plaque density in the hippocampus, while males showed decreased detergent-soluble Aβ in the neocortex following sleep fragmentation.
Discussion: Chronic sleep fragmentation exacerbates AD-related pathology in APPSAA mice in a sex-dependent manner, with females showing greater vulnerability to Aβ accumulation and astrocyte reactivity following sleep disruption. These findings suggest that environmental sleep disruptions may contribute to the higher prevalence of AD in women and highlight the importance of addressing sleep fragmentation as a modifiable risk factor for AD.
Keywords: neuroinflammation, astrocyte reactivity, circadian rhythm, sexual dimorphism, amyloidosis, environmental stressors
Introduction
Sleep disturbances are a hallmark of Alzheimer’s disease (AD), and disrupted sleep can both result from and contribute to AD pathology.1,2 This interaction may create a self-reinforcing process in which early sleep disruption accelerates amyloid-beta (Aβ) accumulation and neuroinflammation, further degrading sleep quality and contributing to cognitive decline. In animal models, circadian disruptions can occur prior to Aβ plaque deposition, suggesting a role for soluble Aβ or APP in regulating sleep.3,4 Mechanistically, sleep disruption influences AD progression through multiple pathways. For example, disrupted sleep impairs clearance mechanisms, potentially leading to Aβ accumulation.5 Beyond Aβ dynamics, sleep fragmentation triggers neuroinflammatory responses. Sleep deprivation and fragmentation stimulate microglial activation and increase expression of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6).6–8 This heightened inflammatory state could directly contribute to AD pathology or accelerate progression from preclinical to symptomatic AD.
While multiple types of sleep disturbances can trigger these pathological mechanisms,sleep fragmentation – characterized by frequent awakenings and reduced sleep consolidation rather than solely decreased total sleep duration – has been specifically identified as a significant risk factor for AD and cognitive decline in older adults.2,9,10 Sleep fragmentation arises from numerous medical and environmental factors, including obstructive sleep apnea, insomnia, circadian rhythm disorders, and environmental disruptors such as noise and light pollution. Crucially, environmental factors and disruptions caused by caregiving responsibilities can also lead to significant sleep fragmentation.11 Moreover, sleep disorders and disruptions disproportionately affect women from caregiving and during the menopausal transition, and this sex disparity may partly underlie the higher prevalence of AD in women.9 However, the sex-specific effects of sleep fragmentation on AD-related pathology remain inadequately characterized.
Much of our mechanistic understanding of the interplay between sleep disruptions and AD pathology comes from animal models and the majority of studies have focused on males.5–8,12–17 However, traditional transgenic mouse models overexpressing human amyloid precursor protein (APP) and/or Presenilin-1 (PS1) have inherent limitations. These include potential artifactual phenotypes due to APP overexpression and effects of PS1 that are independent of Aβ. Moreover, commonly used sleep disruption protocols often involve total sleep deprivation7,12,13 rather than the intermittent fragmented sleep patterns more relevant to human experience.
The current study aims to address these critical gaps by investigating how sleep fragmentation, specifically modeling environmental disruptions rather than total sleep restriction, impacts neuroinflammation and amyloid-beta accumulation. Our early investigations of sleep fragmentation used the 3xTg-AD) model that overexpresses APP, and were restricted to females (Duncan et al, 2022). Here we investigated sex differences in the APPSAA knock-in mouse model, which harbors humanized Aβ mutations while expressing APP at normal physiological levels, avoiding the confounds of overexpression. We induced chronic sleep fragmentation using an automated motorized sweeper system that mimics the intermittent arousals characteristic of human sleep fragmentation, and assessed sleep-wake dynamics using non-invasive PiezoSleep monitoring.18 We hypothesized that chronic sleep fragmentation would exacerbate AD-related pathology, including Aβ burden and neuroinflammatory markers, and that these effects would be more pronounced in female mice, consistent with the higher prevalence of both sleep disorders and AD in women. This research provides crucial insights into the specific contribution of sleep fragmentation to AD pathogenesis, potentially identifying avenues for targeted interventions to mitigate AD risk and progression, particularly in the context of common environmental sleep disturbances.
Materials and Methods
Animals and Housing
All animal procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Protocol: 2018–3066). APPSAA mice (B6.Cg-Apptm1.1Dnli/J, RRID:IMSR_JAX:034711) of both sexes (N = 8 per sex per strain; mean age = 8.9 ± 0.5 months SD) were used in this study. The APPSAA strain is a knock-in AD model that expresses three familial AD-linked APP mutations on a C57BL/6J background, exhibiting amyloid-beta plaques and microglial activation while maintaining normal expression of APP.19 All mice were maintained on a 12-hour light:12-hour dark cycle with food and water available ad libitum. Mice were singly housed during week 1 and week 5 for sleep recording, and pair-housed with littermates during week 2 through week 4 to reduce isolation-induced stressors.
Sleep Fragmentation Protocol
Mice were randomly assigned to either undisturbed sleep (US) or chronic sleep fragmentation (SF) conditions using a stratified randomization approach to ensure balanced distribution of sex and genotype across treatment groups. The SF protocol was administered using a mechanized system (Model 80391, Lafayette Instruments, Lafayette, IN) with a sweeping bar that moved horizontally across the cage floor at a speed of 10 cm/second to induce arousal without causing stress or restricting movement. The system was programmed to sweep once every 30 seconds during the designated SF periods.
The SF condition consisted of four 1-hour intervals of enforced wakefulness distributed throughout the 12-hour light phase (ZT0-12) at ZT2-3, ZT4:30–5:30, ZT7-8, and ZT9:30–10:30, providing intermittent disruption while allowing opportunity for recovery sleep between intervals. This protocol was administered Monday through Friday for 5 consecutive weeks, with mice allowed undisturbed sleep during weekends to prevent habituation to the stimulus. This schedule resulted in a total of 25 days of sleep fragmentation (100 hours of enforced wakefulness) over the experimental period.
In the undisturbed sleep condition, the mice were housed in new cages with identical dimensions but no sweeping bar apparatus. All mice were acclimated to new cages for one week prior to experiment start. This controlled for potential confounding variables such as novel object exposure or changes in housing environment. Room temperature was maintained at 22±1°C, and staff activity in the housing room was minimized during the light phase to prevent any potential unplanned sleep disruption in both groups.
Sleep and Circadian Rhythm Monitoring
Physiology parameters to determine sleep patterns were monitored using the non-invasive piezoelectric system, which detects vibration patterns characteristic of sleep versus wake states, with sleep characterized as almost exclusively breathing rhythms.18,20 Sleep recordings were collected continuously during the experimental period when mice were in individual cages. Physiology parameters to determine sleep were quantified as the percentage of time spent asleep during the light phase, dark phase, and total 24-hour period. In addition, the data collected by the piezoelectric system were exported to the circadian rhythm analysis program ClockLab (Actimetrics V 6.1) for analysis of circadian rhythm parameters. Data is converted to an activity measure from 0 to 3, with 0 representing sleep and 3 representing continuous high activity.20 Cosinor analysis is used to fit a standard sine wave to the data to determine amplitude (difference between the peak and the trough of the rhythm, which ranges from 0 to 3), and mean estimating statistic of the rhythm (MESOR), or the mean of the model, which ranges from 0 to 3. Also in ClockLab, Non-Parametric Circadian Rhythm Analysis was used to determine two additional circadian parameters; intradaily variability (measure of fragmentation, which ranges from 0–3), and interdaily stability (measure of day-to day rhythm stability, which ranges from 0–2).21
Tissue Collection and Processing
As previously published 23,24, mice were euthanized using CO2 inhalation followed by rapid decapitation. Brains were rapidly removed, with the left hemibrain fixed in 4% paraformaldehyde for 24 hours, then transferred to 30% sucrose for cryoprotection. The right hemibrain was dissected into hippocampus and neocortex regions and flash-frozen for biochemical analyses. Fixed tissue was coronally sectioned at 30 µm thickness using a microtome, and sections were stored at −20°C in cryoprotectant until use. Mice were euthanized at 8:00 ZT.
Immunohistochemistry
Immunohistochemical staining was performed on free-floating brain sections following established protocols.22,23 Every 10th section between 1.3 mm and 2.5 mm posterior to bregma was selected for analysis to ensure comprehensive sampling. The following primary antibodies were used: rabbit anti-glial fibrillary acidic protein (GFAP; 1:10,000, Dako, catalog no. Z0334, RRID: AB_10013382) to label astrocytes; rabbit anti-ionized calcium binding adapter molecule 1 (IBA1; 1:10,000, Wako, catalog no. 019–19741) to label microglia; rat anti-Dectin-1 (1:1000, InvivoGen, catalog no. mabg-mdect, RRID: AB_2753143) for disease-associated microglia; rat anti-CD45 (1:1000, BioLegend, catalog no. 103102, RRID: AB_312967) to detect microglia/macrophages and lymphocytes; rat anti-I-A/I-E (1:1000, BioLegend, catalog no. 107601, RRID: AB_313317) for MHC class II; and mouse anti-Aβ (clone 6E10; 1:3000, BioLegend, catalog no. 803007, RRID: AB_2564657) for detection of amyloid-beta.
To assess the immunoreactivity, entire slides were digitally scanned using a Zeiss AxioScan Z.1, and quantification was performed using HALO software (Indica Labs, Albuquerque, NM). Regions of interest, including the neocortex, corpus callosum, and hippocampus, were manually outlined on each section. Positive staining was quantified using the HALO area fraction algorithm, which normalized positive pixel counts to the outlined area, thereby accounting for regional size variation and enabling standardized comparisons across samples. For Aβ immunohistochemistry, plaque density (number/mm²) was further stratified by size using the HALO object colocalization algorithm with predefined thresholds: small (10–99 μm²), medium (100–399 μm²), and large (≥400 μm²) plaques, as previously described.22,23 Slides were analyzed in batches using identical algorithm parameters, and color markup outputs were visually inspected to confirm appropriate signal detection. Quantification was conducted by observers blinded to treatment condition and genotype.
Biochemical Endpoint Analyses
As previously published,22,23 neocortical and hippocampal tissues were extracted to separate protein fractions with different solubility characteristics. Tissue was homogenized using an Omni Bead Ruptor 24 in PBS-based lysis buffer (1:20 w/v) containing 1 mM EDTA and protease inhibitors (1 mM PMSF, 1 μg/mL leupeptin). Following centrifugation at 12,000 × g for 20 minutes at 4°C, the supernatant was collected as the PBS-soluble fraction. The pellet was rehomogenized in detergent-containing buffer (tissue protein extraction reagent with Halt Protease and Phosphatase Inhibitor Cocktail; Thermo Scientific) at the same volume as the initial homogenization. After centrifugation at 12,000 × g for 20 minutes at 4°C, the supernatant was collected as the detergent-soluble fraction. The remaining pellet was processed with 70% formic acid and centrifuged at 12,000 × g at 4°C for 20 minutes. The supernatant was neutralized 1:20 with 1 M Tris (pH 11) and used as the formic acid-soluble fraction, which contains highly aggregated forms of Aβ.
Brain Cytokine and Chemokine Quantification
Neuroinflammatory cytokine and chemokine levels were measured using custom Meso Scale Discovery (MSD) V-PLEX panels (K15245D and K15048D) according to manufacturer’s instructions, as previously described.22,23 The custom panels quantified the following analytes: IL-1β, IL-6, IL-10, TNF-α, IL-33, IP-10 (CXCL10), MCP-1 (CCL2), MIP-1α (CCL3), MIP-2 (CXCL2), and KC/GRO (CXCL1). The PBS-soluble fraction (50 μL containing 100–200 μg protein) was incubated in pre-coated MSD plates overnight at 4°C. All incubation steps were performed using a VWR® Microplate Shaker at 300 rpm. Plates were washed using a BioTek Microplate Washer 50TS12 with PBS containing 0.05% Tween-20. Following addition of detection antibodies and subsequent incubation, plates were analyzed on a MESO QuickPlex SQ 120MM instrument. Standard curves were generated using recombinant protein standards (8-point standard curve with 4-fold dilutions) and sample concentrations were interpolated from these curves. All measurements were normalized to total protein content as determined by BCA Protein Assay (Thermo Scientific).
Aβ Protein Quantification
Sandwich ELISA assays were performed to quantify amyloid-beta levels as previously described.20 Immunoassay wells (96- or 384-well Immunolon 4HBX plates) were coated overnight with 1.0 μg of human-specific Aβ1-16 monoclonal antibody 42.5 in standard PBS, then blocked with SynBlock (Serotec). Samples from all three fractions (PBS-soluble, detergent-soluble, and formic acid-soluble) were diluted in antigen capture buffer (20 mM Na2HPO₄, 0.4 M NaCl, 2 mM EDTA, 0.4% BlockAce, 0.2% BSA, 0.05% CHAPS, and 0.05% NaN₃; pH 7.0). Dilution ranges were determined empirically through preliminary testing of a subset of samples. Recombinant Aβ42 (rPeptide) was used to establish standard curves in the same buffer.
Following overnight antigen capture at 4°C, plates were washed multiple times with PBS containing 0.5% Tween-20, followed by PBS alone. Plates were then probed with 1.0 μg/mL of biotinylated monoclonal antibody 4G8 (targeting Aβ17-24, Biolegend) in detection buffer (20 mM Na2HPO₄, 0.4 M NaCl, 2 mM EDTA, 1% BSA, and 0.02% thimerosal; pH 7.0). After approximately 2 hours of incubation at room temperature, plates were washed as described above and probed with 0.2 μg/mL of Neutravidin-HRP (ThermoFisher) in detection buffer. Following a final series of washes, plates were developed using TMB substrate (Kirkegaard & Perry Laboratories), and the reaction was stopped with an equal volume of 6% o-phosphoric acid. Optical densities were measured at 450 nm using a BioTek plate reader. Protein concentration was estimated via BCA assay relative to BSA standards.
Statistical Analysis
Data were analyzed using two-way analysis of variance (ANOVA) to assess the effects of biological sex and sleep condition (undisturbed sleep vs sleep fragmentation) on outcome measures including sleep parameters, neuroinflammatory markers, and Aβ levels. Age was included as a covariate in all models. Following the initial two-way ANOVA, data were stratified by sex and brain region (or by time point for sleep measures), and planned comparisons were conducted to evaluate the effect of sleep fragmentation within each subgroup, while continuing to adjust for age. Assumptions for ANOVA were verified prior to analysis. Normality of residuals was assessed using the Shapiro–Wilk test, and homogeneity of variance was evaluated using Levene’s test. When normality assumptions were violated, data were transformed accordingly. Cytokine and chemokine concentrations were normalized using z-score transformations to account for differences in dynamic range across analytes. MHCII and Dectin-1 immunoreactivity data were Box-Cox transformed to achieve normality. A cumulative inflammatory z-score was calculated to reflect overall inflammatory burden across cytokines. Effect sizes were calculated using Cohen’s d, defined as the difference between group means divided by the pooled standard deviation, within each sex and brain region. Percent change was calculated as the difference between group means (sleep fragmentation vs undisturbed sleep), divided by the undisturbed sleep mean, and multiplied by 100. Statistical significance was set at p < 0.05. All analyses were performed using JMP Pro 17 (SAS Institute, Cary, NC). Graphs were generated using GraphPad Prism version 10.4.2 (GraphPad Software, San Diego, CA). Data are presented as mean ± standard error of the mean (SEM), with individual data points shown in figures, unless noted in the figure.
Results
Sleep Fragmentation Effectively Disrupts Sleep-Wake Patterns and Circadian Rhythms in APPSAA Mice
Continuous monitoring of sleep-wake patterns in APPSAA mice using piezoelectric sensors demonstrated clear alterations in the 24-hour sleep profile in both sexes, showing that our sleep fragmentation protocol effectively induced sleep disruption (Figure 1A). As expected, during the light phase, when the sleep was fragmented, there was a main effect of sleep fragmentation at week 1 (F = 2350.93, p < 0.0001) and week 5 (F = 958.23, p < 0.0001) and an interaction of sleep fragmentation and sex at week 5 (F = 57.53, p = 0.048). Stratifying the data by sex and week to evaluate the effect of sleep fragmentation showed that mice with sleep fragmentation had significantly less sleep across all groups (p < 0.01).
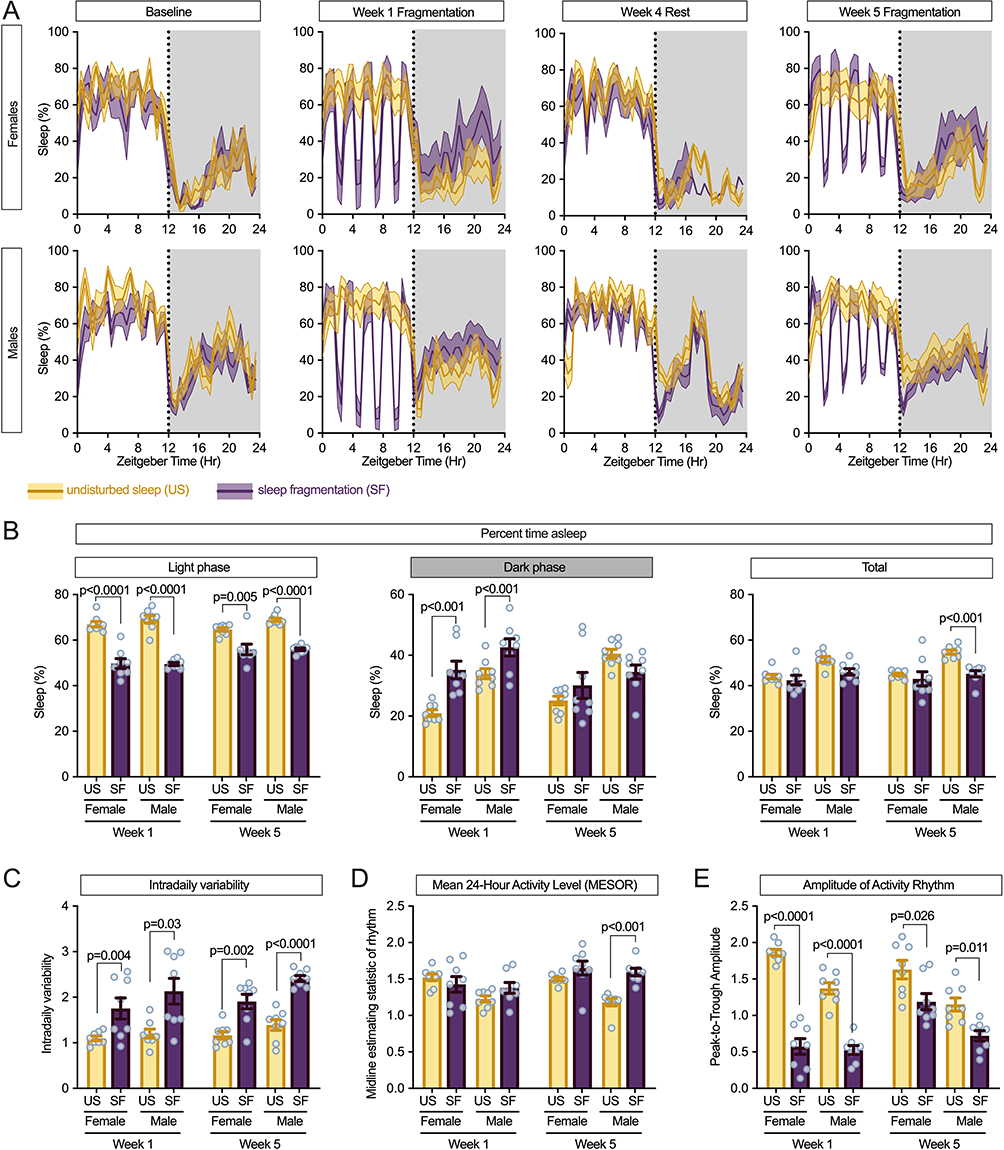 |
Figure 1 Sleep fragmentation disrupts sleep-wake patterns and circadian rhythms in APPSAA mice. (A) Twenty-four-hour sleep profiles showing percentage of time spent asleep throughout the day (Zeitgeber time) in female (top) and male (bottom) mice at baseline, week 1 of fragmentation, week 4 rest period, and week 5 of fragmentation. Yellow lines represent undisturbed sleep (US) controls; purple lines represent sleep fragmentation (SF) groups. Shaded areas indicate the dark phase (active period). Data shown as mean with shaded regions representing SD. Vertical dotted lines indicate light-dark transitions. (B) Percentage of time spent asleep during the light phase (left), dark phase (middle), and total 24-hour period (right) at weeks 1 and 5. Sleep fragmentation significantly decreased sleep during the light phase while increasing sleep during the dark phase, particularly in females at week 1. (C) Intradaily variability (IV), a measure of rhythm fragmentation, showing significant increases with sleep fragmentation in both sexes at weeks 1 and 5. Higher values indicate more disrupted and fragmented daily rhythms, a characteristic associated with neurodegenerative conditions. (D) Mean 24-hour activity level (MESOR) showing increased overall activity with sleep fragmentation, with a significant sex-by-treatment interaction at week 5. (E) Amplitude of the activity rhythm (peak-to-trough difference) showing pronounced reduction with sleep fragmentation, indicating flattening of the circadian rhythm. The effect was more persistent in males at week 5. Data in (A) are presented as mean ± SD. Data in (B-E) are presented as mean ± SEM with individual data points (n = 8 per group). P-values from post-hoc analyses between US and SF conditions are indicated. |
Conversely, in the dark phase (no sleep fragmentation), a main effect of sleep fragmentation (F = 1051.98, p < 0.0001) and sex (F = 794.01, p = 0.0002) was seen at week 1. By week 5, a main effect of sex (F = 792.81, p = 0.001) but not sleep fragmentation was found. During the dark phase at week 1, both female and male mice slept more after sleep fragmentation (p < 0.001) (Figure 1B). The overall 24-hour sleep time showed a significant main effect of sleep fragmentation at week 5 (F = 10.81, p = 0.0028), with a significant interaction between sex and sleep fragmentation (F = 5.18, p = 0.0310), reflecting greater resilience to chronic sleep disruption in females compared to males. At both week 1 (F = 12.71, p = 0.0014) and week 5 (F = 11.80, p = 0.0019), there was a main effect of sex with male mice sleeping more than female mice.
To assess circadian rhythm integrity, we analyzed several key parameters of the daily activity pattern. Intradaily variability (IV), a direct measure of rhythm fragmentation, was significantly increased by the sleep fragmentation protocol in both sexes at week 1 (F = 13.29, p = 0.0011) and became even more pronounced by week 5 (F = 53.85, p < 0.0001), indicating progressive deterioration of rhythm consolidation (Figure 1C). This effect was observed in both females (week 1: p = 0.004; week 5: p = 0.002) and males (week 1: p = 0.03; week 5: p < 0.0001).
Mean 24-hour activity level (MESOR) showed significant main effects of both sex (week 1: F = 5.39, p = 0.0280; week 5: F = 9.20, p = 0.0053) and sleep fragmentation condition (week 5: F = 20.19, p = 0.0001), with a significant interaction at week 5 (F = 4.74, p = 0.0383) (Figure 1D). Post-hoc analysis revealed that sleep fragmentation significantly increased overall activity levels in both females and males by week 5, with a more pronounced effect in males (p < 0.001).
The amplitude of the daily activity rhythm, which reflects the robustness of circadian oscillation, was dramatically reduced by sleep fragmentation (Figure 1E). Significant main effects of sleep fragmentation on rhythm amplitude were observed at both week 1 (F = 167.47, p < 0.0001) and week 5 (F = 15.35, p = 0.0006), along with significant main effects of sex (week 1: F = 12.44, p = 0.0015; week 5: F = 20.80, p < 0.0001) and a significant interaction at week 1 (F = 7.92, p = 0.0090). Sleep fragmentation reduced rhythm amplitude in both females (week 1: p < 0.0001; week 5: p = 0.026) and males (week 1: p < 0.0001; week 5: p = 0.011).
Sleep Fragmentation Alters Aβ Levels and Plaque Characteristics in a Sex- and Brain Region-Dependent Manner
Sleep fragmentation exacerbated Aβ pathology in female APPSAA knock-in mice, revealing important sex differences in AD-related mechanisms. Unlike previous studies that used transgenic overexpression models, our examination of APPSAA mice, which express Aβ from the endogenous promoter at physiological levels, tests whether sleep fragmentation affects Aβ dynamics.
We used ELISA to measure different fractions of Aβ in the brain (Figure 2A). In the PBS-soluble fraction, which represents primarily monomeric and small oligomeric Aβ species, sleep fragmentation significantly increased Aβ levels in the neocortex of female mice (p = 0.015), while no significant changes were observed in males or in the hippocampus of either sex (Figure 2A, left panel), and no main effect or interactions. The detergent-soluble fraction, which contains larger oligomeric Aβ aggregates, showed significant effects of sleep fragmentation in both sexes, but with divergent patterns. In the neocortex, sleep fragmentation increased Aβ levels in females (p = 0.05) but decreased them in males (p = 0.017). In the hippocampus, a significant main effect of sleep fragmentation was observed for detergent-soluble Aβ (F = 14.67, p = 0.0007), with post-hoc analyses showing consistent decreases in this Aβ fraction in both females (p = 0.01) and males (p = 0.057) (Figure 2A). For the formic acid-soluble fraction, which contains highly insoluble fibrillar Aβ typically aggregated in plaques, a significant sex-by-sleep fragmentation interaction was found in the neocortex (F = 6.33, p = 0.0181). Sleep fragmentation significantly increased Aβ levels in the neocortex of female mice (p = 0.001) but had no significant effect in the male neocortex or in the hippocampus of either sex (Figure 2A).
 |
Figure 2 Sleep fragmentation alters Aβ levels and plaque characteristics in APPSAA mice. (A) ELISA measurements of Aβ in PBS-soluble (left), detergent-soluble (middle), and formic acid-soluble (right) fractions from neocortex and hippocampus. Sleep fragmentation increased PBS-soluble and formic acid-soluble Aβ in female neocortex while decreasing detergent-soluble Aβ in the hippocampus of both sexes. (B) Representative images of 6E10 immunostaining showing Aβ plaques in male and female APPSAA mice with undisturbed sleep (US) or sleep fragmentation (SF). Scale bars: 25 μm. (C) Quantification of total Aβ plaque burden (% area) measured by digital pathology using the HALO area fraction algorithm. (D) Plaque density (number/mm²) by size category: small (10–99 μm²), medium (100–399 μm²), and large (≥400 μm²) plaques, quantified using the HALO object colocalization algorithm with size exclusions. Sleep fragmentation significantly increased medium-sized plaque density in female hippocampus. Data presented as mean ± SEM with individual data points (n = 8 per group). |
Immunohistochemical analysis of Aβ plaques using the 6E10 antibody revealed morphological differences in plaque characteristics between experimental groups (Figure 2B). While total Aβ plaque burden (% area) showed no significant main effects of sleep fragmentation in either brain region, a significant main effect of sex was detected in the hippocampus (F = 5.59, p = 0.0255), with females exhibiting higher plaque burden than males (Figure 2C). A trend for a sleep fragmentation effect on plaque burden was also seen in the hippocampus (F = 3.701, p = 0.065).
Further analysis of plaque characteristics by size revealed differences in specific plaque populations. For medium-sized plaques (100–399 μm²), sleep fragmentation significantly increased plaque density in the hippocampus of female mice (p = 0.034), while no significant changes were observed in other size categories (Figure 2D). Sex was associated with differences in plaque counts in multiple size categories, including small plaques in both cortex (F = 6.01, p = 0.0210) and hippocampus (F = 4.88, p = 0.0358), and medium plaques in both cortex (F = 4.82, p = 0.0369) and hippocampus (F = 10.44, p = 0.0032). These findings suggest that sleep fragmentation differentially affects Aβ processing and aggregation in male and female APPSAA mice, with female mice showing increased soluble and insoluble Aβ in the neocortex, while both sexes showed decreased oligomeric Aβ in the hippocampus following sleep fragmentation.
Sleep Fragmentation Increases Neuroinflammatory Cytokines and Chemokines in APPSAA Mice
MSD multiplex analysis of brain cytokines and chemokines showed a pattern of greater neuroinflammatory responses to sleep fragmentation than undisturbed sleep in APPSAA mice, along with sex and regional differences (Figure 3A). In the neocortex, analysis revealed sleep fragmentation-induced increases in IL-1β (F = 5.17, p = 0.0312), CCL2 (F = 11.04, p = 0.0026), CXCL2 (F = 11.53, p = 0.0021), CXCL1 (F = 4.58, p = 0.0414), and CXCL10 (F = 4.41, p = 0.0451). A significant sex-by-sleep fragmentation interaction was also detected for CXCL2 in the neocortex (F = 5.05, p = 0.0330). In the neocortex, significant main effects of sex were observed for TNF-α (F = 9.39, p = 0.0049), CCL2 (F = 8.47, p = 0.0072), and CXCL10 (F = 6.80, p = 0.0147). In the hippocampus, fewer markers were significantly altered, with only CXCL2 showing a significant main effect of sleep condition (fragmentation-induced increase, F = 8.98, p = 0.0058). Of all the cytokines measured, only IL-33 was decreased by sleep fragmentation in the hippocampus (F = 5.85, p = 0.0226), with this effect being more pronounced in male mice.
 |
Figure 3 Sleep fragmentation increases neuroinflammatory cytokines and chemokines in APPSAA mice. (A) Heatmap displaying z-score normalized levels of cytokines and chemokines in the neocortex and hippocampus of female and male APPSAA mice with undisturbed sleep (US) or sleep fragmentation (SF). Yellow-Orange indicates higher expression while purple-black indicates lower expression relative to the mean across all groups. Bold and asterisk in the cytokine name indicate the main effect of sleep fragmentation (*p<0.05, **p<0.01). In the heatmap boxes, asterisks indicate significant differences (p < 0.05) between US and SF conditions within each sex and brain region. (B) Cumulative inflammatory z-score representing the overall cytokine/chemokine burden in neocortex and hippocampus. Sleep fragmentation significantly increased the inflammatory index in both female and male neocortex. (C–E) Absolute concentrations (pg/mg total protein) of representative inflammatory mediators: IL-1β (C), CCL2 (D), and CXCL2 (E). Data presented as mean ± SEM with individual data points (n = 8 per group). |
To assess the overall inflammatory burden, we calculated a cumulative z-score across all measured cytokines and chemokines. Two-way ANOVA revealed robust effects of both sleep fragmentation (F = 16.96, p = 0.0003) and sex (F = 9.28, p = 0.0051) on the cumulative inflammatory score in the neocortex, without a significant interaction between these factors (F = 1.94, p = 0.1749). In contrast, the hippocampus showed no significant main effects of either sleep fragmentation (F = 0.69, p = 0.4149) or sex (F = 2.18, p = 0.1515) on the cumulative score. Post-hoc analysis demonstrated that sleep fragmentation significantly increased the inflammatory index in both female (p = 0.009) and male mice (p = 0.019) in the neocortex (Figure 3B).
To further evaluate the changes in neuroinflammation, we focused on three of the cytokines and chemokines altered by sleep fragmentation. IL-1β showed a substantial increase in the neocortex of male mice (49.4% increase, Cohen’s d = 1.21) but only modest changes in females (12.7% increase, Cohen’s d = 0.45) (Figure 3C). CCL2 demonstrated pronounced increases in both female (30.0% increase, Cohen’s d = 1.02) and male (53.6% increase, Cohen’s d = 1.58) neocortex (Figure 3D). CXCL2 levels were consistently elevated across both sexes and brain regions following sleep fragmentation, with particularly strong and statistically significant effects in the hippocampus of male mice (42.1% increase, Cohen’s d = 1.63) (Figure 3E).
Sleep Fragmentation Increases Astrocyte Reactivity
Immunohistochemical analysis of GFAP expression revealed significant effects of both sex and sleep condition on astrocyte reactivity in APPSAA knock-in mice (Figure 4). In general, higher levels of GFAP expression were associated with the sleep fragmentation condition and female sex, in both neuroanatomical regions examined. Concerning the neocortex, two-way ANOVA indicated significant main effects of sex (F = 77.45, p < 0.0001) and sleep fragmentation (F = 32.54, p < 0.0001) as well as a significant interaction between sex and sleep fragmentation on GFAP-positive area (F = 4.87, p = 0.0361). Thus, female sex appeared to exacerbate the stimulatory effect of sleep fragmentation on astrocyte reactivity in the neocortex.
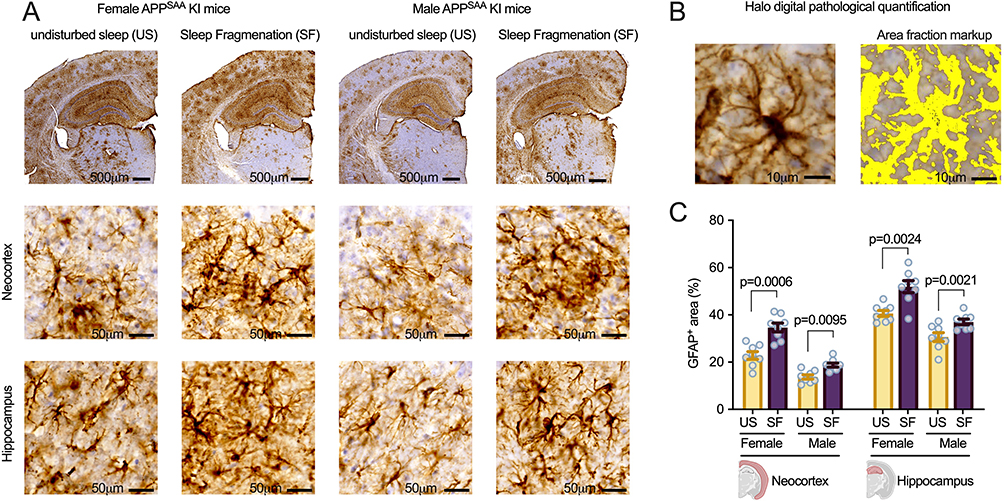 |
Figure 4 Sleep fragmentation increases astrocyte reactivity in both male and female APPSAA mice. (A) Representative images of GFAP immunostaining showing distribution across brain sections, with higher magnification images of the neocortex and hippocampus from male and female APPSAA knock-in mice with undisturbed sleep (US) or sleep fragmentation (SF). Scale bars: 500 μm (top row), 50 μm (middle and bottom rows). (B) Example of HALO digital pathology quantification showing area fraction markup of GFAP-positive staining (yellow). Scale bars: 10 μm. (C) Quantification of GFAP-positive area (%) in the neocortex and hippocampus of female and male APPSAA mice with US or SF (n = 8 per group). Quantification was performed at 300 μm intervals throughout each brain region using HALO digital pathology software. Data are presented as mean ± SEM with individual data points shown. Sleep fragmentation increased GFAP immunoreactivity in the neocortex of females (52.2% increase, Cohen’s d = 2.47) and males (36.2% increase, Cohen’s d = 1.97), and in the hippocampus of females (28.1% increase, Cohen’s d = 2.13) and males (20.9% increase, Cohen’s d = 1.44). P-values from planned post-hoc analyses between US and SF conditions are indicated. |
In the hippocampus, two-way ANOVA similarly revealed significant main effects of sex (F = 54.77, p < 0.0001) and sleep fragmentation (F = 29.68, p < 0.0001) on GFAP-positive area. However, no significant interaction between sex and sleep fragmentation was detected in this region (F = 0.39, p = 0.5372).
Post-hoc analyses examining the effect of sleep fragmentation within each sex showed that sleep fragmentation significantly increased GFAP-positive area in both males and females across both brain regions examined. In the neocortex, sleep-fragmented female mice showed a significant increase in GFAP immunoreactivity compared to undisturbed sex-matched controls (F = 20.04, p = 0.0006, Cohen’s d = 2.47, 52.2% increase), as did sleep-fragmented males (F = 9.24, p = 0.0095, Cohen’s d = 1.97, 36.2% increase). In the hippocampus, sleep fragmentation similarly increased GFAP immunoreactivity in both females (F = 14.15, p = 0.0024, Cohen’s d = 2.13, 28.1% increase) and males (F = 14.65, p = 0.0021, Cohen’s d = 1.44, 20.9% increase).
Sleep Fragmentation Decreases Markers of Reactive Microglia/Macrophages in Male Mice
APPSAA mice displayed robust microglial and macrophage signatures that were selectively affected by sleep fragmentation (Figure 5A–D). IBA1-positive microglia were abundant throughout the neocortex and hippocampus, with intensified labeling surrounding amyloid plaques. These plaque-associated microglia displayed reactive morphology characterized by enlarged cell bodies and thickened processes, with similar patterns observed in both sexes. Despite this robust microglial presence, quantitative analysis revealed no significant effects of sleep fragmentation on IBA1-positive area in either the neocortex (F = 0.19, p = 0.6651) or hippocampus (F = 1.05, p = 0.3145), and no significant sex-by-sleep condition interactions were detected (Figure 5E).
 |
Figure 5 Sleep fragmentation selectively reduces MHCII and Dectin-1 expression in male APPSAA mice. (A) Representative low-magnification images show the pattern of IBA1, CD45, MHCII, and Dectin-1 staining across the neocortex, hippocampus, and deeper structures such as the thalamus, with the most robust staining observed in the neocortex and hippocampus. (B) Widefield epifluorescence images show that MHCII colocalizes with IBA1⁺ microglia but not GFAP⁺ astrocytes. (C) Dectin-1 also colocalizes with IBA1⁺ microglia but not GFAP⁺ astrocytes. (D) Representative images illustrate distinct microglial/macrophage phenotypes in APPSAA mice by treatment group. IBA1 reveals plaque-associated microglia with hypertrophic morphology; CD45 identifies potential infiltrating leukocytes, particularly in the female hippocampus; MHCII labels antigen-presenting cells, predominantly in the male hippocampus under undisturbed sleep; and Dectin-1 marks phagocytic cells associated with damage-responsive states. Arrow highlights a Dectin-1⁺ cell that appears to be engulfing another cell. Halo digital pathological quantification of IBA1 (E), CD45 (F), MHCII (G), and Dectin-1 (H) in neocortex and hippocampus. Scale bars: A = 500 μm; B–D = 25 μm. |
CD45 immunoreactivity displayed a unique pattern of staining, particularly in female mice, where small round CD45-positive cells were observed in the hippocampus that were not detected with other microglial markers. These cells were morphologically distinct from typical microglia and appeared to be absent or less abundant in male mice. Given that CD45 is a leukocyte common antigen, these cells may represent infiltrating lymphocytes present in the APPSAA mice, which were present regardless of sleep condition. For CD45, quantitative analysis showed a significant main effect of sex in the neocortex (F = 7.37, p = 0.0114, Cohen’s d = 0.97), with females exhibiting higher CD45 immunoreactivity than males. However, sleep fragmentation did not significantly affect CD45-positive area in either brain region, and no significant sex-by-sleep fragmentation interactions were observed (Figure 5F).
MHCII immunostaining, indicative of antigen-presenting capacity, labeled large bushy cells with a distinctive morphology. In the hippocampus of male APPSAA mice with undisturbed sleep, there was notably intense MHCII staining compared to other groups, suggesting enhanced microglial priming in these animals. These MHCII-positive cells appeared to be concentrated in specific hippocampal subregions, particularly in the CA1 region (Figure 5A and D), rather than being uniformly distributed. Interestingly, in the hippocampus of male mice, sleep fragmentation significantly decreased MHCII immunoreactivity (p = 0.015, Cohen’s d = 0.74), with mean values decreasing from 0.84% in undisturbed sleep to 0.25% with sleep fragmentation (70.2% decrease). This effect was specific to males, as female mice showed minimal MHCII expression regardless of sleep condition (Figure 5G).
Dectin-1 is a C-type lectin receptor on myeloid cells, linked to phagocytosis and pro-inflammatory cytokine production, and is also a damage-associated (DAM) microglia gene (Clec7A).24,25 Dectin-1 positive cells were less abundant compared to the other markers but displayed a distinctive morphology (Figure 5A and D). Some Dectin-1 positive cells appeared to be in the process of engulfing other cells, consistent with the role of this receptor in phagocytosis. The distribution of these cells was patchy rather than uniform throughout the tissue. Similarly to MHCII, sleep fragmentation significantly reduced hippocampal Dectin-1 immunoreactivity in male mice (p = 0.018, Cohen’s d = 0.67), with mean values decreasing from 0.55% to 0.25% (54.5% decrease). For Dectin-1 in the neocortex, two-way ANOVA revealed a significant main effect of sleep fragmentation (F = 4.99, p = 0.0339, Cohen’s d = 0.51), with sleep fragmentation generally reducing Dectin-1 immunoreactivity. Additionally, a significant sex-by-sleep fragmentation interaction was observed for hippocampal Dectin-1 (F = 6.60, p = 0.0160, Cohen’s d = 0.73), indicating that the effect of sleep fragmentation on this marker differed between males and females (Figure 5H). Collectively, these results demonstrate that sleep fragmentation selectively reduces markers associated with microglial activation and phagocytosis in male APPSAA mice, with minimal effects on overall microglial presence.
Discussion
Here, we show that chronic sleep fragmentation alters neuroinflammation and Aβ pathology in a sex-specific manner in APPSAA knock-in mice. Our protocol successfully disrupted the normal daily sleep pattern, reducing sleep during the light (rest) phase and increasing it during the dark (active) phase. Circadian rhythm disturbances, including increased intradaily variability and reduced rhythm amplitude, accompanied the sleep fragmentation protocol. Increased intradaily variability and decreased amplitude have both been identified as risk factors for AD.26 These changes in the daily sleep-wake rhythm were associated with different effects on AD-related pathology in males and females. In females, sleep fragmentation increased PBS-soluble and formic acid–soluble Aβ in the neocortex, while in males, sleep fragmentation decreased detergent-soluble Aβ levels, suggesting sex-dependent differences in Aβ processing and/or aggregation. After sleep fragmentation, pro-inflammatory cytokines and chemokines (IL-1β, CCL2, and CXCL2) were elevated in the neocortex of both sexes, but hippocampal cytokine levels increased only in males. Unexpectedly, markers of microglial activation (MHCII and Dectin-1) were reduced in males, indicating a distinct microglial response to sleep fragmentation. GFAP immunoreactivity was elevated in both sexes following sleep fragmentation, with females showing consistently higher astrocyte reactivity across conditions. Together, these results highlight sex differences in the brain’s response to sleep fragmentation and point to possible mechanisms by which sleep fragmentation could influence AD progression.
Sleep fragmentation is a common feature of human life, often driven by environmental factors such as caregiving responsibilities, shift work, or noise.27 In our mouse model, by 5 weeks of daily sleep fragmentation, female mice preserved total daily sleep time by increasing sleep immediately after the intervals of enforced wakefulness and during the active phase. Males, by contrast, failed to fully compensate, resulting in reduced total sleep and increased activity, which are features suggestive of elevated arousal or inadequate sleep pressure.28–30 Sex differences in response to sleep fragmentation may be biologically significant, as persistent arousal has been linked to immune dysregulation and metabolic stress in both animal and human studies.31–33 Our data also suggest that the circadian rhythm disruption induced by sleep fragmentation, particularly in female mice, may influence AD-related pathology independent of total sleep loss. This is consistent with an earlier finding in elderly humans that fragmentation of the daily rest-activity rhythm was associated with increased incidence of AD, even when the total daily rest time was accounted for.34 Although the relationship between circadian disruption and AD in humans is likely bidirectional,2,9,10 our results support a model in which environmental perturbations to circadian rhythms can contribute to AD-related pathology. While this connection remains correlative, the parallels to human studies underscore the importance of considering rhythm integrity, not just sleep quantity, as a potentially modifiable factor in disease progression. Interestingly, although males showed less behavioral compensation for sleep disruption, they exhibited reduced levels of oligomeric Aβ, in contrast to the increased Aβ accumulation observed in females. This paradox highlights complex, sex-specific relationships between sleep, circadian function, and Aβ metabolism that are not easily explained by sleep duration alone.
Our current findings in the APPSAA mice of both sexes expand upon our previous studies in the 3x-Tg AD female and revealed some differences. In the 3x-Tg AD female, sleep fragmentation selectively elevated Aβ levels and neuroinflammatory markers in the detergent-soluble fraction of the hippocampus without significant effects in the neocortex. Overexpression of APP and other genotype differences may modulate the effects of sleep fragmentation on neuropathological changes, as we have also observed with studies of other AD mouse strains.6
The contrasting effects of sleep fragmentation on Aβ pathology in males and females may reflect intrinsic variations in how male and female brains respond to sleep disruption at a molecular level, potentially involving sex-dependent alterations in glymphatic clearance, Aβ production, or aggregation dynamics. In female APPSAA mice, sleep fragmentation consistently increased Aβ burden, evident across both soluble and insoluble fractions and confirmed by histological evidence for a trend of greater plaque density. This suggests that sleep disruption promotes both enhanced Aβ production and aggregation in females. In contrast, males exhibited a selective decrease in detergent-soluble Aβ, typically enriched for oligomeric species, which are thought to be particularly neurotoxic in AD.35–37 This reduction occurred without changes in other Aβ pools or plaque burden, raising the possibility that males activate distinct compensatory mechanisms in response to sleep disruption. One potential explanation for this difference lies in the baseline microglial state, as males under control conditions showed higher expression of MHCII and Dectin-1 (markers linked to antigen presentation and phagocytosis24,38) suggesting a more activated immune profile that could enhance Aβ clearance.39 Although sleep fragmentation reduced these markers in males, their overall clearance capacity may still exceed that of females, who showed lower baseline expression. Astrocyte reactivity may also contribute to these differences, with females exhibiting higher GFAP immunoreactivity, which was further elevated by sleep fragmentation. Increased astrocytic reactivity has been associated with mislocalization of aquaporin-4, a key component of the glymphatic system, potentially impairing waste clearance during sleep, further reducing glymphatic efficiency associated with sleep fragmentation in females, compounding their susceptibility to Aβ accumulation.40–43 Yet, hormonal influences may also play a role, as estrogen modulates Aβ metabolism and inflammatory responses.44–46 These sex-specific responses highlight that sleep and Aβ pathology are linked through multiple overlapping factors, including immune activity, waste clearance, and hormones, which warrant further research.
While our study offers new insight into how sleep fragmentation differentially affects AD-related pathology in male and female APPSAA mice, there are several limitations. First, the cross-sectional design restricted our ability to assess the temporal progression of pathology or identify early markers of vulnerability. We focused on a single mid-life time point (8 months), but the effects of sleep fragmentation may differ across the lifespan, particularly in relation to hormonal transitions and age-related changes in clearance mechanisms. We also did not directly measure glymphatic function (eg, via in vivo imaging) or track real-time Aβ dynamics (eg, via microdialysis), which limited our mechanistic insight. While we observed sex differences in neuroinflammatory markers, our study was not designed to test whether these immune changes causally drive Aβ accumulation. Lastly, we did not assess functional outcomes such as behavior, synaptic integrity, or persistent circadian disruption following the cessation of sleep fragmentation.
Our findings highlight several important directions for future research. First, longitudinal investigations tracking sleep architecture, neuroinflammation, and Aβ pathology could identify critical sequences and early vulnerability indicators. Second, mechanistic studies should incorporate direct assessments of glymphatic clearance to evaluate whether impaired waste removal contributes to sex-dependent Aβ accumulation, and if targeting astrocytic reactivity may reveal whether elevated GFAP expression impairs glymphatic function or serves a compensatory role. Third, targeted interventions that modulate microglial activation or cytokine signaling could help determine whether neuroinflammation contributes to pathological or functional outcomes. Finally, it will be critical to test whether interventions that improve sleep quality or stabilize circadian rhythms can mitigate Aβ pathology, particularly in females. These could include pharmacological agents that enhance slow-wave sleep, chronotherapeutic approaches to strengthen circadian timing, or environmental modifications such as thermoregulation and reducing nighttime light exposure. Such strategies, if effective, may offer low-cost, non-invasive options to reduce the impact of sleep and circadian disruption on AD risk.
Conclusion
In sum, our findings demonstrate that chronic sleep fragmentation induces sex-specific changes in Aβ pathology, neuroinflammation, and circadian rhythm disruption in an AD-relevant mouse model. These results highlight the importance of considering both biological sex and circadian integrity when evaluating the impact of environmental sleep disturbances on neurodegenerative disease processes. While additional work is needed to clarify underlying mechanisms and long-term outcomes, this study provides a foundation for future investigations into how modifiable sleep and circadian factors may influence AD vulnerability and progression.
Declaration of Generative AI Usage in Scientific Writing
During the preparation of this work, the author(s) used ChatGPT and Claude to assist in evaluating the original text for clarity, completeness, and style. These tools provided editorial suggestions and recommendations; however, the author(s) reviewed and edited all content as necessary. The author(s) take full responsibility for the final content of the published article.
Abbreviations
AD, Alzheimer’s disease; Aβ, Amyloid-beta; APP, Amyloid precursor protein; APPSAA, Alzheimer’s disease mouse model with APP mutations; ANOVA, Analysis of variance; BCA, Bicinchoninic acid assay; BSA, Bovine serum albumin; CA1, Cornu Ammonis 1 region of hippocampus; CCL2, C-C Motif Chemokine Ligand 2 (MCP-1); CCL3, C-C Motif Chemokine Ligand 3 (MIP-1α); CD45, Cluster of Differentiation 45 (leukocyte common antigen); CXCL1, C-X-C Motif Chemokine Ligand 1 (KC/GRO); CXCL2, C-X-C Motif Chemokine Ligand 2 (MIP-2); CXCL10, C-X-C Motif Chemokine Ligand 10 (IP-10); DAM, Damage-associated microglia; EDTA, Ethylenediaminetetraacetic acid; ELISA, Enzyme-linked immunosorbent assay; GFAP, Glial fibrillary acidic protein; HRP, Horseradish peroxidase; IBA1, Ionized calcium binding adapter molecule 1; IHC, Immunohistochemistry; IL-1β, Interleukin-1 beta; IL-6, Interleukin-6; IL-10, Interleukin-10; IL-33, Interleukin-33; IP-10, Interferon gamma-induced protein 10 (CXCL10); IV, Intradaily variability; KC/GRO, Keratinocyte chemoattractant/growth-regulated oncogene (CXCL1); MCP-1, Monocyte chemoattractant protein-1 (CCL2); MESOR, Midline estimating statistic of rhythm; MHCII, Major histocompatibility complex class II; MIP-1α, Macrophage inflammatory protein-1 alpha (CCL3); MIP-2, Macrophage inflammatory protein-2 (CXCL2); MSD, Meso Scale Discovery; PBS, Phosphate-buffered saline; PMSF, Phenylmethylsulfonyl fluoride; PS1, Presenilin-1; SEM, Standard error of the mean; SF, Sleep fragmentation; TMB, 3,3′,5,5′ -Tetramethylbenzidine; TNF-α, Tumor necrosis factor alpha; US, Undisturbed sleep; ZT, Zeitgeber time.
Acknowledgments
Equipment used in this study was supported by the Office of the Vice President for Research at the University of Kentucky (SS and MJD). Research was supported by the National Institutes of Health (NIH R01 AG068215; MPM, MJD, ADB, BFO, and SS).
Author Contributions
MRH, HRW, CEJ, TM, MFC, MGL, KNR, HMH, EGD, ADB conducted the research. MPM, MJD, BFO, SS, and ADB provided funding and contributed to study design. ADB led the data analysis and wrote the initial draft of the manuscript. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
BFO is a co-owner of Signal Solutions, LLC, which manufactures the piezoelectric sleep monitoring equipment used in this study. He also reports NIH grants to Signal Solutions, typically with 50% subawarded to UK, but not directly related to this manuscript (but potentially synergistic). On his current STTR award, he is the PI through his UK faculty position, even though the award is to Signal Solutions. He also reports multiple conflict of interest management plans, and a UK COI committee, to help him manage these potential conflicts of interest. He receives a portion of his yearly income from Signal Solutions, that is variable, but averages roughly 25% of his yearly salary. He attends most scientific meetings paid by Signal Solutions, even when presenting UK related research, mainly because it is easier; non-financial support includes admin support from his company, to free up his time for both UK and the company; reports patent “Stimulation System Based on Mechanical Vibration for Modification and Characterization of Sleep and Behavior in Rodents”, not directly relevant to the manuscript. MJD reports honoraria for reviews of grants related to research on Alzheimer’s disease from State of Florida Department of Health. All other authors report no conflicts of interest related to this work.
References
1. Dufort-Gervais J, Mongrain V, Brouillette J. Bidirectional relationships between sleep and amyloid-beta in the hippocampus. Neurobiol Learn Mem. 2019;160:108–117. doi:10.1016/j.nlm.2018.06.009
2. Yes J, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology-a bidirectional relationship. Nat Rev Neurol. 2014;10(2):115–119. doi:10.1038/nrneurol.2013.269
3. Sterniczuk R, Dyck RH, Laferla FM, Antle MC. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes. Brain Res. 2010;1348:139–148. doi:10.1016/j.brainres.2010.05.013
4. Ambree O, Touma C, Gortz N, et al. Activity changes and marked stereotypic behavior precede Abeta pathology in TgCRND8 Alzheimer mice. Neurobiol Aging. 2006;27(7):955–964. doi:10.1016/j.neurobiolaging.2005.05.009
5. Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326(5955):1005–1007. doi:10.1126/science.1180962
6. Duncan MJ, Guerriero LE, Kohler K, et al. Chronic fragmentation of the daily sleep-wake rhythm increases amyloid-beta levels and neuroinflammation in the 3xTg-AD mouse model of Alzheimer’s disease. Neuroscience. 2022;481:111–122. PMID: 34856352. doi:10.1016/j.neuroscience.2021.11.042
7. Parhizkar S, Gent G, Chen Y, et al. Sleep deprivation exacerbates microglial reactivity and AP deposition in a TREM2-dependent manner in mice. Sci Trans med. 2023;15(693):eade6285. doi:10.1126/scitranslmed.ade6285
8. Zhu Y, Zhan GX, Fenik P, et al. Chronic sleep disruption advances the temporal progression of tauopathy in P301S mutant mice. J Neurosci. 2018;38(48):10255–10270. doi:10.1523/jneurosci.0275-18.2018
9. Johnson CE, Duncan MJ, Murphy MP. Sex and sleep disruption as contributing factors in Alzheimer’s disease. J Alzheimers Dis. 2024;97(1):31–74. doi:10.3233/jad-230527
10. Lucey BP, Bateman RJ. Amyloid-beta diurnal pattern: possible role of sleep in Alzheimer’s disease pathogenesis. Neurobiol Aging. 2014;35 (Suppl 2):S29–34. doi:10.1016/j.neurobiolaging.2014.03.035
11. Gottesman RF, Lutsey PL, Benveniste H, et al. Impact of sleep disorders and disturbed sleep on brain health: a scientific statement from the American heart association. Stroke. 2024;55(3):E61–E76. doi:10.1161/str.0000000000000453
12. Cankar N, Beschorner N, Tsopanidou A, et al. Sleep deprivation leads to non-adaptive alterations in sleep microarchitecture and amyloid-b accumulation in a murine Alzheimer model. Cell Rep. 2024;43(11):114977. doi:10.1016/j.celrep.2024.114977
13. Di Meco A, Joshi YB, Praticò D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol Aging. 2014;35(8):1813–1820. doi:10.1016/j.neurobiolaging.2014.02.011
14. Holth JK, Fritschi SK, Wang C, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019;363(6429):880–883. doi:10.1126/science.aav2546
15. Kincheski GC, Valentim IS, Clarke JR, et al. Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-β oligomers in mice. Brain Behav Immun. 2017;64:140–151. doi:10.1016/j.bbi.2017.04.007
16. Martin SC, Joyce KK, Lord JS, et al. Sleep disruption precedes forebrain synaptic tau burden and contributes to cognitive decline in a sex-dependent manner in the P301S tau transgenic mouse model. Eneuro. 2024;11(6):e242024. doi:10.1523/eneuro.0004-24.2024
17. Niu L, Zhang F, Xu XJ, et al. Chronic sleep deprivation altered the expression of circadian clock genes and aggravated Alzheimer’s disease neuropathology. Brain Pathol. 2022;32(3):e13028. doi:10.1111/bpa.13028
18. Yaghouby F, Donohue KD, O’Hara BF, Sunderam S. Noninvasive dissection of mouse sleep using a piezoelectric motion sensor. J Neurosci Meth. 2016;259:90–100. doi:10.1016/j.jneumeth.2015.11.004
19. Xia D, Lianoglou S, Sandmann T, et al. Novel app knock-in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol Neurodegener. 2022;17(1):41. doi:10.1186/s13024-022-00547-7
20. Turton SM, Padgett S, Maisel MT, et al. Interactions between daily sleep-wake rhythms, γ-secretase, and amyloid-β peptide pathology point to complex underlying relationships. Biochimica et Biophysica Acta. 2025;1871(6):167840. doi:10.1016/j.bbadis.2025.167840
21. Witting W, Kwa IH, Eikelenboom P, Mirmiran M, Swaab DF. Alterations in the circadian rest-activity rhythm in aging and Alzheimer’s disease. Biol Psychiatry. 1990;27(6):563–572. doi:10.1016/0006-3223(90)90523-5
22. Duncan MJ, Farlow H, Tirumalaraju C, et al. Effects of the dual orexin receptor antagonist DORA-22 on sleep in 5XFAD mice. Alzheimers Dement. 2019;5(1):70–80. doi:10.1016/j.trci.2019.01.003
23. Macheda T, Roberts K, Lyons DN, et al. Chronic intermittent hypoxia induces robust astrogliosis in an Alzheimer’s disease-relevant mouse model. Neuroscience. 2019;398:55–63. doi:10.1016/j.neuroscience.2018.11.040
24. Goodridge HS, Reyes CN, Becker CA, et al. Activation of the innate immune receptor Dectin-1 upon formation of a “phagocytic synapse”. Nature. 2011;472(7344):471–475. doi:10.1038/nature10071
25. Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169(7):1276–1290e17. doi:10.1016/j.cell.2017.05.018
26. Li P, Gao L, Gaba A, et al. Circadian disturbances in Alzheimer’s disease progression: a prospective observational cohort study of community-based older adults. Lancet Healthy Longev. 2020;1(3):e96–e105. doi:10.1016/s2666-7568(20)30015-5
27. Johnson DA, Billings ME, Hale L. Environmental Determinants of Insufficient Sleep and Sleep Disorders: implications for Population Health. Curr Epidemiol Rep. 2018;5(2):61–69. doi:10.1007/s40471-018-0139-y
28. Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68(6):1023–1042. doi:10.1016/j.neuron.2010.11.032
29. Meerlo P, Sgoifo A, Suchecki D. Restricted and disrupted sleep: effects on autonomic function, neuroendocrine stress systems and stress responsivity. Sleep Med Rev. 2008;12(3):197–210. doi:10.1016/j.smrv.2007.07.007
30. Han KS, Kim L, Shim I. Stress and sleep disorder. Exp Neurobiol. 2012;21(4):141–150. doi:10.5607/en.2012.21.4.141
31. Irwin MR. Sleep and inflammation: partners in sickness and in health. Nat Rev Immunol. 2019;19(11):702–715. doi:10.1038/s41577-019-0190-z
32. Hong S, Lee DB, Yoon DW, Yoo SL, Kim J. The effect of sleep disruption on cardiometabolic health. Life. 2025;15(1):60. doi:10.3390/life15010060
33. Garbarino S, Lanteri P, Bragazzi NL, Magnavita N, Scoditti E. Role of sleep deprivation in immune-related disease risk and outcomes. Commun Biol. 2021;4(1):1304. doi:10.1038/s42003-021-02825-4
34. Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep. 2013;36(7):1027–1032. doi:10.5665/sleep.2802
35. Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–6453. doi:10.1073/pnas.95.11.6448
36. McLean CA, Cherny RA, Fraser FW, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46(6):860–866. doi:10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m
37. Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–842. doi:10.1038/nm1782
38. Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. 2015;15(4):203–216. doi:10.1038/nri3818
39. Heneka MT, Van der Flier WM, Jessen F, et al. Neuroinflammation in Alzheimer disease. Nat Rev Immunol. 2025;25(5):321–352. doi:10.1038/s41577-024-01104-7
40. Simon M, Wang MX, Ismail O, et al. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid beta plaque formation in mice. Alzheimers Res Ther. 2022;14(1):59. doi:10.1186/s13195-022-00999-5
41. Kress BT, Iliff JJ, Xia M, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76(6):845–861. doi:10.1002/ana.24271
42. Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra111. doi:10.1126/scitranslmed.3003748
43. Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342(6156):373–377. doi:10.1126/science.1241224
44. Yue X, Lu M, Lancaster T, et al. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci U S A. 2005;102(52):19198–19203. doi:10.1073/pnas.0505203102
45. Villa A, Vegeto E, Poletti A, Maggi A. Estrogens, neuroinflammation, and neurodegeneration. Endocr Rev. 2016;37(4):372–402. doi:10.1210/er.2016-1007
46. Loiola RA, Wickstead ES, Solito E, McArthur S. Estrogen promotes pro-resolving microglial behavior and phagocytic cell clearance through the actions of annexin A1. Front Endocrinol. 2019;10:420. doi:10.3389/fendo.2019.00420
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.