Back to Journals » Drug Design, Development and Therapy » Volume 14
Chronic Rhinosinusitis with Nasal Polyps: Targeting IgE with Anti-IgE Omalizumab Therapy
Authors Kariyawasam HH, James LK ![]()
Received 30 September 2020
Accepted for publication 24 November 2020
Published 10 December 2020 Volume 2020:14 Pages 5483—5494
DOI https://doi.org/10.2147/DDDT.S226575
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Harsha H Kariyawasam,1– 3 Louisa K James4
1Specialist Allergy and Clinical Immunology, Royal National ENT Hospital, University College London Hospitals NHS Foundation Trust, London, UK; 2Department of Rhinology, Royal National ENT Hospital, University College London Hospitals NHS Foundation Trust, London, UK; 3University College London, London, UK; 4Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK
Correspondence: Harsha H Kariyawasam
Specialist Allergy and Clinical Immunology, Royal National ENT Hospital, University College London Hospitals NHS Foundation Trust, 47-49 Huntley Street, London WC1E 6DG, UK
Email [email protected]
Abstract: Chronic rhinosinusitis with nasal polyps (CRSwNP) is a complex, clinically heterogeneous and persistent inflammatory disorder of the upper airway. Detailed mechanistic insights into disease pathogenesis are lacking, but it is now accepted that local tissue IgE driven T2-high inflammatory pathways are critical to disease. The recent CRSwNP Phase 3 POLYP1 and POLYP2 replicate studies of blocking IgE with omalizumab confirmed rapid improvements in all clinical parameters of sinonasal disease, confirming a pivotal role for IgE driven inflammatory pathways in CRSwNP. This review summarises the biology of IgE in relation to CRSwNP. Insight into how IgE may drive CRSwNP is evaluated in the context of clinical improvements seen with omalizumab. The need for further studies using a broader patient and biomarker specific groups to aid more precise drug-patient selection alongside more detailed mechanistic studies of omalizumab in CRSwNP is highlighted.
Keywords: IgE, nasal polyps, omalizumab, rhinosinusitis
Introduction
Chronic rhinosinusitis (CRS) is the broad term used to define a set of symptoms that are driven by an inflammatory sinonasal system. CRS is clinically characterised by the existence of two or more symptoms, one of which should be either nasal obstruction or anterior/posterior nasal mucus and ± facial pain/pressure ± reduction or loss of smell. Chronicity is arbitrarily defined by the presence of symptoms for at least 12 weeks.1 Different immune pathways drive specific CRS subtypes termed endotypes, explaining heterogeneity of clinical traits, disease severity and treatment response. Current “one size fits all treatment” approaches have, unsurprisingly, often been ineffective. As such, CRS is universally accepted as a difficult to treat disease syndrome. There is, therefore, an urgent need to identify disease endotypes and match this to disease mechanism-specific targeted therapy. This approach is now termed precision medicine.
Chronic Rhinosinusitis with Nasal Polyps
CRS is conventionally and conveniently divided into CRS with and without nasal polyps (CRSwNP and CRSsNP, respectively). Nasal polyps appear as visible sinonasal mass-like tissue structures that can progressively obstruct sinus drainage and nasal airflow. In one centre prevalence study of CRS using data on n=5525 patients, 80% had CRSsNP and approximately 20% had CRSwNP.2 The tissue inflammatory profile in CRSwNP may vary with ethnicity, with neutrophilic dominant forms in China to a mixed population of both neutrophilic and eosinophilic inflammation to highly eosinophilic states in the West.3 Particularly in more Caucasian populations, CRSwNP is a more distinct immunological disease entity similar to that found in allergic or eosinophilic asthma.4 CRSwNP and asthma are commonly co-associated.4
Inflammation and CRSwNP
Airway inflammation is complex and only aspects directly relevant to understanding IgE driven inflammation are considered here. In the simplest form, airway inflammation can be viewed as the mucosal response to injury. A rapid innate and adaptive immune response in health leads to elimination of the inciting agent and tissue repair back to health.5 In disease, inflammation and tissue injury continues unchecked in a dysregulated and amplified manner (Figure 1). In the context of CRSwNP, the inciting agent is still speculative but probably a combination of infection, allergen and environmental stress in the context of genetic predisposition. In CRSwNP, the epithelial barrier lacks structural integrity and is disrupted.5 This increases sinonasal mucosal surface vulnerability and promotes an active epithelial inflammatory state. Allergen-specific IgE on mast cell and basophil surfaces on binding antigen undergo cross-linking and leads to rapid cell degranulation. The release of preformed immune mediators and key cytokines IL-4 (Th2 drive and immunoglobulin class switching to IgE production), IL-5 (eosinophil recruitment and survival factor) and IL-13 (Th2 drive and mucus production) further amplify and sustain existing inflammation. DCs will present allergen to naïve T cells which in the IL-4 cytokine rich environment promotes Th2 cell development. Th2 cells further secrete IL-4, IL-5 and IL-13 thus setting up auto-inflammatory loops that further amplify and sustain existing inflammation. Superantigens from nasal Staphylococcus (Staph.) aureus colonies lead to polyclonal T and B cell activation. ILC2 induction by epithelial cytokines such as TSLP, IL-25 and IL-33 leads to further immune amplification with release of IL-5 and IL-13/IL-4. Th2 cells can directly respond to IL-25 and IL-33,6 adding a further layer of inflammatory amplification. Consistently high numbers of B cells and plasma cells along with high concentrations of IgA, IgG and IgE are found in nasal polyp tissue.7 Local production at germinal centre-like pseudofollicles occurs. With antigen stimulus, IgE production occurs via immunoglobulin class-switching, where B cell production of one immunoglobulin can be changed for another. As a result, B-cell activation, division, somatic hypermutation and selection occur with class switching to local IgE production.
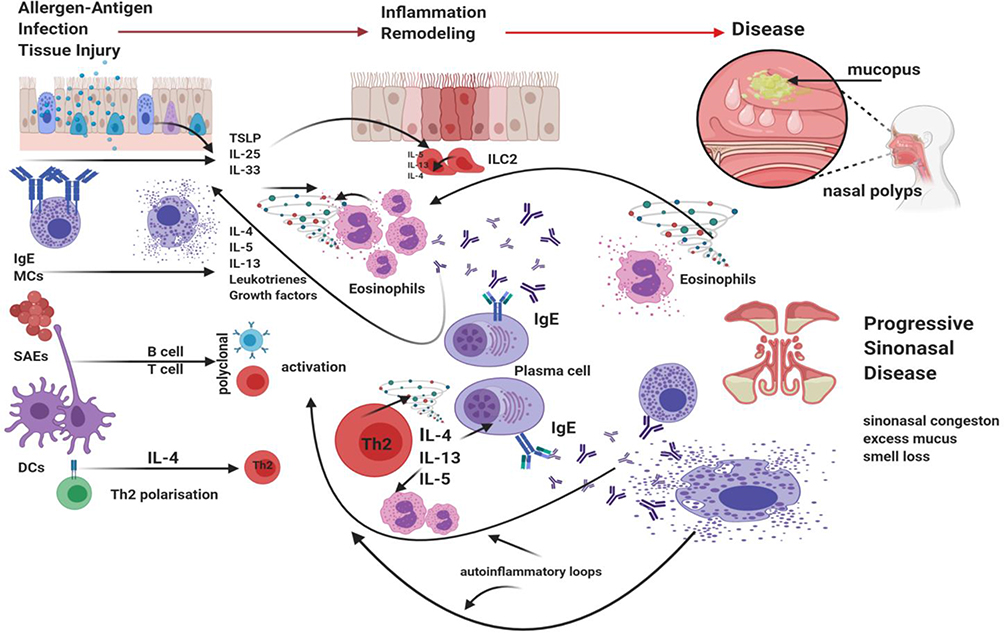 |
Figure 1 Summary of T2-high inflammatory pathways presumed to drive CRSwNP. Acute immune signalling leading to inflammation is activated following antigen-specific IgE on MCs and basophils binding antigen, cross-linking and activating calcium-dependent signalling with rapid degranulation and subsequent release of key immune mediators and cytokines such as IL-4, IL-5 and IL-13. Inflammatory cytokines are in fact secreted by an array of inflammatory and structural cells. In airway disease, SAEs further act as super-antigens leading to polyclonal T and B cell activation. Epithelial derived cytokines TSLP, IL-25 and IL-33 stimulate ILC2s to release large amounts of IL-5 and IL-13 but also IL-4. Th2 cells are now driven in the inflammatory cytokine milieu to further amplify inflammation with induction of auto-inflammatory loops. The lack of T and B cell regulation promotes such excessive inflammation. As such further T2 inflammatory mediators including IL-4, IL-5 and IL-13 are formed and released, with further production of specific IgE. Chronic inflammation becomes of established. Created with BioRender.com. |
CRSwNP with Non-Steroidal Anti-Inflammatory Drug Sensitivity
CRSwNP associated with aspirin or non-steroidal anti-inflammatory drug (NSAID) exacerbated respiratory disease (NERD) is a distinct clinical and inflammatory subset. In the USA, it is estimated that 7.2% out of 19 million Americans have NERD.8 Compared to CRSwNP non-NERD group, it represents a more severe CRSwNP form, difficult to treat disease with conventional medical treatment, with often rapid recurrence of disease despite comprehensive surgery and thus NERD patients often have a history of having recurrent surgery. Inflammation in CRSwNP-NERD is amplified. The pathogenesis of NERD is related to dysregulation of eicosanoid synthesis. Excess LTE4 is characteristic but precise immune mechanisms are still unclear. Up to 90% have colonisation with Staph. aureus with markedly higher amounts of polyp tissue total IgE, specific IgE to inhalants but also IgE to SAEs with excess eosinophilic inflammation.9 Such excessive tissue inflammation is often associated with high blood eosinophils, a reflection of a tendency to more enhanced systemic inflammation. Such patients also have more severe and difficult to treat asthma.
CRSwNP and IgE-Mediated Aeroallergen Sensitisation
The average age of onset for CRSwNP is around 42 years with range from 40 to 60 years.1 Such midlife presentation is usually as a part of late onset eosinophilic asthma. IgE-mediated sensitisation to aeroallergens is probably not directly relevant to this combined upper and lower airway syndrome, as it is rare for allergen sensitisation to develop in adulthood, and the actual prevalence of IgE-mediated allergic rhinitis decreases after 60 years.10 In one case series, only 50% of patients with CRS demonstrated specific aeroallergen sensitisation.11 Such allergen sensitisation (IgE-based immune memory) does not necessarily imply direct allergen driven immunological activation (allergy). As such, allergic sensitisation from childhood does not increase the risk of CRS in adults.12,13 Thus, patients with allergic rhinitis have similar prevalence of CRSwNP to the normal population.14–16 Skin prick test positivity had no predictive value of increased nasal IgE. Total IgE (ie, IgE to non-defined antigen as well as allergen-specific IgE) levels in nasal polyp tissue were present to a similar amount in both atopic and non-atopic patients.17 Patients can therefore have both allergic rhinitis and CRS, but this association is more coincidental rather than causal. Also the presence of allergen sensitisation does not correlate with objective measures of CRSwNP disease severity such as nasal polyp score (NPS) (a measure of polyp volume), SNOT-22 scores (a measure of sinonasal disease severity and associated general well-being), radiological severity scores or the rate of polyp recurrence.18 Over simplistic interpretation of IgE biology in CRSwNP lead to the past incorrect assumption that local tissue IgE itself has no functional role in CRSwNP. Only now has it become accepted that local mucosal IgE, often unrelated to inhaled aeroallergens, has a pivotal role in CRSwNP.
IgE Structure
IgE conforms to the basic immunoglobulin Y-shaped structure with two ε heavy chains and two light chains (Figure 2) as seen, for example, with IgG. Like IgM, the IgE molecule ε heavy chain contains four constant domains (Cε1-Cε4) and lacks a hinge region between Cε1-Cε2.19 Antibody specificity is determined by the variable regions. The variable region can be further subdivided into hypervariable (HV) and framework (FR) regions. It is the hypervariable (HV) region within the variable regions that has direct contact with the specific antigen. The HV regions are also termed the complementarity determining regions (CDRs). The FR regions act like a scaffold structure to enable the HV regions to be held in place to allow contact with the antigen. It is the IgE distal Fc region, comprised of only the two heavy chains, that binds cellular receptors. There are two principal receptors, the high-affinity receptor FcεRI and the low-affinity FcεRII/CD23.
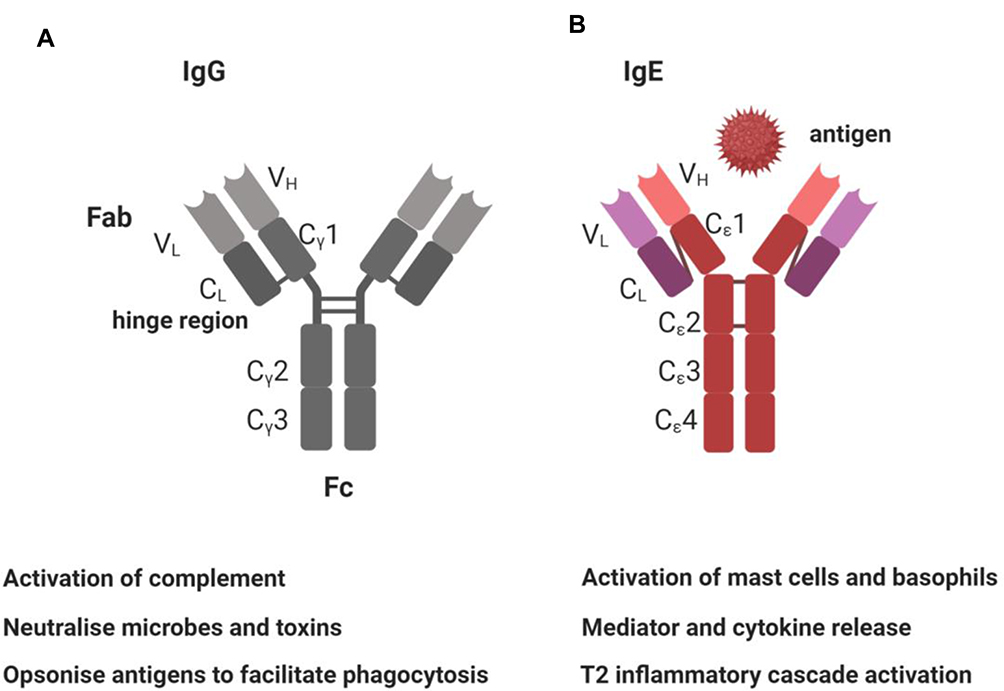 |
Figure 2 Summary of IgG and IgE structure and function. (A) IgG antibody structure is of four polypeptide chains, with two identical light (L) chains and two identical heavy (H) chains, that conform to a flexible Y-shaped structure. The two heavy chains are linked to each other by disulphide bonds. Each heavy chain is also linked to a light chain by a disulphide bond. There are five types of mammalian immunoglobulin heavy chain: γ, δ, α, μ and ε for IgG, IgD, IgA, IgM and IgE, respectively. Each chain has a variable (VH) region at the amino terminus, that forms the specific antigen-binding site, and a constant (CH) region, which determines the antibody isotype. The IgG heavy chain is comprised of CH1, hinge region, CH2, CH3 domains and the VH region. The light chains κ or λ chains also have variable (VL) and constant (CL) regions. (B) IgE is also monomeric but with two ε-heavy chains and two light chains. IgE thus also has capacity to also simultaneously bind two antigens. The heavy chain C-terminal regions are composed of four Cε dimers, Cε 1–4. The specific molecular structure of these dimers determines specific IgE binding capacity for cellular receptors FcεRI and CD23. IgE does not have a hinge region but instead a C-ε2 domain. This allows IgE more flexibility during receptor interaction. Created with BioRender.com. |
Blood and Tissue IgE
The high affinity of IgE for FcεRI (Ka ≈ 1010 M−1) ensures that most tissue IgE is cell bound.20 Serum IgE is present at very low levels in the serum of healthy individuals and despite being elevated in people with allergic disease, still remains 1000-fold lower than serum IgG. Blood IgE is often raised in CRSwNP, particularly with more severe disease.21 This IgE is usually not against known aeroallergens and thus classed as non-specific, and considered tissue overspill, where unbound possibly excess IgE is washed out into blood. Smaller amounts may still be from secreted IgE from circulating IgE+ plasma cells.22 It thus becomes apparent that local tissue IgE may be more relevant or functional in the local tissue environment. The exact allergen drive for the production of IgE in CRSwNP is currently uncertain but microbes, classic aeroallergens and even nasal autoantigens are likely triggers. The factors that promote the survival and rapid expansion of IgE+ B cells are unknown. It is now certain, however, regardless of obvious allergen sensitisation, IgE synthesis is local in nasal polyp tissue.23
IgE Synthesis
Isotype class-switching, a process of somatic recombination that joins the antigen-specific variable region (VH) gene locus of an antibody to a separate immunoglobulin heavy chain (CH) locus, is a pre-requisite for the production of IgE, IgG and IgA. IgE class-switching is a tightly regulated process dependent upon IL-4 and IL-13 production and CD40 ligand (CD40L) expression. IL-4 is produced by both tissue MCs and Th2 cells, whereas IL-13 production is restricted to mainly Th2 cells.19 CD40L is constitutively expressed by mast cells and upregulated on T cells following antigen-induced activation. The first step of IgE class-switching is the activation of the intervening epsilon (Iε) promoter which is upstream of the IgE locus (Cε). IL-4 and IL-13 signalling induce phosphorylation of STAT6, a transcription factor that can bind to and activate the Iε promoter. CD40L signalling results in the activation of the transcription factor (NF-κB), which co-operates with STAT-6 to activate Cε transcription. Transcription of the Iε promoter is essential for class switch recombination to IgE and results in a sterile germline Cε transcript lacking the variable (VDJ) region. Transcription of sterile germline transcript enables targeting by a complex array of transcription factors of the switch region of the IgE locus (Sε). DNA cleavage at Sε allows physical recombination of the Cε heavy chain region to the variable region genes. Looped out regions of DNA that result from the splicing out of the intervening immunoglobulin constant region genes, termed switch circles, are a by-product of this process and can be utilised as markers of productive IgE class-switching.
CD40L/IL-4 mediated activation of STAT-6 and NF-κB also results in the expression of activation-induced cytidine deaminase (AID), a critical enzyme required for class switch recombination. Following class-switch recombination, re-arranged IgE is expressed as a membrane immunoglobulin. Further selection and affinity maturation result in either positive (clonal expansion) or negative selection (cell death). Positively selected cells undergo differentiation into either memory cells or plasma cells allowing production and secretion of specific IgE. Such IgE-switched B cells preferentially favour the plasma cell fate, whereas the existence of IgE+ memory B cells has been a source of debate.24 IgE+ B cells are overall low in numbers.
Class switch recombination, affinity maturation and B cell differentiation are all elements of the germinal centre reaction that occurs within secondary lymphoid organs. Here specialised T follicular helper cells (TFH) regulate the development of germinal centre B cells through expression of CD40 and the secretion of cytokines (eg, IL-21 and IL-4) and chemokines.25
Differentiated, class-switched plasma cells migrate out of germinal centres into bone marrow where immunoglobulin secretion occurs. It is increasingly evident, particularly in autoimmune disease and allergy, that germinal centre-like reactions may also occur within mucosal tissue at sites of immune assault. Compelling evidence demonstrates that the respiratory mucosa is not only the primary site for IgE synthesis17,26 but also that the local environment of the respiratory mucosa in allergic rhinitis, asthma and nasal polyps supports class-switch recombination and clonal expansion of IgE-producing B cells,23,27,28 the numbers of which positively correlate with TFH cell numbers in nasal polyp tissue.29 This supports the hypothesis that local mucosal plasma cells represent the primary source of IgE and that circulating IgE may be the result of excess “spill-over” into the circulation.
IgE Driven Inflammation
It is now clear that IgE is a key driver of T2 inflammation, yet the exact immune signalling cascades that promote IgE responses but also regulate such pathways are still unknown in CRSwNP. The memory B cell pool in nasal tissue has a diverse transcriptional heterogeneity and understanding the different antibody functional repertoires is still very limited.
IgE-mediated inflammation is characterised by i) immediate release from tissue mast cells of inflammatory mediators following exposure to relevant allergens/antigen and ii) subsequent infiltration of Th2 cells and eosinophils to mucosal surfaces at the site of allergen exposure (Figures 1 and 3). The immediate hypersensitivity response is a powerful immune event, as evident from the ability to lead to the induction of rapid anaphylaxis and death. The effects of IgE-mediated inflammation are dependent upon the expression of low and high-affinity IgE receptors. FcεRI is expressed constitutively on mast cells and basophils and can be induced on dendritic cells by exposure to Th2 cytokines. Mucosal exposure to allergen results in cross-linking of specific IgE bound to FcεRI on tissue mast cells and basophils, triggers rapid degranulation and release of inflammatory mediators into the immediate local environment. IgE can be against SAE, and such IgE may act as super antibodies.30 Here, SAEs may bind such IgE bound cells both in the classic allergen manner via CDRs but also act as ‘superantigens’ by binding to the anti-SAE IgE framework regions that are already present as anti-SAE IgE-FcεRI complexes bound on the cell surface. The mediators released during this early phase response induce the recruitment of T cells, eosinophils and APCs. The low-affinity IgE receptor, CD23 (FcεRII), is expressed on APCs and B cells. CD23 binding of allergen-IgE complexes enables more efficient capture of allergen by APCs. This lowers the concentration threshold of allergen required for activation of an immune response by a factor of 100-fold. Subsequent cytokine production by activated T cells provides positive feedback for further IgE production. It thus becomes evident that blocking the function of IgE in CRSwNP has the potential to attenuate inflammation and regulate several key inflammatory pathways.
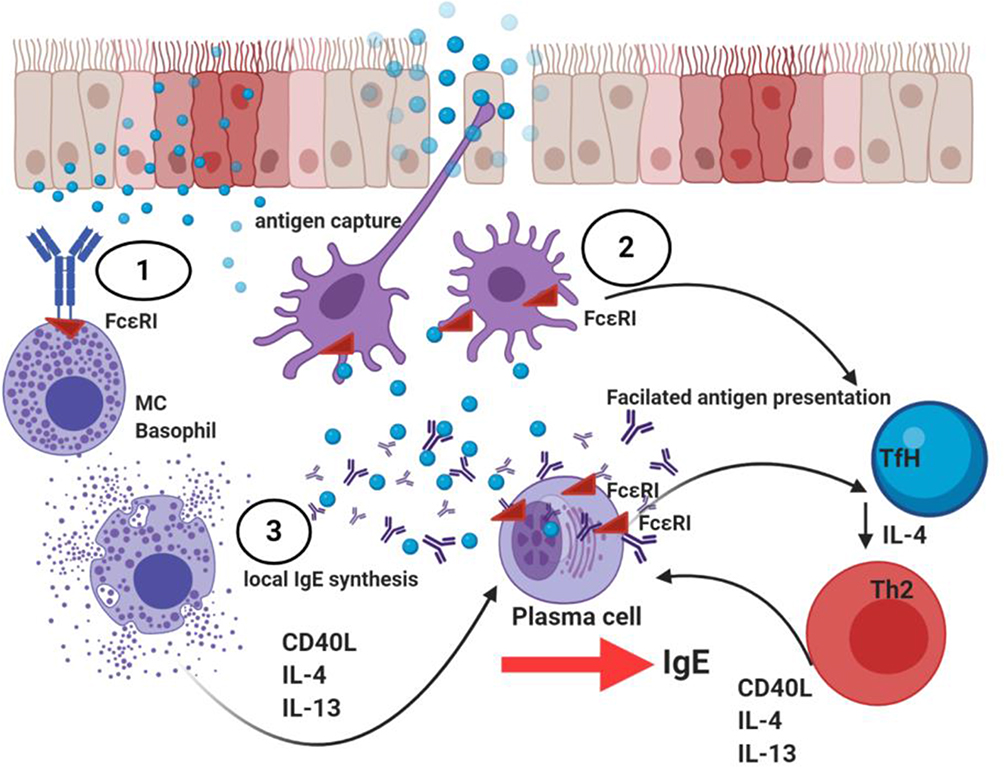 |
Figure 3 Summary of local IgE driven inflammation in nasal polyp tissue. 1. The epithelium in CRSwNP is disrupted and there is increased and sustained epithelial activation. Allergen impaction of the nasal mucosa epithelial surface leads to its solubilisation and diffusion to sites of mast cell (MC)-basophil residence where cross-linking of two or more high affinity (FcεRI) IgE receptors lead to MC and basophil activation, subsequent degranulation and release of mediators such as histamine, leukotrienes, key T2 cytokine such as IL-4, IL-13 and IL-5 that activates key inflammatory signalling pathways with induction of autoinflammatory loops. 2. Antigen-presenting cells and particularly dendritic cells (DCs) also take up allergen and present processed allergen peptide in the context of MHC Class II to naïve Th0 cells in a T2 cytokine micromilieu of IL-4. Th0 cells undergo activation and differentiation to Th2 cells leading to the further release of Th2 cytokines. Th2 and TfH derived IL-4 in particular is essential for B cell class switching to IgE production in secondary lymphoid tissue. Local synthesis of IgE occurs in nasal polyp tissue. Such increased local IgE induce FcεRI expression on DCs and further stabilise FcεRII on B cells. This now enhances the ability of both cells to take up antigen-IgE complexes and undertake allergen presentation to T cells. FcεRII+ (CD23) B cells present allergen in a non-cognate manner termed facilitated antigen presentation. CD23+ epithelial cells allow epithelial transcytosis of IgE-allergen complexes. As such, IgE-allergen complexes on the airway surface migrate into the mucosa, and now engage with tissue resident FcεRI-mast cells/basophils. 3. Local IgE throughout the nasal tissue binds to cells expressing both the high affinity (mast cells, basophils) and low-affinity IgE receptor (B cells, eosinophils, monocytes and T cells). With such IgE-receptor association these cells are now sensitised for activation on encountering specific allergen. Further allergen trapping and focussing by antigen-presenting cells leading to CD4+ T cell activation generating a Th2 pattern of cytokines (IL-4, IL-5, IL-9 and IL-13), sustains and amplifies inflammation. A complex inflammatory cascade leading to eosinophilic and other inflammatory cell infiltration, elevated serum IgE levels and mucus hypersecretion becomes established. Tissue remodeling programmes become activated. Created with BioRender.com. |
Omalizumab
Omalizumab (Xolair®, Novartis) is a murine humanised anti-human-IgE monoclonal IgG1 antibody. The complementarity determining regions (CDRs) of the protein is still comprised of approximately 5% murine amino acid residues.31 Omalizumab binds unbound IgE at the Cɛ3 domain.32 Then, by steric hindrance, omalizumab prevents IgE from interacting with the high-affinity IgE receptor FcɛRI on the cell surface preventing subsequent mast cell and basophil activation and degranulation.33 Free IgE is effectively neutralised and thus inhibition of IgE-mediated signalling rapidly occurs. Omalizumab first received FDA approval for severe allergic asthma in 2003. All patients had to demonstrate IgE sensitisation to a perennial aeroallergen such as house dust mite or cat to receive omalizumab for severe asthma. Almost 17 years later, after later realisation that IgE may have a functional role in CRSwNP regardless of classic aeroallergen atopic status and that active T2 inflammatory pathways in CRSwNP must involve IgE related inflammation, rigorous Phase 3 clinical studies with omalizumab have finally been completed in CRSwNP.
Clinical Studies
Phase 3 Studies: POLYP1 and POLYP2
POLYP1 and POLYP2 studies are replicate Phase 3 studies undertaken to confirm the clinical efficacy of omalizumab.34 All patients were placed on the intranasal steroid mometasone furoate in the 5-week run-in period and throughout the study. Volunteers continued on standard treatment of nasal lavage but long-term antibiotics beyond 14 days were not permitted. Total blood IgE was measured. Allergen skin prick testing nor blood IgE to specific aeroallergens was not undertaken. Volunteers with severe CRSwNP (mean SNOT-22=59.8 and 59.2 in active arms of Polyp1 and Polyp2, respectively) were randomised 1:1 to either 2 or 4 weekly subcutaneous omalizumab or placebo, the dose calculated from a standard chart based on serum IgE (minimum-maximum 30–1500 IU/mL) vs weight in kilograms (minimum 30.1–150 kg). Treatment duration was short at only 24 weeks. The dose range for omalizumab was 75–600 mg. The main primary study endpoint of nasal polyp score (NPS) was evaluated before commencement and then at 4 weeks, 8 weeks, 16 weeks and 24 weeks of treatment. The other primary endpoint was the nasal congestion score (NCS), which was recorded as part of the daily eDiary along with subjective sense of smell, postnasal drip, runny nose. These were scored individually and also as a combined value termed the Total Nasal Symptom Score (TNSS). The secondary endpoints of SNOT-22 and UPSIT (objective smell function score) were evaluated at baseline and above time points. Asthma related quality of life (AQLQ) at baseline and then at 16 and 24 weeks was recorded.
Impressively, rapid improvement in NPS and NCS was seen as early as after 4–8 weeks of treatment, along with the SNOT-22, UPSIT scores and TNSS. After 8 weeks there was no further marked improvements in the measured clinical parameters. NPS and NCS for omalizumab versus placebo at week 4 showed a mean change from baseline NPS for omalizumab versus placebo of –0.92 (95% CI, –1.37, –0.48) and –0.52 (95% CI, –0.94, –0.11) in POLYP1 and 2, respectively. NCS for omalizumab versus placebo was –0.25 (95% CI, –0.46, –0.04) and –0.26 (95% CI, –0.45, –0.07) in POLYP1 and 2, respectively.
At study completion at week 24, for omalizumab versus placebo, compared to baseline values the mean change in NPS was –1.08 versus +0.06 (treatment arm differences (95% CI), –1.14 (–1.59, –0.69); p < 0.0001) and –0.90 versus –0.31 (treatment arm differences (95% CI), –0.59 (–1.05, –0.12); p=0.0140) in POLYP1 and 2, respectively. The mean change in NCS was –0.89 versus –0.35 (treatment arm differences (95% CI), –0.55 (–0.84, –0.25); p=0.0004) and –0.70 versus –0.20 (treatment arm differences (95% CI), –0.50 (–0.80, –0.19); p=0.0017) in POLYP1 and 2, respectively. No real further improvements were seen after 16 weeks of treatment. Surprisingly, the NERD subgroup did not do any better to the non-NERD cohort with improvements in NPS and NCS.
In terms of secondary study endpoints, the mean week 4 improvement in SNOT-22 score with omalizumab versus placebo was –10.43 (95% CI –15.08, –5.79) and –8.84 (–13.84, –3.84) in POLYP1 and 2, respectively. Improvements in olfaction were not impressive. The mean difference in change from baseline at week 8 in UPSIT for omalizumab versus placebo was 3.78 (95% CI 1.56–6.00) and 3.44 (1.03–5.85) in POLYP1 and POLYP2, respectively. Noting the short duration of the study, there was a relative reduction of 62.5% in oral rescue systemic steroid use in the omalizumab group and none of the treatment arms proceeded to surgical polypectomy at study completion.
As expected, co-associated asthma improved, consistent with overlapping shared upper and lower airway mucosal-IgE driven inflammatory pathways driving both CRSwNP and asthma. The omalizumab group was at least 4 times more likely to improve by at least ≥0.5-point on the AQLQ compared to placebo [OR], 3.7; 95% 297 CI, 1.0–13.7; p=0.0492 in POLYP1 and OR 4.0; 95% CI, 1.1–15.3; p=0.0396 in POLYP2.
Previous Proof of Concept Studies and Real-Life Data Studies
The overwhelmingly positive sinonasal outcomes in POLYP1 and POLYP2 are somewhat expected, given that several smaller, mainly observational studies demonstrated a beneficial effect for omalizumab in CRS. The Vennera et al study confirmed a decrease in polyp size, and the important cost saving of decreased frequency of surgery and less need for INS in such patients.35 Here, the group were not just CRSwNP but also with severe co-associated asthma. A small but still relevant study by Pinto et al using a DBPC trial design of CRSwNP patients confirmed decreasing SNOT-20 scores over 6 months of omalizumab treatment.36 The SNOT-20 fails to consider nasal obstruction and sense of smell. The patients here, however, failed to improve on objective tests of airflow, endoscopic NPS, sino-nasal imaging and olfaction. The patients here were only selected for their CRS status and thus a heterogenous population. In a small but well-designed POC study of 15 volunteers with CRSwNP and coexistent allergic or non-allergic asthma, 2 weekly or monthly injections of omalizumab, compared to 4 placebo volunteers, led to a reduction in NPS at 4 months.37 Improvement was seen regardless of atopic status. In an observational study, our own group selected only patients with both severe CRSwNP and asthma.38 Using the SNOT-22 and ACQ-7 score, we reported marked clinical improvements in both CRSwNP and severe allergic asthma together. Improvement was seen as early as after the first dose of omalizumab at the 4-week assessment and continued to show sustained improvement with therapy until the last time point assessment at 16 weeks of therapy. Here, all the patients had severe CRSwNP with a median SNOT-22 at baseline of 52/110. The total serum IgE (median) was 396.5 IU/L, eosinophil count (median) 0.52 x 109/L, 77% had aspirin sensitivity and all had asthma with aeroallergen sensitisation. Thus, this group was a group of both severe CRSwNP and co-associated severe asthma. What was interesting was that the improvement with omalizumab was similar to the group that instead underwent sinonasal surgery. This was despite the surgery group having more severe disease with a median SNOT-22 of 70 and as expected high T2 biomarkers of total blood IgE of 268.0 IU/L, eosinophil count of 0.55 x109/L. Indeed, the SNOT-22 improvements after FESS have been shown through meta-analysis of 15 studies to approach the improvements seen with omalizumab in our study, POLYP1 and POLYP2.39 The overwhelming conclusion from clinical studies is that omalizumab is effective in treating CRSwNP. The effects are seen early on and benefits are sustained with continued treatment. The implications of such findings for modelling health-related cost-savings and resource utilisation are discussed later.
Safety and Adverse Events
Omalizumab was shown as remarkably safe in the current Phase 3 CRSwNP studies, with more adverse events reported in the placebo group. Such safety is similar to that published for omalizumab in severe asthma studies over more than a decade.40 There were no safety concerns of any form attributed to omalizumab.
Mechanism of Omalizumab
The efficacy of omalizumab in allergic asthma and now CRSwNP highlights the central role of IgE in regulating the recruitment and function of effector cells that mediate T2 inflammation. The omalizumab studies confirm that aeroallergen IgE-mediated sensitisation is irrelevant to CRSwNP and associated asthma clinical outcomes and supports a role for local mucosal IgE in eosinophilic airway disease. Omalizumab binds to the Cε3 domain of IgE, a site which overlaps with the binding sites for both FcεRI and FcεRII. Importantly, whilst omalizumab can prevent binding of free IgE to the high and low-affinity IgE receptors, it cannot cross-link receptor-bound IgE which is essential to avoid receptor-mediated activation of effector cells. Despite this omalizumab can still accelerate the dissociation of IgE from FcεRI on mast cells and basophils.41 FcεRI expression correlates closely with serum IgE levels due to the accelerated loss of unoccupied FcεRI at the cell surface. For this reason, omalizumab treatment results in a rapid reduction in the level of FcεRI expression by FcεRI-expressing mast cells, basophils and antigen-presenting cells.42,43 Depletion of IgE and parallel reductions in FcεRI expression limit effector cell activation and thus reduce IgE-mediated symptoms. For example, in patients with allergic asthma, omalizumab results in depletion of tissue eosinophils in the lung44 which may be attributed to reductions in the recruitment of inflammatory cells following allergen exposure. Reduction in blood eosinophils has also been demonstrated following omalizumab therapy in CRSwNP.45
Other off-label use of omalizumab in various “non-allergic” conditions has found similar findings with respect to clinical improvements. Similar to observations in allergic asthma, reductions in the numbers of blood and tissue eosinophils were reported in patients with eosinophilic gastroenteritis following omalizumab treatment.46 Likewise, a clinical trial of omalizumab in patients with severe non-atopic asthma demonstrated reductions in FcεRI expression on basophils and dendritic cells along with clinical improvement in lung function.47 A loss of IgE-mediated mast and basophil cell signalling will decrease IL-4, IL-13 and IL-5 production and cellular release.48 Th2 lineage differentiation and maintenance will lessen as IL-4 concentrations decrease. IL-4/IL-13 driven autoinflammatory loops are no longer sustained. With the decrease in levels of IL-5 blood and tissue eosinophil depletion occurs.49–51 A general knockdown of the T2 inflammatory axis is seen.44 Viral rhinitis is a potent drive for CRSwNP52 and omalizumab may prevent viral immune amplification of inflammatory pathways in CRSwNP. An important and novel finding with omalizumab treatment is improvement in innate viral defence. Omalizumab has been shown to increase interferon-α release from PBMCs and plasmacytoid DCs.53
Outstanding Questions
As with all clinical studies relating to a clinical intervention, the most pressing question is have the studies been undertaken in the correct patient population. In Polyp1 and Polyp2 there were only n=16 (22.2%) and n=24 (38.7%) patients, respectively, with NERD, the more clinically challenging, difficult to treat subset of patients that dominate tertiary rhinology clinics. NERD patients often have high blood eosinophilia. Such eosinophilia often indicates more severe disease.54 In Polyp1 and Polyp2 the mean blood eosinophil counts were 0.3344 x 109 L and 0.310.8 x 109/L respectively. These are within normal blood eosinophil ranges of 0.4–0.5 x109/L. Furthermore, in CRSwNP high eosinophils indicate high mucosal IgE concentrations, as IL-5 is active alongside IL4/IL-13 active signalling that drive local tissue IgE synthesis.17,55
Given the diversity of CRS subtypes with regards to patterns of cellular infiltrate, inflammatory mediators and remodeling programmes,56 it is obvious that not all patients will respond to omalizumab,36 and there will always be heterogeneity in clinical outcomes in those that do respond. Given limited healthcare resources, omalizumab cannot be just given to anyone or everyone with CRSwNP. Thus, it is most disappointing that no attempt was made by the Polyp1 and Polyp2 study team to evaluate what clinical traits or biomarkers of disease predicted disease response. As more biologics that target CRSwNP become available,57,58 such data to guide clinicians will become essential.
Our own group analysed what factors predicted CRS clinical response in a real-life study.45 Here, all patients had severe asthma, were sensitised to at least one perennial aeroallergen and had a median eosinophil count of 0.45 x 109/L. Importantly, 75% patients had NERD and therefore represented the more severe, difficult to treat CRSwNP subtype. Omalizumab treatment response data for allergic CRSsNP patients were also included. Using a linear modeling approach, we showed that it was the serum eosinophil count and more frequent two weekly omalizumab dosing frequency, not the serum IgE level that predicted treatment response with omalizumab.45 Interestingly, blood eosinophil counts not IgE also predict better clinical response in asthma with omalizumab.59,60 Twice weekly omalizumab is more effective in urticaria.61 This latter observation can be explained to some extent on the basis of drug pharmacodynamics/kinetics as discussed in the next paragraph.
Omalizumab related improvements in asthmatics are only evident when unbound serum-free IgE falls below 20 IU/mL.62 Falls in serum-free IgE is in turn dependent on the omalizumab dose.63 The serum elimination half-life of omalizumab is on average 26 days,64 whilst the half-life of IgE binding to FcɛRI is 14–21 days. Following such time, half the IgE is again free and has the potential to bind new receptors, and upon new allergen binding theoretically at least lead to re-activated IgE-mediated cell signalling.20,65 Thus, more frequent omalizumab will capture any released IgE and limit IgE-FcɛRI binding. In addition, the allosteric properties of omalizumab in relation to IgE binding may also be relevant. High concentrations of omalizumab leads to a phenomenon termed accelerated dissociation.66 Here, IgE-FcɛRI complexes become destabilized; as a result, rapid dissociation of IgE from FcɛRI occurs. IgE- FcɛRI mediated cellular activation is lost and downstream inflammatory cascades are not activated. Future omalizumab studies in more severe patient subgroups, with investment to evaluate which biomarkers, clinical traits and dosing schedules that predict optimal clinical response are urgently needed. Only then can true precision medicine be delivered.
As more biologics become available for CRSwNP, in the current era of limited healthcare resources, treatment cost will undoubtedly factor into which drug is chosen. There is a real concern that health economists will fail to factor in the several complex variables that will determine overall cost-benefit. Firstly, the association of CRSwNP with severe asthma must be recognised. The reduction in the need for frequent ENT and pulmonology visits along with reduction in surgery and polypharmacy with associated cost-reductions must also be considered. The long-term health benefits from less systemic steroid exposure must be incorporated into any health modeling undertaken. It is only after such detailed healthcare utilisation analysis can the true cost of any biologic regime be evaluated.
Unfortunately, the inability to so far clearly define the more detailed immune endotypes in CRSsNP means that up to 80% of patients with CRS have been neglected in terms of trials of biologics. Given that CRSsNP and CRSwNP are equally severe, studies evaluating T2-high CRSsNP immune endotype matched against appropriate biologics including omalizumab are now urgently needed.
Conclusion
Omalizumab can effectively treat CRSwNP. Rapid and sustained clinical improvement is seen. In addition, omalizumab treats associated severe asthma. At last, clinicians can treat two difficult co-associated airway diseases with one drug. Obtaining complete airway control must become the goal. It is now important that the incorrect assumption that a single identical biological pathway leads to the diverse inflammatory disease of CRSwNP is immediately abandoned. Eosinophilic CRSwNP with or without aeroallergen sensitisation should also be considered for omalizumab treatment. Larger, broad biomarker-based studies evaluating clinical response in more severe CRSwNP groups are needed, as clinicians need to urgently understand how to match the right patients to omalizumab. Clinical trials in patients with high-T2 inflammation driven CRS without nasal polyps are also needed. Studies must continue to assess the upper and lower outcomes together. Going forward, ENT surgeons and pulmonologists will need to work together. Only then can true integrated airway management and patient care be provided.
Abbreviations
ACQ-7, asthma control questionnaire 7; AQLQ, asthma quality of life questionnaire; CRS, chronic rhinosinusitis; CRSsNP, chronic rhinosinusitis without nasal polyps; CRSwNP, chronic rhinosinusitis with nasal polyps; DBPC, double-blind placebo-controlled; DCs, dendritic cells; FESS, functional endoscopic sinus surgery; IgE, immunoglobulin E; IL, interleukin; ILCs, innate lymphoid cells; LT, leukotriene; NERD, non-steroidal anti-inflammatory exacerbated respiratory disease; NF-κ, nuclear factor-kappa-B; PBMC, peripheral blood mononuclear cell; POC, proof of concept; SAEs, Staphylococcus aureus enterotoxins; STAT6, signal transducer and activator of transcription 6; Tfh, T follicular helper; TSLP, thymic stromal lymphopoietin; UPSIT, university of Pennsylvania smell identification test.
Funding
This manuscript was not funded.
Disclosure
HHK has undertaken paid consultancy work for Sanofi and Novartis, paid lecture commitment work for AstraZeneca, received lecture fees and support for conference attendance from GSK.HHK has no other financial or non-financial conflicts of interest to declare in relation to this work. LKJ reports no financial or non-financial conflicts of interests.
References
1. Fokkens WJ, Lund VJ, Hopkins C, et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology. 2020;58(1):1–464. doi:10.4193/Rhin20.401
2. Benjamin MR, Stevens WW, Li N, et al. Clinical characteristics of patients with chronic rhinosinusitis without nasal polyps in an academic setting. J Allergy Clin Immunol Pract. 2019;7(3):1010–1016. doi:10.1016/j.jaip.2018.10.014
3. Wei B, Liu F, Zhang J, et al. Multivariate analysis of inflammatory endotypes in recurrent nasal polyposis in a Chinese population. Rhinology. 2018;56(3):216–226. doi:10.4193/Rhin17.240
4. Kariyawasam HH, Rotiroti G. Allergic rhinitis, chronic rhinosinusitis and asthma: unravelling a complex relationship. Curr Opin Otolaryngol Head Neck Surg. 2013;21(1):79–86. doi:10.1097/MOO.0b013e32835ac640
5. Samitas K, Carter A, Kariyawasam HH, Xanthou G. Upper and lower airway remodelling mechanisms in asthma, allergic rhinitis and chronic rhinosinusitis: the one airway concept revisited. Allergy. 2018;73:993–1002.
6. Lam EP, Kariyawasam HH, Rana BM, et al. IL-25/IL-33-responsive T2 cells characterize nasal polyps with a default T17 signature in nasal mucosa. J Allergy Clin Immunol. 2015.
7. Tan BK, Li QZ, Suh L, et al. Evidence for intranasal antinuclear autoantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2011;128(6):1198–1206.e1191. doi:10.1016/j.jaci.2011.08.037
8. White AA, Stevenson DD. Aspirin-exacerbated respiratory disease. N Engl J Med. 2018;379(11):1060–1070. doi:10.1056/NEJMra1712125
9. Van Zele T, Gevaert P, Watelet JB, et al. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol. 2004;114(4):981–983. doi:10.1016/j.jaci.2004.07.013
10. Baptist AP, Nyenhuis S. Rhinitis in the elderly. Immunol Allergy Clin North Am. 2016;36(2):343–357. doi:10.1016/j.iac.2015.12.010
11. Tan BK, Zirkle W, Chandra RK, et al. Atopic profile of patients failing medical therapy for chronic rhinosinusitis. Int Forum Allergy Rhinol. 2011;1(2):88–94. doi:10.1002/alr.20025
12. Van Lancker JA, Yarnold PA, Ditto AM, et al. Aeroallergen hypersensitivity: comparing patients with nasal polyps to those with allergic rhinitis. Allergy Asthma Proc. 2005;26:109–112.
13. Chang EH, Stern DA, Willis AL, et al. Early life risk factors for chronic sinusitis: a longitudinal birth cohort study. J Allergy Clin Immunol. 2018;141(4):1291–1297.e1292. doi:10.1016/j.jaci.2017.11.052
14. Settipane GA, Chafee FH. Nasal polyps in asthma and rhinitis. A review of 6037 patients. J Allergy Clin Immunol. 1977;59(1):17–21. doi:10.1016/0091-6749(77)90171-3
15. Bunnag C, Pacharee P, Vipulakom P, Siriyananda C. A study of allergic factor in nasal polyp patients. Ann Allergy. 1983;50:126–132.
16. Johansson L, Akerlund A, Holmberg K, et al. Prevalence of nasal polyps in adults: the Skovde population-based study. Ann Otol Rhinol Laryngol. 2003;112(7):625–629. doi:10.1177/000348940311200709
17. Bachert C, Gevaert P, Holtappels G, et al. Total and specific IgE in nasal polyps is related to local eosinophilic inflammation. J Allergy Clin Immunol. 2001;107(4):607–614. doi:10.1067/mai.2001.112374
18. Pearlman AN, Chandra RK, Chang D, et al. Relationships between severity of chronic rhinosinusitis and nasal polyposis, asthma, and atopy. Am J Rhinol Allergy. 2009;23(2):145–148. doi:10.2500/ajra.2009.23.3284
19. Gould HJ, Sutton BJ, Beavil AJ, et al. The biology of IgE and the basis of allergic disease. Annu Rev Immunol. 2003;21(1):579–628. doi:10.1146/annurev.immunol.21.120601.141103
20. Sutton BJ, Davies AM. Structure and dynamics of IgE-receptor interactions: fcepsilonRI and CD23/FcepsilonRII. Immunol Rev. 2015;268(1):222–235. doi:10.1111/imr.12340
21. Johns CB, Laidlaw TM. Elevated total serum IgE in nonatopic patients with aspirin-exacerbated respiratory disease. Am J Rhinol Allergy. 2014;28(4):287–289. doi:10.2500/ajra.2014.28.4054
22. Eckl-Dorna J, Pree I, Reisinger J, et al. The majority of allergen-specific IgE in the blood of allergic patients does not originate from blood-derived B cells or plasma cells. Clin Exp Allergy. 2012;42(9):1347–1355. doi:10.1111/j.1365-2222.2012.04030.x
23. Gevaert P, Nouri-Aria KT, Wu H, et al. Local receptor revision and class switching to IgE in chronic rhinosinusitis with nasal polyps. Allergy. 2013;68(1):55–63. doi:10.1111/all.12054
24. Davies JM, Platts-Mills TA, Aalberse RC. The enigma of IgE+ B-cell memory in human subjects. J Allergy Clin Immunol. 2013;131(4):972–976. doi:10.1016/j.jaci.2012.12.1569
25. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50(5):1132–1148. doi:10.1016/j.immuni.2019.04.011
26. Smurthwaite L, Walker SN, Wilson DR, et al. Persistent IgE synthesis in the nasal mucosa of hay fever patients. Eur J Immunol. 2001;31(12):3422–3431. doi:10.1002/1521-4141(200112)31:12<3422::AID-IMMU3422>3.0.CO;2-T
27. Takhar P, Smurthwaite L, Coker HA, et al. Allergen drives class switching to IgE in the nasal mucosa in allergic rhinitis. J Immunol. 2005;174(8):5024–5032. doi:10.4049/jimmunol.174.8.5024
28. Takhar P, Corrigan CJ, Smurthwaite L, et al. Class switch recombination to IgE in the bronchial mucosa of atopic and nonatopic patients with asthma. J Allergy Clin Immunol. 2007;119(1):213–218. doi:10.1016/j.jaci.2006.09.045
29. Zhang YN, Song J, Wang H, et al. Nasal IL-4(+)CXCR5(+)CD4(+) T follicular helper cell counts correlate with local IgE production in eosinophilic nasal polyps. J Allergy Clin Immunol. 2016;137(2):462–473. doi:10.1016/j.jaci.2015.07.025
30. Chen JB, James LK, Davies AM, et al. Antibodies and superantibodies in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2017;139(4):1195–1204.e1111. doi:10.1016/j.jaci.2016.06.066
31. Presta LG, Lahr SJ, Shields RL, et al. Humanization of an antibody directed against IgE. J Immunol. 1993;151:2623–2632.
32. Hook WA, Zinsser FU, Berenstein EH, Siraganian RP. Monoclonal antibodies defining epitopes on human IgE. Mol Immunol. 1991;28(6):631–639. doi:10.1016/0161-5890(91)90132-4
33. Wright JD, Chu HM, Huang CH, et al. Structural and physical basis for anti-IgE therapy. Sci Rep. 2015;5(1):11581. doi:10.1038/srep11581
34. Gevaert P, Omachi TA, Corren J, et al. Efficacy and safety of omalizumab in nasal polyposis: two randomized Phase III trials. J Allergy Clin Immunol. 2020;146(3):595–605. doi:10.1016/j.jaci.2020.05.032
35. Vennera Mdel C, Picado C, Mullol J, et al. Efficacy of omalizumab in the treatment of nasal polyps. Thorax. 2011;66(9):824–825. doi:10.1136/thx.2010.152835
36. Pinto JM, Mehta N, DiTineo M, et al. A randomized, double-blind, placebo-controlled trial of anti-IgE for chronic rhinosinusitis. Rhinology. 2010;48(3):318–324. doi:10.4193/Rhin09.144
37. Gevaert P, Calus L, Van ZT, et al. Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. J Allergy Clin Immunol. 2013;131(1):110–116. doi:10.1016/j.jaci.2012.07.047
38. Bidder T, Sahota J, Rennie C, et al. Omalizumab treats chronic rhinosinusitis with nasal polyps and asthma together-a real life study. Rhinology. 2018;56(1):42–45. doi:10.4193/Rhin17.139
39. Le PT, Soler ZM, Jones R, et al. Systematic review and meta-analysis of SNOT-22 outcomes after surgery for chronic rhinosinusitis with nasal polyposis. Otolaryngol Head Neck Surg. 2018;159(3):414–423. doi:10.1177/0194599818773065
40. Agache I, Rocha C, Beltran J, et al. Efficacy and safety of treatment with biologicals (benralizumab, dupilumab and omalizumab) for severe allergic asthma: a systematic review for the EAACI guidelines - recommendations on the use of biologicals in severe asthma. Allergy. 2020;75(5):1043–1057. doi:10.1111/all.14235
41. Serrano-Candelas E, Martinez-Aranguren R, Valero A, et al. Comparable actions of omalizumab on mast cells and basophils. Clin Exp Allergy. 2016;46(1):92–102. doi:10.1111/cea.12668
42. Prussin C, Griffith DT, Boesel KM, et al. Omalizumab treatment downregulates dendritic cell FcepsilonRI expression. J Allergy Clin Immunol. 2003;112(6):1147–1154. doi:10.1016/j.jaci.2003.10.003
43. Lin H, Boesel KM, Griffith DT, et al. Omalizumab rapidly decreases nasal allergic response and FcepsilonRI on basophils. J Allergy Clin Immunol. 2004;113(2):297–302. doi:10.1016/j.jaci.2003.11.044
44. Djukanovic R, Wilson SJ, Kraft M, et al. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med. 2004;170(6):583–593. doi:10.1164/rccm.200312-1651OC
45. Sahota J, Bidder T, Livingston R, et al. Chronic rhinosinusitis and omalizumab: eosinophils not IgE predict treatment response in real-life. Rhinology. 2018;1(1):147–153. doi:10.4193/RHINOL/18.077
46. Foroughi S, Foster B, Kim N, et al. Anti-IgE treatment of eosinophil-associated gastrointestinal disorders. J Allergy Clin Immunol. 2007;120(3):594–601. doi:10.1016/j.jaci.2007.06.015
47. Garcia G, Magnan A, Chiron R, et al. A proof-of-concept, randomized, controlled trial of omalizumab in patients with severe, difficult-to-control, nonatopic asthma. Chest. 2013;144(2):411–419. doi:10.1378/chest.12-1961
48. Takaku Y, Soma T, Nishihara F, et al. Omalizumab attenuates airway inflammation and interleukin-5 production by mononuclear cells in patients with severe allergic asthma. Int Arch Allergy Immunol. 2013;161(Suppl s2):107–117. doi:10.1159/000350852
49. Skiepko R, Zietkowski Z, Lukaszyk M, et al. Changes in blood eosinophilia during omalizumab therapy as a predictor of asthma exacerbation. Postepy Dermatol Alergol. 2014;31:305–309. doi:10.5114/pdia.2014.40973
50. Noga O, Hanf G, Brachmann I, et al. Effect of omalizumab treatment on peripheral eosinophil and T-lymphocyte function in patients with allergic asthma. J Allergy Clin Immunol. 2006;117(6):1493–1499. doi:10.1016/j.jaci.2006.02.028
51. van Rensen EL, Evertse CE, van Schadewijk WA, et al. Eosinophils in bronchial mucosa of asthmatics after allergen challenge: effect of anti-IgE treatment. Allergy. 2009;64(1):72–80. doi:10.1111/j.1398-9995.2008.01881.x
52. Golebski K, van Tongeren J, van Egmond D, et al. Specific induction of TSLP by the viral RNA analogue poly(I:C) in primary epithelial cells derived from nasal polyps. PLoS One. 2016;11(4):e0152808. doi:10.1371/journal.pone.0152808
53. Gill MA, Liu AH, Calatroni A, et al. Enhanced plasmacytoid dendritic cell antiviral responses after omalizumab. J Allergy Clin Immunol. 2018;141(5):1735–1743.e1739. doi:10.1016/j.jaci.2017.07.035
54. Aslan F, Altun E, Paksoy S, Turan G. Could Eosinophilia predict clinical severity in nasal polyps? Multidiscip Respir Med. 2017;12(1):21. doi:10.1186/s40248-017-0102-7
55. Haughney J, Morice A, Blyth KG, et al. A retrospective cohort study in severe asthma describing commonly measured biomarkers: eosinophil count and IgE levels. Respir Med. 2018;134:117–123. doi:10.1016/j.rmed.2017.12.001
56. Tomassen P, Vandeplas G, Van Zele T, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. 2016;137(5):1449–1456.e1444. doi:10.1016/j.jaci.2015.12.1324
57. Kariyawasam HH. Chronic rhinosinusitis with nasal polyps: insights into mechanisms of disease from emerging biological therapies. Expert Rev Clin Immunol. 2019;15(1):59–71. doi:10.1080/1744666X.2019.1541738
58. Kariyawasam HH, James LK, Gane SB. Dupilumab: clinical efficacy of blocking IL-4/IL-13 signalling in chronic rhinosinusitis with nasal polyps. Drug Des Devel Ther. 2020;14:1757–1769. doi:10.2147/DDDT.S243053
59. Casale TB, Chipps BE, Rosen K, et al. Response to omalizumab using patient enrichment criteria from trials of novel biologics in asthma. Allergy. 2018;73(2):490–497. doi:10.1111/all.13302
60. Humbert M, Taille C, Mala L, et al. Omalizumab effectiveness in patients with severe allergic asthma according to blood eosinophil count: the STELLAIR study. Eur Respir J. 2018;51.
61. Turk M, Kocaturk E, Cure K, Yilmaz I. Two-week intervals during omalizumab treatment may provide better symptom control in selected patients with chronic urticaria. J Allergy Clin Immunol Pract. 2018;6(4):1389–1390. doi:10.1016/j.jaip.2018.01.027
62. Hochhaus G, Brookman L, Fox H, et al. Pharmacodynamics of omalizumab: implications for optimised dosing strategies and clinical efficacy in the treatment of allergic asthma. Curr Med Res Opin. 2003;19(6):491–498. doi:10.1185/030079903125002171
63. Casale TB. Anti-immunoglobulin E (omalizumab) therapy in seasonal allergic rhinitis. Am J Respir Crit Care Med. 2001;164(supplement_1):S18–21. doi:10.1164/ajrccm.164.supplement_1.2103023
64. Lowe PJ, Tannenbaum S, Gautier A, Jimenez P. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE-mediated) asthma. Br J Clin Pharmacol. 2009;68(1):61–76. doi:10.1111/j.1365-2125.2009.03401.x
65. Geha RS, Helm B, Gould H. Inhibition of the Prausnitz-Kustner reaction by an immunoglobulin epsilon-chain fragment synthesized in E coli. Nature. 1985;315(6020):577–578. doi:10.1038/315577a0
66. Davies AM, Allan EG, Keeble AH, et al. Allosteric mechanism of action of the therapeutic anti-IgE antibody omalizumab. J Biol Chem. 2017;292(24):9975–9987. doi:10.1074/jbc.M117.776476
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.