Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Chronic Pseudomonas aeruginosa infection-induced chronic bronchitis and emphysematous changes in CCSP-deficient mice
Authors Matsumoto T, Fujita M ![]() , Hirano R, Uchino J
, Hirano R, Uchino J ![]() , Tajiri Y, Fukuyama S, Morimoto Y, Watanabe K
, Tajiri Y, Fukuyama S, Morimoto Y, Watanabe K
Received 27 May 2016
Accepted for publication 4 July 2016
Published 20 September 2016 Volume 2016:11(1) Pages 2321—2327
DOI https://doi.org/10.2147/COPD.S113707
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Takemasa Matsumoto,1 Masaki Fujita,1 Ryosuke Hirano,1 Junji Uchino,1 Yukari Tajiri,2 Satoru Fukuyama,2 Yasuo Morimoto,3 Kentaro Watanabe1
1Department of Respiratory Medicine, Faculty of Medicine, Fukuoka University, 2Research Institute for Diseases of the Chest, Graduate School of Medical Sciences, Kyushu University, Fukuoka, 3Department of Occupational Pneumology, Institute of Industrial and Ecological Sciences, University of Occupational and Environmental Health, Kitakyushu, Japan
Abstract: The club cell secretory protein (CCSP) is a regulator of lung inflammation following acute respiratory infection or lung injury. Recently, the relationship between CCSP and COPD has been reported. Since COPD results from an abnormal inflammatory response, we hypothesized that CCSP could have a protective role against chronic inflammation-induced lung damage. To address this issue, the pathophysiology of chronic lung inflammation induced by Pseudomonas aeruginosa in CCSP-deficient mice was determined. A tube of 5 mm in length was soaked in a fluid containing P. aeruginosa (PAO01 strain) for 1 week and inserted into the trachea of CCSP-deficient mice. One week later, P. aeruginosa was administered into the trachea. Five weeks after insertion of tube, the mice were sacrificed. Bronchoalveolar lavage fluids were collected to determine the bacterial growth, and the lung histology and physiology were also examined. P. aeruginosa was continuously detected in bronchoalveolar lavage fluids during the study. Neutrophils were increased in the bronchoalveolar lavage fluids from the CCSP-deficient mice in comparison to wild-type mice. A histological study demonstrated chronic inflammation around bronchus, serious bronchial stenosis, and alveolar enlargement in the CCSP-deficient mice. The lung physiology study demonstrated an increase in the lung compliance of the CCSP-deficient mice. Chronic P. aeruginosa inflammation resulted in chronic bronchitis and emphysematous changes in the CCSP-deficient mice. CCSP could play an important role in protecting the host from the chronic inflammation-induced lung damage.
Keywords: chronic bronchitis, animal models, emphysema, inflammation
Introduction
The club cell secretory protein (CCSP or CC-10/uteroglobin) is produced by the nonciliary bronchial epithelium as well as the uterine and urethral ducts.1 CCSP exhibits high homology with rabbit uteroglobin, which has multiple activities, including immunosuppressive, anti-inflammatory, antiproteinase, and antiphospholipase A2 activities.2 This suggests that CCSP might play a role in the anti-inflammatory response.3,4 CCSP-deficient mice (CCSP−/−) have been reported to demonstrate more severe inflammation than wild-type controls in response to viral infection.5 With regard to human diseases, correlations between smoking-related disease and serum or bronchoalveolar levels of CCSP have been well demonstrated.1,6 In addition, club cells have been found to be reduced in the lungs of asthmatics,7 and CCSP concentrations were also reduced in the sera of asthmatics.8 In large amounts, the protein can play an important role in lung homeostasis.
Although CCSP could play a role in the acute inflammatory response, reports concerning the relationship between CCSP and chronic lung disease have been quite limited. Recently, characterization of COPD has revealed that CCSP was strongly related to COPD progression.9,10 COPD is a major cause of chronic morbidity and mortality throughout the world. It is characterized by airflow limitation, which is not fully reversible. The disease results from an abnormal inflammatory response to inhaled particles and gases (mainly from smoking) in the lung.11 COPD consists of pulmonary emphysema, destruction of alveoli, and chronic bronchitis – diseases that mainly involve the bronchi. Recently, chronic P. aeruginosa infection has been reported to be associated with a greater severity of COPD.12
We hypothesized that CCSP could have a protective role against chronic inflammation-induced lung damage. Thus, CCSP deficiency would lead to more lung inflammation and result in the destruction of lung, the characteristic feature of COPD. To address this issue, we developed a mouse model of chronic P. aeruginosa infection using CCSP−/− mice, and the pathophysiology of the mouse model was determined.
Materials and methods
Animal model
A bacterial culture of P. aeruginosa (PAO01 strain) was used to cause infection in this study. For animal model, CCSP-deficient mice (CCSP−/−) of the 129 strains background and 129 mice (wild-type) were used.13 First, we made the chronic respiratory infection model of P. aeruginosa with modification of the method previously described.14 An intravenous catheter of external diameter 1 mm was purchased from the Atom Medical Co. (Saitama, Japan). A tube of length 5 mm, which was soaked in fluid containing P. aeruginosa (PAO01 strain) at a concentration of 1×106 CFU/mL for 1 week, was inserted into the trachea of the mice. One week later, P. aeruginosa (5×104 CFU/animal) was administered intranasally; an untreated tube was inserted into the trachea of the control mice. Four weeks after intranasal administration of P. aeruginosa, the mice were sacrificed (Figure 1A). Bacterial growth was examined in the bronchoalveolar lavage (BAL) fluids. The cell count in the BAL fluids was determined and the lung histology was also investigated. This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Fukuoka University, and followed the Guidelines for Animal Experimentation, Fukuoka University (based on the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology). The IACUC is charged with protecting the safety and welfare of animals used in research at or in conjunction with Fukuoka University.
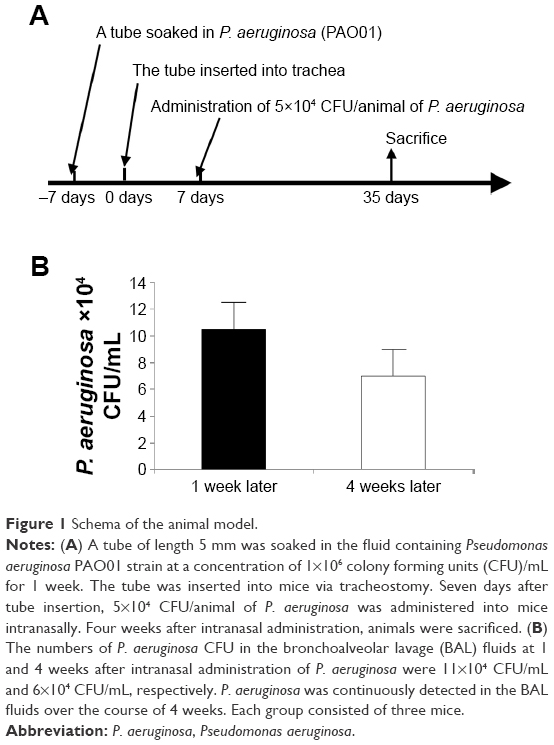 | Figure 1 Schema of the animal model. |
Lung histology and morphometry
The lungs were inflated at 25 cm H2O static pressure by intratracheal instillation of 4% paraformaldehyde in phosphate-buffered saline (PBS) prior to the staining of 4 μm thick tissue sections with hematoxylin and eosin (H&E), alcian blue staining, and Sirius red staining. The mean linear intercept (Lm), an indicator of air space size, was calculated for each mouse as described previously.15–17 The destructive index (DI) was calculated to evaluate the destruction.18 Stenosis was measured as the index of bronchial stenosis, which is equal to the area of the bronchial lumen divided by the area of the external bronchus. The area was measured using the Win Roof software program (version 5.5; Mitani Corporation, Tokyo, Japan).
BAL fluids
In all the mice the lungs were lavaged with 0.5 mL aliquots of PBS ten times for a total of 5 mL. The total cell counts were determined with a hemocytometer. Differential counts of BAL fluid were performed on 200 cells from smears stained with a modified Wright’s stain (DiffQuik; American Scientific Products, McGaw Park, IL, USA). BAL fluid supernatants were analyzed using an enzyme-linked immunosorbant assay (ELISA) kit according to the manufacturer’s instructions (R&D Systems, Inc., Minneapolis, MN, USA). Inflammatory cytokines/chemokines such as interferon (IFN)-γ, keratinocyte-derived chemokine (KC), and tumor necrosis factor (TNF)-α were measured.
Pulmonary physiology
A variety of pulmonary physiological parameters such as the pressure–volume curve (P–V) were examined according to previously described methods.15–17,19 From the P–V curve, static compliance was calculated using the slope of the early phase of the P–V curve.
Statistics
The data were expressed as the mean ± standard error. The Mann–Whitney test was used to compare the two groups. A P-value of <0.05 was considered to indicate a statistically significant difference. All the statistical analyses were performed using the JMP software program (version 7.0; SAS Institute Inc., Cary, NC, USA).
Results
Chronic respiratory infection model
The numbers of P. aeruginosa colony forming units (CFU) in the BAL fluids at 1 and 4 weeks after intranasal administration were 11×104 CFU/mL and 6×104 CFU/mL, respectively. P. aeruginosa was continuously detected in BAL fluids during the 4 weeks (Figure 1B). Inflammatory cell infiltrates were observed in the lung, mainly around the bronchus and small numbers were observed around the alveoli (Figure 2A). Most of the mice in the experiment survived until sacrifice. These results indicated that a consistent chronic respiratory infection model of P. aeruginosa had been established.
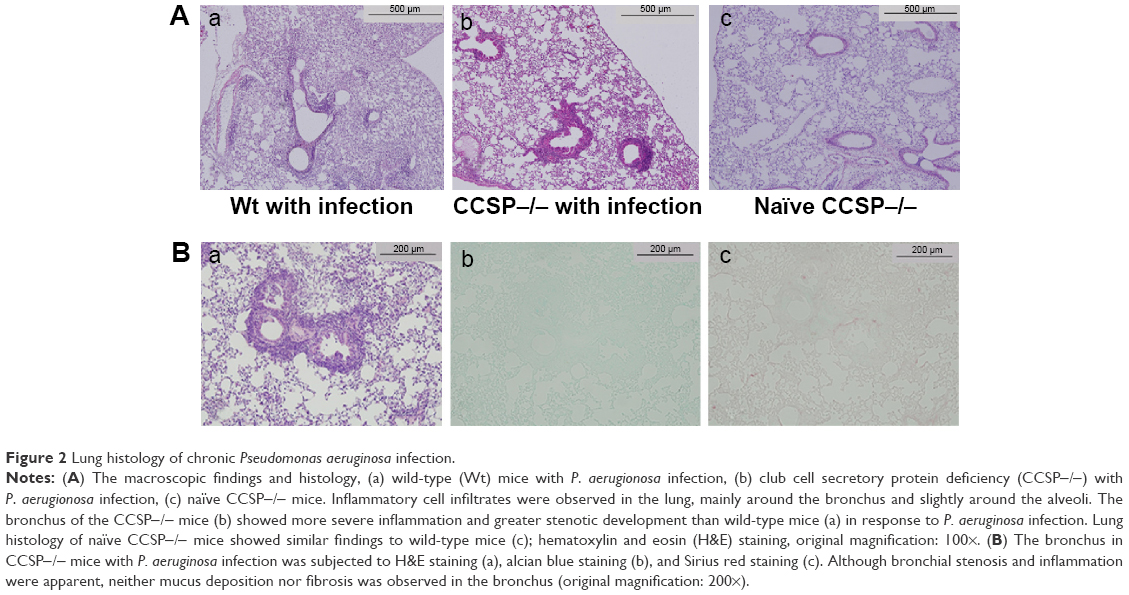 | Figure 2 Lung histology of chronic Pseudomonas aeruginosa infection. |
CCSP−/− mice of the chronic infection model
The macroscopic findings and histology of the naïve CCSP−/− mice were similar to the descriptions previously reported for naïve wild-type mice.13 The lungs of the chronic infection mouse model were subjected to H&E staining. Inflammatory cell infiltration was observed around the alveoli and the bronchus. In comparison to wild-type mice, the bronchus of the CCSP−/− mice showed more severe inflammation and greater stenotic development in response to P. aeruginosa infection (Figure 2A). The bronchi of P. aeruginosa-infected CCSP−/− mice were subjected to H&E, alcian blue, and Sirius red staining. Although bronchial stenosis and inflammation were apparent, neither mucus deposition nor fibrosis was observed in the bronchus (Figure 2B).
The cell counts in the BAL fluids were also investigated. The number of neutrophils, but not the macrophages or lymphocytes, was significantly increased in the BAL fluids of the CCSP−/− mice in comparison to the wild-type mice (Figure 3A). Cytokines in the BAL fluids were measured by the cytokine ELISA kit. The augmentation of the inflammatory cytokine, KC, was observed; however, it was not significant in IFN-γ and TNF-α (Figure 3B).
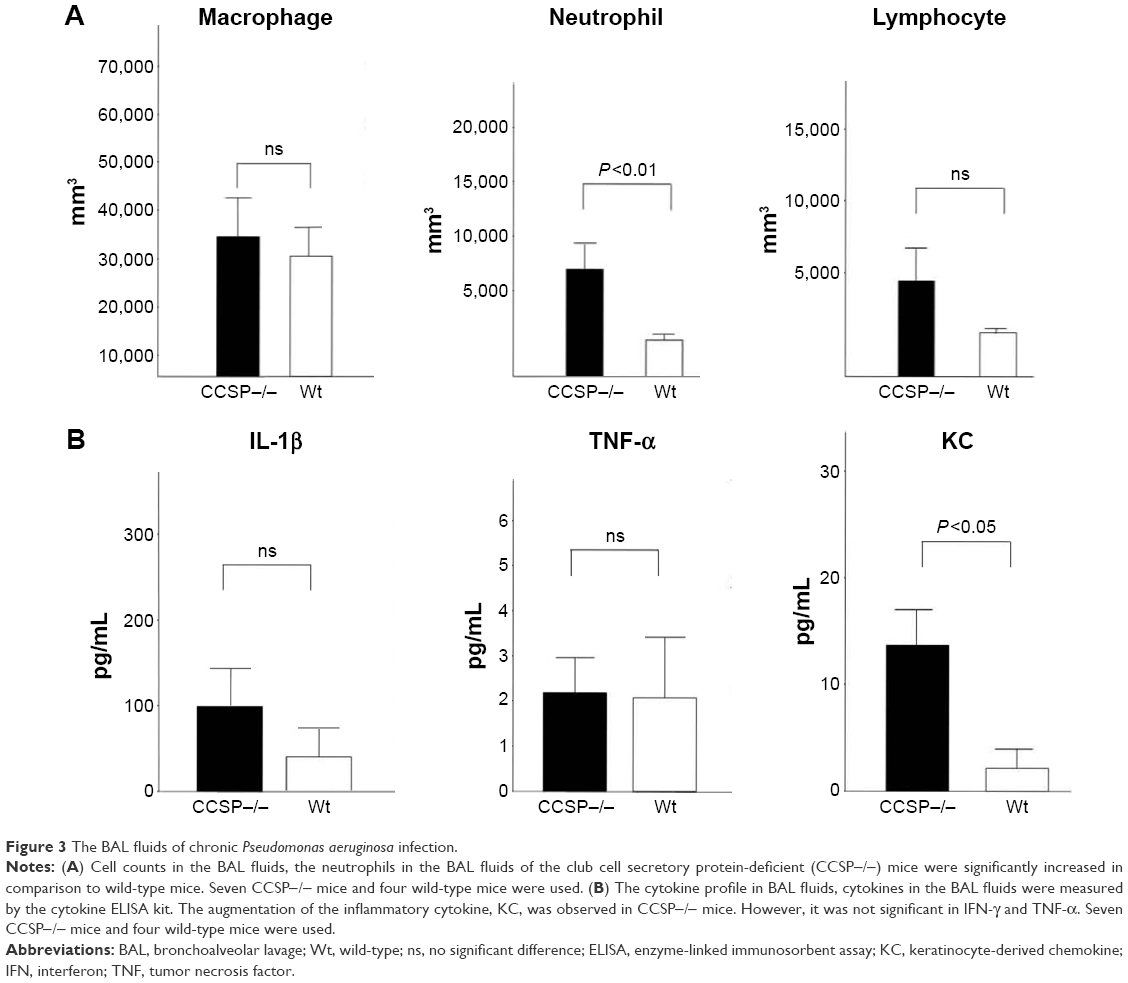 | Figure 3 The BAL fluids of chronic Pseudomonas aeruginosa infection. |
Characterization of the animal model
The histologic and physiologic findings of the naïve CCSP−/− mice were also similar to those of the naïve wild-type mice (data not shown). The index of bronchial stenosis was investigated. The bronchus of the CCSP−/− mice showed significantly greater stenotic development in comparison to wild-type mice (Figure 4). The Lm was measured by previously reported methods to evaluate the emphysematous changes of the lung. The Lm in the CCSP−/− mice was significantly higher than in the wild-type mice (Figure 5A). The DI was measured by previously reported methods to evaluate the destruction of the alveolus wall of the lung. The DI in the CCSP−/− mice was significantly higher than in the wild-type mice (Figure 5B). P–V curves were drawn using the lung physiology data. The lung compliance was then calculated using the slope of the early phase of the P–V curve. The CCSP−/− mice showed significantly higher lung compliance than the wild-type mice (Figure 5C).
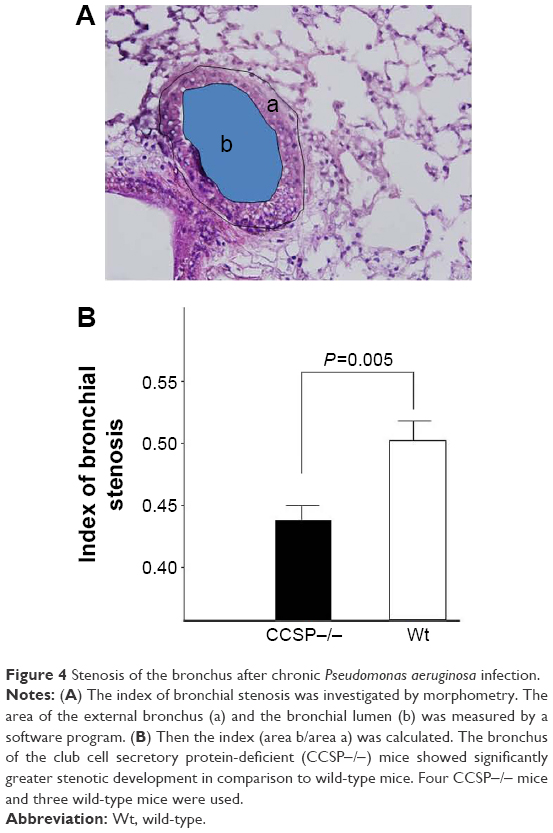 | Figure 4 Stenosis of the bronchus after chronic Pseudomonas aeruginosa infection. |
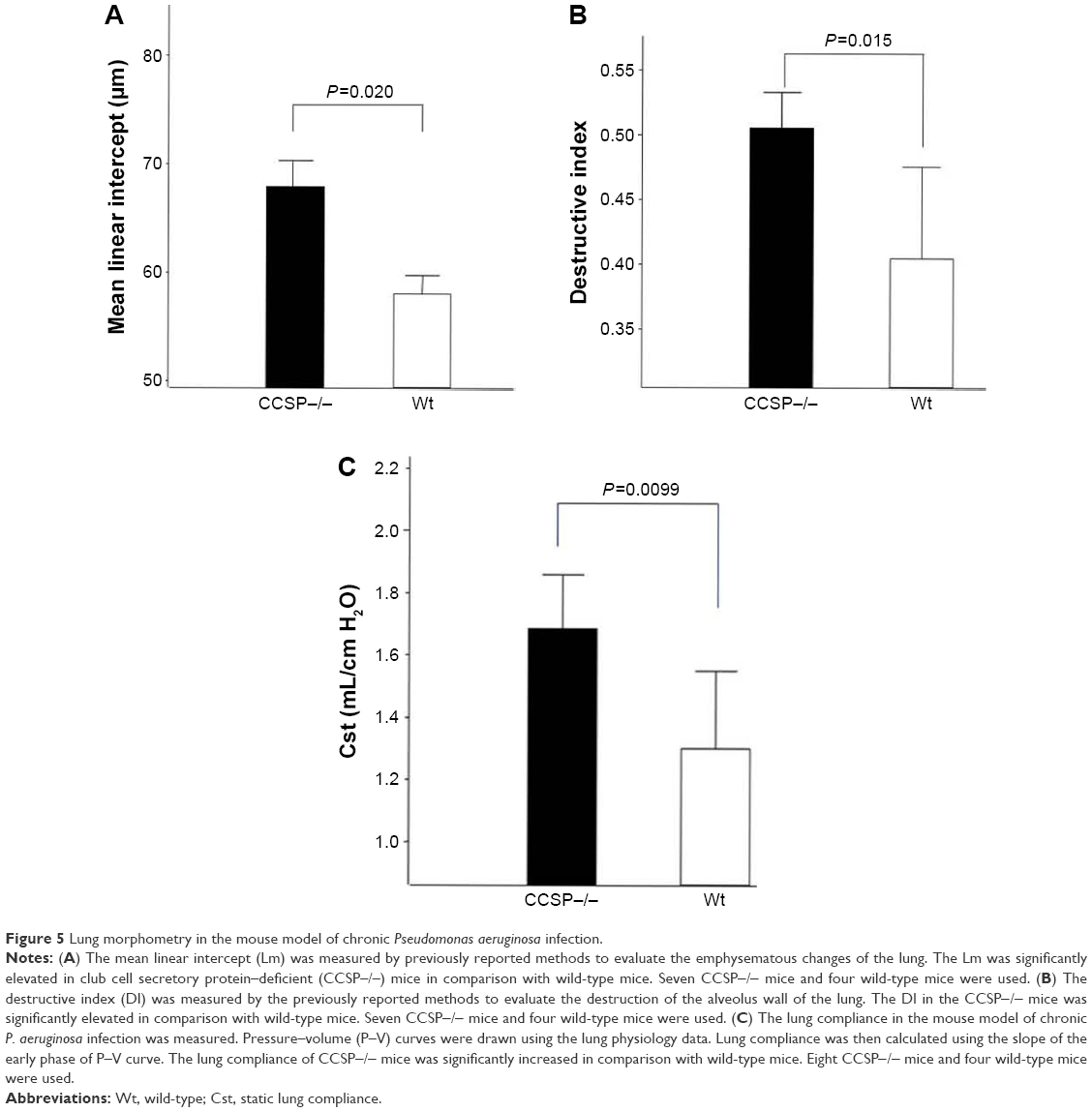 | Figure 5 Lung morphometry in the mouse model of chronic Pseudomonas aeruginosa infection. |
Discussion
In this study, we investigated the pathophysiological characterization in persistent P. aeruginosa infection in CCSP−/− mice. Chronic respiratory infection model of P. aeruginosa has been reported previously.14 However, the procedure was too difficult to perform. The present procedure is simple and reproducible compared to the method reported previously. Insertion of the tube soaked in P. aeruginosa-containing fluids did not result in persistent infection. We found that the additional administration of P. aeruginosa at 7 days after insertion of the tube was a key factor for the establishment of chronic persistent infection.
Chronic inflammation of P. aeruginosa in CCSP−/− mice demonstrated the aggravated inflammation around the small bronchus and resulted in bronchial stenosis and emphysematous changes. This pathological condition is similar to that of chronic bronchitis and pulmonary emphysema, which are the characteristic findings of COPD. The relationship between CCSP and P. aeruginosa has been investigated previously.20 However, the previous study used a model of acute P. aeruginosa infection. CCSP deficiency was associated with enhanced pulmonary inflammation and improved killing of bacteria after an acute pulmonary infection. Concentrations of the proinflammatory cytokines such as interleukin-1β and TNF-α were modestly increased after 6 and 24 hours, respectively, in CCSP−/− mice. CCSP plays a role in the modulation of pulmonary inflammation during infection and recovery. On the contrary, we used chronic P. aeruginosa inflammation model and clarified further the role of CCSP. Deletion of CCSP leads to chronic bronchitis and emphysematous change. The important role of CCSP in chronic inflammation was first identified in the present study.
Interestingly, we found that chronic P. aeruginosa inflammation produced chronic bronchitis and emphysematous change in the CCSP−/− mice. This model is therefore considered to be a chronic bronchitis-related emphysematous model. In humans, the pathology of COPD is characterized by small airway inflammation.21 Recent studies have attempted to characterize COPD and demonstrated that CCSP is strongly related to COPD progression.9,10 In another study, chronic P. aeruginosa infection in COPD patients was reported to be associated with severe obstruction pattern.12 In the present study, severe inflammation around small airways was remarkably observed. Chronic inflammation affects lung morphogenesis and causes several pathological involvements including COPD.22 King described that chronic bronchitis resulted in bronchial stenosis and led to alveolar emphysema.23 Moreover, CCSP has been demonstrated to be influenced by cytokines such as TNF-α, KC, or IFN-γ.24,25 These cytokines were found to be essential for the pathogenesis of COPD.15–17,26 Especially, KC levels increased after administration of elastase with increase of inflammation.16 It is a plausible explanation that, in response to chronic inflammation by P. aeruginosa, the deletion of CCSP led to an increase in KC levels, resulted in severe inflammation around small airways, and finally caused destruction of lung architecture, findings which are similar to those observed in COPD patients. The limitation was that this model was chronic inflammation model induced by P. aeruginosa, but not human COPD model such as smoking. In future, we should investigate chronic smoking model using CCSP−/− mice.
CCSP widely distributes to the ducts, such as bronchioles and uterine or urinary ducts. We speculate that CCSP could play an important role in keeping ducts patent against chronic inflammation. Given the important role of CCSP, Gamez et al27 have recently indicated that it might be a target for new treatments for bronchitis-related emphysema. The supplementation of CCSP will be achieved in future using this model. We should also seek to elucidate the role of CCSP in human disease.
Conclusion
Chronic P. aeruginosa inflammation resulted in chronic bronchitis and emphysematous changes in the CCSP-deficient mice. CCSP could play an important role in protecting the host from the chronic inflammation-induced lung damage.
Acknowledgments
The authors appreciate the assistance of Dr Brian Quinn with language editing. The study was supported by a grant-in-aid for scientific research from the Japan Society for the Promotion of Science (No 14570553 [to MF]), and funding from Astra-Zeneca Research Fellow 2003 (to MF). An abstract of part of the study has been presented in an international conference: Matsumoto T, Fujita M, Hirano R, Tashiro N, Asai Y, Fukuyama S, Morimoto Y, Nakanishi Y, Watanabe K. Chronic Infection of Pseudomonas aeruginosa induced COPD like changes in CCSP-deficient mice. Abstract no A2697. ATS, May 16–21, 2014, San Diego, CA, USA.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Bernard A, Marchandise FX, Depelchin S, Lauwerys R, Sibille Y. Clara cell protein in serum and bronchoalveolar lavage. Eur Respir J. 1992;5(10):1231–1238. | ||
Lesur O, Bernard A, Arsalane K, et al. Clara cell protein (CC16) induces a phospholipase A2-mediated inhibition of fibroblast migration in vitro. Am J Respir Crit Care Med. 1995;152(1):290–297. | ||
Mantile G, Miele L, Cordella-Miele E, Singh G, Katyal SL, Mukherjee AB. Human Clara cell 10-kDa protein is the counterpart of rabbit uteroglobin. J Biol Chem. 1993;268(27):20343–20351. | ||
Boers JE, Ambergen AW, Thunnissen FB. Number and proliferation of Clara cells in normal human airway epithelium. Am J Respir Crit Care Med. 1999;159(5 Pt 1):1585–1591. | ||
Harrod KS, Mounday AD, Stripp BR, Whitsett JA. Clara cell secretory protein decreases lung inflammation after acute virus infection. Am J Physiol. 1998;275(5 Pt 1):L924–L930. | ||
Shijubo N, Itoh Y, Yamaguchi T, et al. Serum and BAL Clara cell 10 kDa protein (CC10) levels and CC10-positive bronchiolar cells are decreased in smokers. Eur Respir J. 1997;10(5):1108–1114. | ||
Shijubo N, Itoh Y, Yamaguchi T, et al. Clara cell protein-positive epithelial cells are reduced in small airways of asthmatics. Am J Respir Crit Care Med. 1999;160(3):930–933. | ||
Shijubo N, Itoh Y, Yamaguchi T, et al. Serum levels of Clara cell 10-kDa protein are decreased in patients with asthma. Lung. 1999;177(1):45–52. | ||
Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–1192. | ||
Braido F, Riccio AM, Guerra L, et al. Clara cell 16 protein in COPD sputum: a marker of small airways damage? Respir Med. 2007;101(10):2119–2124. | ||
Global Initiative for Chronic Obstructive Lung Disease (GOLD). The Global Strategy for the Diagnosis, Management and Prevention of COPD. 2015. Available from: http://www.goldcopd.org. Accessed March 1, 2016. | ||
Boixeda R, Almagro P, Díez-Manglano J, et al. Bacterial flora in the sputum and comorbidity in patients with acute exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:2581–2591. | ||
Watson TM, Reynolds SD, Mango GW, Boe IM, Lund J, Stripp BR. Altered lung gene expression in CCSP-null mice suggests immunoregulatory roles for Clara cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(6):L1523–L1530. | ||
Morinaga Y, Yanagihara K, Nakamura S, et al. In vivo efficacy and pharmacokinetics of tomopenem (CS-023), a novel carbapenem, against Pseudomonas aeruginosa in a murine chronic respiratory tract infection model. J Antimicrob Chemother. 2008;62(6):1326–1331. | ||
Fujita M, Shannon JM, Irvin CG, et al. Overexpression of tumor necrosis factor-a produces an increase in lung volumes and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2001;280(1):L39–L49. | ||
Fujita M, Ye Q, Ouchi H, et al. Retinoic acid failed to reverse emphysema in adult mouse model. Thorax. 2004;59(3):224–230. | ||
Fujita M, Ouchi H, Ikegame S, et al. Critical role of tumor necrosis factor receptor 1 in the pathogenesis of pulmonary emphysema in mice. Int J Chron Obstruct Pulmon Dis. In press 2016. | ||
Koike K, Ishigami A, Sato Y, et al. Vitamin C prevents cigarette smoke-induced pulmonary emphysema in mice and provides pulmonary restoration. Am J Respir Cell Mol Biol. 2014;50(2):347–357. | ||
Irvin CG, Tu YP, Sheller JR, Funk CD. 5-lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. Am J Phsyiol. 1997;272(6 Pt 1):L1053–L1058. | ||
Hayashida S, Harrod KS, Whitsett JA. Regulation and function of CCSP during pulmonary Pseudomonas aeruginosa infection in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;279(3):L452–L459. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–498. | ||
King PT. The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis. 2009;4:411–419. | ||
Yao XL, Levine SJ, Cowan MJ, Logun C, Shelhamer JH. Tumor necrosis factor-a stimulates human Clara cell secretory protein production by human airway epithelial cells. Am J Respir Cell Mol Biol. 1998;19(4):629–635. | ||
Yao XL, Ikezono T, Cowan M, Logun C, Angus CW, Shelhamer JH. Interferon-g stimulates human Clara cell secretory protein production by human airway epithelial cells. Am J Physiol. 1998;274(5 Pt 1):L864–L869. | ||
Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534. | ||
Gamez AS, Gras D, Petit A, et al. Supplementing defect in club cell secretory protein attenuates airway inflammation in COPD. Chest. 2015;147(6):1467–1476. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.