Back to Journals » Journal of Blood Medicine » Volume 14
Chronic Myeloid Leukemia, from Pathophysiology to Treatment-Free Remission: A Narrative Literature Review
Received 12 July 2022
Accepted for publication 6 February 2023
Published 6 April 2023 Volume 2023:14 Pages 261—277
DOI https://doi.org/10.2147/JBM.S382090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Ikhwan Rinaldi,1 Kevin Winston2,3
1Division of Hematology and Medical Oncology, Department of Internal Medicine, Cipto Mangunkusumo National General Hospital, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia; 2Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia; 3Hospital Medicine, Bhakti Medicare Hospital, Sukabumi, Indonesia
Correspondence: Ikhwan Rinaldi, Division of Hematology and Medical Oncology, Department of Internal Medicine, Cipto Mangunkusumo National General Hospital, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia, Email [email protected]
Abstract: Chronic myeloid leukemia (CML) is one of the most common leukemias occurring in the adult population. The course of CML is divided into three phases: the chronic phase, the acceleration phase, and the blast phase. Pathophysiology of CML revolves around Philadelphia chromosome that constitutively activate tyrosine kinase through BCR-ABL1 oncoprotein. In the era of tyrosine kinase inhibitors (TKIs), CML patients now have a similar life expectancy to people without CML, and it is now very rare for CML patients to progress to the blast phase. Only a small proportion of CML patients have resistance to TKI, caused by BCR-ABL1 point mutations. CML patients with TKI resistance should be treated with second or third generation TKI, depending on the BCR-ABL1 mutation. Recently, many studies have shown that it is possible for CML patients who achieve a long-term deep molecular response to stop TKIs treatment and maintain remission. This review aimed to provide an overview of CML, including its pathophysiology, clinical manifestations, the role of stem cells, CML treatments, and treatment-free remission.
Keywords: chronic myeloid leukemia, remission, tyrosine kinase inhibitor
Introduction
Chronic myeloid leukemia or chronic myelogenous leukemia (CML) is a hematopoietic stem cell (HSC) disorder, as are all leukemias. In CML, the disorder is characterized by translocation t(9;22)(q34;q11), resulting in the fusion of BCR and ABL1 genes into the pathogenic BCR-ABL1 oncogene, with many subsequent effects on downstream pathways.1,2 The main pathway effect of this fused oncogene is to activate tyrosine kinase pathway constitutively, resulting in a proliferative advantage of the mutant HSCs compared to normal HSCs, and the gradual displacement of normal HSCs.3
CML symptoms range from asymptomatic to overt leukostasis, depending on the stage of CML; leukostasis often occurs in the blast phase. Meanwhile, hyperleukocytosis is the most common symptom, and is often present in all phases (chronic phase, accelerated phase, and blast phase).1 The blast phase is similar to acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL). The diagnosis of CML often requires a basic hematology workup combined with identification of chromosomal abnormalities. Different phases of CML can be differentiated by clinical symptoms and blast percentage.
CML is one of the model diseases for targeted therapy. It is widely accepted that discovery of tyrosine kinase inhibitors (TKIs) has changed the treatment landscape of CML.4 Prior to the introduction of TKIs, CML was universally fatal in the long term. However, since TKIs have become the first-line treatment for CML, the life expectancy of CML patients has become similar to that of people without CML.5,6
While TKIs have indeed improved the survival of CML patients, there are still unresolved issues associated with TKI treatment, such as adverse side effects and impact on quality of life. Furthermore, a very important issue associated with TKI treatment is the need for most patients to continuously take the TKI to prevent relapse. Thus, many patients are on lifelong TKI treatment. However, several studies, such as STIM1 trial, have shown that patients with a deep molecular response (DMR) can achieve treatment-free remission (TFR) with no relapse after discontinuing their TKI.7 This review article aimed to discuss all aspects of CML.
Pathophysiology of CML
The presence of BCR-ABL1 fusion gene in hematopoietic stem cells has been shown to be sufficient for initiation of CML.8 Thus, the pathophysiology of CML revolves around the BCR-ABL1 fusion gene. Approximately 90% to 95% of patients with CML have reciprocal translocation of t(9;22) (q34;q11.2).1,2 This results in a shortened chromosome 22, termed Philadelphia chromosome, containing the BCR-ABL1 oncogene. However, It should be noted that BCR-ABL1 may also be present in other types of leukemia, such as acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML).9 The significance of BCR-ABL1 in ALL and AML is still under investigation.
At the time of writing, the mechanism of Philadelphia chromosome formation and the time from the formation of the chromosome to the occurrence of clinical symptoms of CML are still not known with certainty. The Philadelphia chromosome was previously thought to have been formed due to the influence of radiation from the atomic bombs of Hiroshima and Nagasaki on CML patients in Japan.10 Indeed, in vitro studies have shown that exposure of myeloid cell lines to high-dose radiation causes the expression of BCR-ABL1.10 However, the majority of people are not exposed to chronic radiation. Some experts believe that the Ph chromosome can be formed as a result of spontaneous random translocation. Supporting this is the fact that the BCR-ABL1 gene has also been observed in a small proportion of healthy individuals.11,12 This could also mean that not all BCR-ABL1 fusion genes develop into CML. Further studies are needed to elucidate the origins of the BCR-ABL1 gene; however, it will be very difficult to isolate the exact causes that contribute to the formation of the Philadelphia chromosome due to the relative rareness of the disease and issues with determining the onset of the disease, as it is initially asymptomatic.
As has been described by much of the scientific literature on this topic, the BCR-ABL1 hybrid gene located on the Ph chromosome synthesizes the oncoprotein 210 kD (p210), which plays a role in leukemogenesis of CML.13 The p210BCR-ABL1 protein can continuously activate the tyrosine kinase pathway, resulting in an increase in the signal transduction of unregulated downstream oncogenic pathways such as JAK/STAT, PI3K/AKT, RAF, MYC, and RAS/MEK.3,13 All of these pathways are involved in cell growth, cell survival, and the inhibition of apoptosis. Thus, HSCs with BCR-ABL1 translocation have a proliferative advantage due to their enhanced response to growth factors. Furthermore, proliferation-inhibitory factors are less responsive. Thus, overall, the presence of BCR-ABL1 in hematopoietic stem cells will encourage their transformation into leukemia stem cells (LSCs) and replacement or displacement of normal hematopoietic stem cells.
In CML, there are other types of BCR-ABL1 fusion and breakpoints. For example, p190BCR-ABL1 (e1a2), which encodes a hybrid 190 kDa protein, can be observed in CML, but the frequency is rare. Based on several studies, p190BCR-ABL1 occurs in only 1–2% of CML patients.14,15 Other BCR-ABL1 proteins include p230BCR-ABL1.
A point of interest is that not all CML patients have Philadelphia chromosome. In a minority of CML patients, the Philadelphia chromosome is lacking but identifiable BCR-ABL1 oncogenes are present. Observations have shown that the number of confirmed CML patients with no Philadelphia chromosome is up to 5–10%.16–18 Interestingly, approximately 25–50% of these patients still have BCR-ABL1 gene rearrangement, as confirmed by molecular examinations.18,19 In other words, the BCR-ABL1 genes in this subset of CML patients originate outside of the Philadelphia chromosome. All other patients have no confirmed BCR-ABL gene rearrangement.
According to a study by Wang et al, BCR-ABL-negative CML patients have a worse overall survival and have a higher risk of their leukemia transforming into acute myeloid leukemia.20 Without the presence of BCR-ABL, most TKIs are not indicated as a main treatment, which may be one of the factors contributing to a worse prognosis in BCR-ABL-negative CML patients. Gene mutations commonly found in BCR-ABL-negative CML include SETBP1, ASXL1, NRAS/KRAS, SRSF2, CSF3R, U2AF1, and many others.21 Some of these mutations may be used as potential targets for therapy in BCR-ABL-negative CML, but the evidence to support this is currently lacking.
Pathophysiology of CML Progression
CML is grouped into three phases, namely the chronic phase, the accelerated phase, and the blast phase.1 In the chronic phase, LSCs are confined to hematopoietic tissues. However, in the blast phase, the LSCs gain an invasive capability that enables them to infiltrate other organs, most notably the spleen, resulting in splenomegaly. Furthermore, immature blast cells now dominate in blast phase. The process of transformation from chronic CML to the accelerated phase or blast phase is thought to be caused by amplification of the BCR-ABL1 protein and increased activation of the downstream tyrosine kinase pathway, combined with the emergence of additional chromosomal abnormalities (ACAs).22,23 Oxidative stress and impaired DNA repair have also been implicated.24–26 In addition, mutations such as deletion of the IKZF1 gene and loss of CCAAT protein expression have been reported to cause the immaturity of granulocytes.27,28 Blast-phase patients often have multiple gene mutations.29 Common gene mutations due to genomic instability in blast-phase CML are summarized in Table 1.22,30,31
 |
Table 1 Common Gene Mutations in Blast-Phase CML |
Additional Chromosomal Abnormalities in CML
LSCs are genetically unstable, so they have a tendency to present with other genomic or cytogenetic abnormalities.32,33 Additional cytogenetic abnormalities (ACAs) have been shown to increase in the later phases of CML. For example, in the chronic phase or at diagnosis, only approximately 5–10% of CML patients have ACAs, but this increases to up to 80% in the blast phase.34,35 Cytogenetic changes commonly observed in the blast phase include +8, +19, double Philadelphia chromosome, and isochromosome 17q.36,37
The presence of ACAs in the early stages appears to be associated with disease progression in CML. A study by Clark et al showed poor progression-free survival in CML patients with ACAs.37 Additionally, a study by Chandran et al also found a poor prognostic effect of ACAs in CML.35 The latest CML guidelines from the European LeukemiaNet state that the appearance of ACAs is one of the early indicators of disease progression, and the high-risk type of ACAs are associated with TKI treatment resistance.38 A study by Meggyesi et al showed that 30 out of 65 imatinib-resistant patients (46%) had ACAs.39 Monitoring of ACAs during TKI treatment is important as ACAs are associated with treatment resistance.40
CML Stem Cells
Evidence of the HSC origin of CML comes from the observation that the translocation of BCR-ABL1 is observed in myeloid, erythroid, and megakaryocytic cells. However, the effects of this translocation on erythroid and megakaryocytic cells are still unclear. The suppression of erythroid and megakaryocytic cells in CML is very likely due to the high proliferation of myeloid cells causing disruption. The malignant HSCs, termed leukemia stem cells (LSCs), provide a reservoir of self-replenishing leukemia cells.2 Thus, to completely eradicate CML, LSCs ideally must be targeted and removed to prevent relapse.
LSCs in chronic-phase CML can be identified via positive CD34 markers and negative CD38 markers.2,41–43 However, using only these surface antigen markers provides a heterogenous population, and these markers are not unique to CML LSCs.41 For example, the population of normal HSCs can also be identified using positive CD34 markers and negative CD38 markers as well. CD33 marker is also expressed by both normal stem cells and LSCs. However, there is a greater density of CD33 in LSCs.44 Indeed, LSC identification is also a challenge in other cancers.45
In an article by Hoshmand et al, it is stated that markers such as CD25, CD26, and interleukin-1-receptor accessory protein (IL1RAP) have been reported to be expressed specifically on CML stem cells.41 CD25 is encoded by the IL2RA gene and the CD25 marker has been reported to be expressed by more than 90% of CD34+/CD38− LSCs.46,47 Meanwhile, neither CD26 nor IL-1RAP have been shown to be expressed in LSCs in most other malignancies.48 However, it should be noted that both CD26 and IL-1RAP are expressed in normal mast cells, which may complicate identification.49,50 There are some possible combinations of cell surface markers for reliable identification of CML LSCs such as Lin-negative, CD34-positive, CD38-negative/low, CD45RA-negative, KIT-negative, and CD26-positive but this require further confirmations.41,51
LSCs have a persistent presence, even with tyrosine kinase inhibitor treatment.52 Thus, patients in remission are not “cured” and may technically relapse once TKI treatment is stopped. This suggests that there may be BCR-ABL1-independent pathways that maintain the self-renewal of CML LSCs. A study by Zhao et al showed that the maintenance of cancer stem cells requires activation of Hedgehog signaling.53 Additionally, a study by Chen et al demonstrated that the Alox5 gene is important for LSC survival.54 Other pathways or processes that support CML LSCs persistence include JAK1-STAT3 signaling, PI3K/AKT/FOXO signaling, Wnt signaling, and impaired DNA damage responses.55–59 LSCs also have increased expression of IL-1 receptors, which, if activated, reduce the growth of LSCs.60 Other cytokines that provide support to LSCs include TNF-alpha and IL-6.61 Thus, it is not unexpected that a significant proportion of patients in remission relapse after stopping treatment.
Therapies to target CML LSCs has not been used clinically. One may argue that since long-term sustained molecular responses from TKI produce a significant proportion of patients remaining in molecular response when stopping TKI, therapies to target CML LSCs may not be needed. However, there will always be a relatively high proportion of patients that will not be able to achieve TFR status and thus, therapies targeting CML LSCs may be useful for this population. We the authors speculate that using combination therapies of TKI with one or several drugs targeting LSCs pathways may be the new norm in the future. EZH2 inhibitor and JAK inhibitor combine with TKI seem to be effective in eliminating LSCs and should be further researched.62,63
Clinical Manifestations and Diagnosis
As stated above, the majority of CML patients are asymptomatic, especially in the early stages.1 Patients are often suspected to have CML following a medical check-up revealing leukocytosis and basophilia. In the chronic phase, when the majority of CML patients are diagnosed, the symptoms, if there are any, are fatigue, weight loss, cold sweats, and abdominal pain or feeling full quickly due to splenomegaly. Spleen size should be measured, as it is a component of CML prognosis scores such as the Sokal score and the ELTS score. Other symptoms that can occur, albeit rarely, are low-grade fever, bleeding, thrombosis, gouty arthritis, priapism, and gastric ulcers. The baseline diagnostic workup recommended by the ELN 2020, ESMO 2017, and NCCN 2021 guidelines is presented in Table 2.38,64,65
 |
Table 2 Baseline Diagnostic Workup Recommended by the ELN 2020, ESMO 2017, and NCCN 2021 Guidelines |
Upon routine hematological examination, an increase in the number of leukocytes and a shift in the differential count to the left, and myeloid series cells in various stages of differentiations, are often found.66 Leukocyte counts are often above 25 × 109/L with an increase in basophil count.67 In the chronic phase, the blast percentage is often low or negative. Platelets are also elevated in the majority of patients.
Bone marrow examination reveals a hypercellular marrow with more than 75% cellularity, and a granulocyte-to-erythroid ratio of 10:1 to 30:1.67 The increased cellularity is due to elevated myelopoiesis.64 An increase in cellularity is also observed in other leukemia. In CML, basophilia in bone marrow is often observed.
An increase in megakaryocytopoietic activity with hypolobated nuclei and micromegakaryocytes is often observed in the bone marrow.68 Examinations of bone marrows by Mughal et al showed the presence of megakaryocytic clustering in 60.6% of patients and found that megakaryocytic clustering was correlated with a high Sokal score.69 Elevated reticulin fibrosis in the bone marrow has been observed in approximately 30% of CML patients and can be associated with worse symptoms; however, this has not been incorporated into the current CML prognostic stratification, similarly to megakaryocytic clustering.64,70 The blast proportion in the chronic phase should be <15%, and a number above this indicates progression to the accelerated phase or blast phase.64 Immunohistochemistry through CD34 staining may be used to help blast identification.71
Cytogenetic analysis and qualitative PCR are recommended for CML diagnosis.38,64 In cytogenetic analysis, the Philadelphia chromosome may not be identified in some patients due to abnormal locations of the BCR-ABL1 gene. Hence, a FISH examination should be conducted to identify the location.38 PCR examination of BCR-ABL transcript is the most reliable method of diagnosing CML. However, false-negative qualitative and quantitative PCR results may occur due to atypical BCR-ABL1 transcripts.64
The accelerated CML phase occurs after one to three years of the chronic CML phase.1 This phase can be suspected when there is an enlargement of the spleen that had shrunk previously following therapy, anemia that becomes more severe, or the development of petechiae or ecchymosis. The presence of a fever is usually a sign of infection. The majority of chronic CML patients will enter the accelerated phase before the blast phase.
The blast phase is characterized by symptoms of extramedullary blast infiltration in organs other than the liver and spleen, such as the lymph nodes, central nervous system, and lungs. Indeed, both the WHO and ELN guidelines use this symptom as one of the criteria for blast-phase diagnosis.38,72 A high blast percentage further differentiates this phase from the accelerated phase. According to the ELN 2020 CML guidelines, the blast percentage needed for diagnosis is ≥30% in the peripheral blood or bone marrow.38 This is in contrast with the WHO guideline, which state that a blast percentage of ≥20% is needed.72 Overall, it is recommended to check for ACAs and gene mutations in the blast phase or when TKI treatment does not produce an adequate response. Furthermore, ACAs and gene mutations are useful for determining treatment.
The blast phase of CML resembles acute leukemia. The blast type is myeloid in up to 80% of cases, or lymphoid in up to 30% of cases (Figure 1).73 In lymphoid blast crisis, B-cell lineage is much more common than T-cell lineage.74,75 Lymphoid blast crisis presents a unique diagnostic and therapeutic challenges. Readers are suggested to refer to an article by Yohannan et al for further information.23
 |
Figure 1 Type of blast phase in CML. |
Therapies for CML
Chemotherapies
Busulfan was one of the earliest chemotherapies used for CML. In the past, busulfan was administered until leukocyte counts reduced to below 30×109 /L. As is the case for all chemotherapies, busulfan is associated with serious adverse effects. Those adverse effects include aplasia, pulmonary fibrosis, and severe infection.
Therapy with short-duration hydroxyurea can be given to CML patients with high leukocyte or high platelet counts with the aim of reducing leukocyte and/or platelet levels prior to the administration of a TKI.76 The recommended duration of administration of hydroxyurea is not stated in the ELN’s CML guideline. Another alternative to the use of hydroxyurea is apheresis (in certain conditions, such as hyperleukocytosis with leukostasis), but this is rarely used because the decrease in leukocytes following apheresis is only temporary. Nowadays, chemotherapies for CML have mostly been abandoned in favor of more effective therapies such as TKIs.
Interferon Alpha
Prior to the TKI era, the use of IFNα was dominant as a first-line therapy. Since the emergence of TKIs, IFNα has not been used as a first-line therapy because TKIs provide better survival and a better molecular response. However, several recent studies have shown that the combination of pegylated-IFNα (PEG-IFNα) with imatinib provides a faster and more potent molecular response.77,78 Currently, several clinical studies of the combination of PEG-IFNα with nilotinib or other TKIs are underway.
Stem Cell Therapy
The use of stem cell transplantation as an initial therapy has now been replaced by TKI therapy. Allogeneic stem cell transplantation may be considered for chronic CML patients who fail to respond to treatment with at least two TKIs. According to the European Society for Medical Oncology (ESMO) guidelines, chronic-stage CML patients who have a T315I mutation are also recommended for stem cell transplantation after a ponatinib trial.38 Patients who are at high risk for transformation to the blast phase are candidates for stem cell transplantation because the outcomes of stem cell transplantation in the blast phase are poor.79
Tyrosine Kinase Inhibitors
The first-line therapy for CML is a tyrosine kinase inhibitor (TKI), except in pregnant women. TKI therapy is a targeted therapy that inhibits the tyrosine kinase pathway in CML, which has been proven in clinical trials to provide better outcomes than previous CML treatments such as hydroxyurea, interferon alpha, and busulfan.
The first trial of a TKI in CML was imatinib compared with interferon and low-dose cytarabine in the newly diagnosed chronic-phase chronic myeloid leukemia (IRIS) study, which showed that 76.2% percent of CML patients given imatinib achieved a complete cytogenetic response (CCyR) after a median follow up of 19 months.80 Meanwhile, in the cytarabine+IFN-α group, the CcyR only reached 14.5%. This striking difference indicated that imatinib provided significantly better CcyR outcomes in the CRF patients. In addition, imatinib provided better survival rates than previous therapies. Therefore, after the publication of this study, TKIs became the first-line therapy used for CML, replacing IFN-α and chemotherapy.
Imatinib works via competitive inhibition of the ATP-binding site in the kinase domain, which subsequently blocks the ability of BCR-ABL to phosphorylate tyrosine residues.81 Side effects of TKI treatment include hypertension, thrombosis, and pleural effusion.82,83 Currently, the mechanisms that cause these side effects are unknown and are the subject of ongoing investigation.
With continuous treatment, approximately 90% of CML patients treated with imatinib have a similar survival to with people without CML. However, the remaining 10% of patients display resistance to imatinib, shown by failure to achieve a molecular response; these patients can often be treated with a second-generation TKI such as dasatinib or nilotinib.38 It should be noted that even with TKI treatments, there is a continuous presence of LSCs, as TKI usually does not eliminate quiescent stem cells.52 A strategy that is currently being developed to eliminate LSCs is the combination of a TKI with thiazolidinediones. Thiazolidinediones, in a study by Prost et al, were shown to impair hematopoiesis through PPAR-Y-STAT5 signaling activation, which may be of benefit in destroying quiescent stem cells.84
The second-generation TKIs consist of dasatinib, nilotinib, bosutinib, and radotinib. They tend to have a higher potency in inhibiting BCR-ABL1 mutants.85 The DASISION trial showed that dasatinib 100 mg once daily was superior to imatinib 400 mg once daily in the outcomes of a MMR.86 The outcome of MR4.5 was also higher from dasatinib than imatinib. The recent DASCERN study also showed a better MMR outcome from dasatinib.87 Similarly, other second-generation TKIs (nilotinib, bosutinib, and radotinib) have higher MMR responses than imatinib.88–90
Ponatinib is a third-generation TKI which has been shown to be more efficacious than other TKIs (Approximately 500 times more potent than imatinib) and is a TKI that has activity against T31UI BCR-ABL1 point mutation.38,91,92 Recommended dose for ponatinib is 45 mg daily but this can be lowered into 30mg daily or 15 mg daily if side effects of thrombotic events, vascular occlusions, and heart failure are of major concern.1,38,93
TKI Resistance
CML patients with TKI resistance should be treated with a second-line TKI and be examined for BCR-ABL1 mutations as the most common cause of TKI resistance is due to point mutations in the kinase domain of BCR-ABL1.94 These point mutations cause a decrease binding affinity of TKI.95 Recommended method to detect BCR-ABL1 resistance mutations is next generation sequencing (NGS).38
The type of TKI given depends on the BCR-ABL1 resistance mutations found. For example, patients with a T315I mutation are recommended to be treated with ponatinib only as patients harboring this mutation have resistance to other TKIs.38,96 In contrast, patients with a Y253H, E255V/K, F359V/I/C mutations can be treated with dasatinib, bosutinib, or ponatinib.38 ELN 2020 CML guideline has a list of recommended tyrosine kinase inhibitors for BCR-ABL1 resistance mutations.38 In general, second-generation TKIs are recommended when there is BCR-ABL1 point mutations. The selection of second-generation or third-generation TKIs should be based on a balance between efficacy and side effects.1
Treatment Response
The goal of therapy in CML is to achieve a hematological response, a cytogenetic response, and a long-term major molecular response. A complete hematological response (CHR) is defined as a normal leukocyte count, a normal platelet level, a normal differential result, and the disappearance of CML symptoms.97 A partial hematological response is defined as a decrease in leukocyte count of less than 50% of the leukocyte level before treatment, or normalization of the leukocyte level with persistent splenomegaly or the presence of blasts in the peripheral blood. Meanwhile, a complete cytogenetic response (CCR) is defined as the absence of metaphase Ph+ in the bone marrow cells.97
Currently, the major molecular response is more often used to monitor therapy, especially for TKI therapy.97 Treatments such as chemotherapies and interferon alpha rarely achieve a major molecular response. Molecular responses should be quantified on an international scale using the ratio of BCR-ABL1 transcripts to ABL1 transcripts.98 A molecular response of BCR-ABL1 1% is equivalent to that of CCR. The molecular response of BCR-ABL1 0.1% is defined as a major molecular response (MMR) or MR3. The molecular response of BCR-ABL1 0.01% is defined as MR4 (Table 3). There is also the term deep molecular response (DMR), defined as MR4 and above.99 CD26, perhaps as a marker of LSCs, can be used as an alternative to detect response, especially for BCR-ABL-independent CML cells; however, it is also difficult to detect small numbers of CD26-positive LSCs.100 Thus, currently, the monitoring of CML patients revolves around the molecular response.
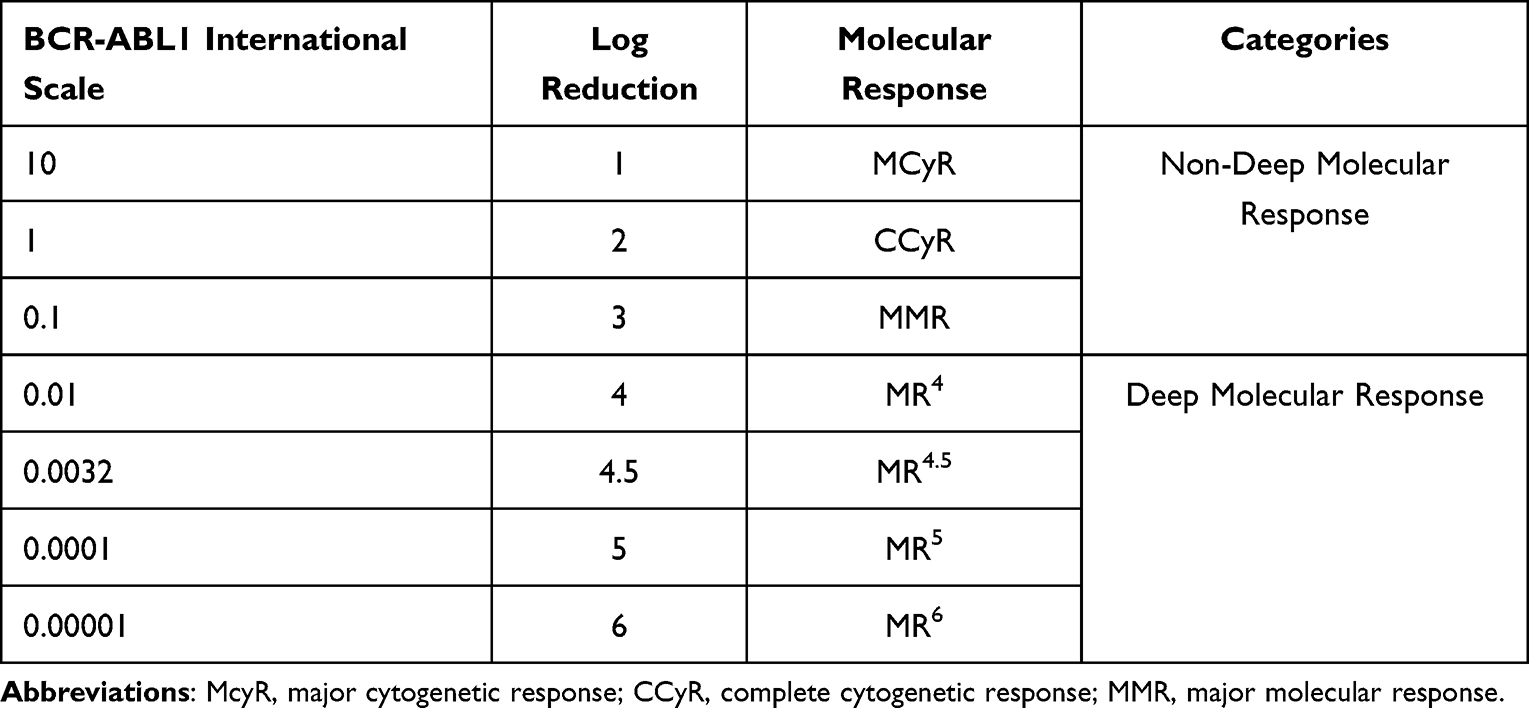 |
Table 3 BCR-ABL1 International Scale and Molecular Response |
Prognosis
There are currently four prognostic systems for CML: (1) Sokal, (2) Euro, (3) EUTOS, and (4) ELTS.101,102 However, it should be noted by clinicians that the Sokal score was developed in the chemotherapy era. Meanwhile, the Euro score was developed in the interferon alpha (IFNα) era. Thus, the eras in which these scoring systems were developed may reduce the validity of the Sokal and Euro scores, because almost all CML patients today are treated with a TKI. Indeed, it is now recommended that the EUTOS and ELTS scores be used instead.
The European treatment and outcome study (EUTOS) scoring system predicts CCR at 18 months after TKI therapy. In contrast to other scoring systems, the EUTOS long-term survival (ELTS) score predicts the mortality risk of CML patients given a TKI therapy using the same variables as the Sokal scoring system. Currently, the ELN recommends the use of the ELTS scoring system compared to the other scoring systems.38
Treatment-Free Remission
Previously, the aim of CML therapy was to delay progression to the blast phase, minimize symptoms, and increase overall survival. These aims are mostly achieved in most patients today. Hence, new outcomes for CML patients, such as treatment-free remission with TKI discontinuation, are now being pursued, especially since there are concerns due to the long-term toxicity of TKI therapies. Another important reason is the cost associated with TKI therapy. Currently, research on CML is focused on achieving DMR to achieve treatment-free remission (TFR).
In general, CML patients require lifelong therapy to prevent recurrence, even when the patient has achieved an MMR status for several years; however, this trend is now changing. The antiproliferative effects of TKIs on LSCs have been shown to be strong; however, the effect on apoptosis is weaker.103 The reason for this is that quiescent LSCs are resistant to apoptosis due to TKIs. A further problem is associated with the BCR-ABL-independent pathway, which maintains the survival of CML stem cells. A study by Jeanpierre et al showed that quiescent stem cells activated BMPR1B, Stat3, and BMP4-niche signals to maintain their survival.104 These signals may not be targeted by TKIs, especially early-generation TKIs.
Recently, it has become known that CML patients who successfully achieve a long-lasting DMR can achieve complete recovery. Such patients have a lower risk of CML relapse when TKI therapy is discontinued. However, only a small proportion of CML patients can achieve a DMR status with their current therapy, due to the presence of CML stem cells left over after therapy. Several trials have shown that a significant proportion of CML patients (approximately 60%) with undetectable minimal residual disease still have a relapse, shown by the identification of BCFR-ABL1-positive cells within the first 2 years after imatinib discontinuation.7,105,106
Based on the results of the stop imatinib (STIM) clinical trial, with a median follow-up of 77 months, 39% of CML patients who achieved MR4 status for two years were able to maintain their remission status without TKI therapy.105 A meta-analysis by Campiotti et al (2017) of 15 cohort studies of patients with undetectable BCR-ABL1 transcript criteria showed that the mean molecular recurrence rate was 51% (95% CI 44–58%; I2: 55%).107 The percentage of TFR in patients who achieved an MMR alone without a DMR was lower than that in patients who achieved a DMR. This result can be interpreted as showing that a significant proportion of patients may achieve TFR.
From all of the available studies, it can be inferred that only CML patients who achieve DMR are candidates for treatment-free remission (Table 4). The factors associated with successful TFR have not yet been established. However, a study by Irani et al demonstrated that CML patients who achieved TFR had higher levels of natural killer (NK) cells.108 This finding was similar to the results of a sub-analysis of EURO-SKI.109 It should be noted that even in patients who achieve a DMR, LSCs are not removed completely. It can be speculated that the patients that achieve TFR have an adequate immunological response in suppressing residual LSCs.
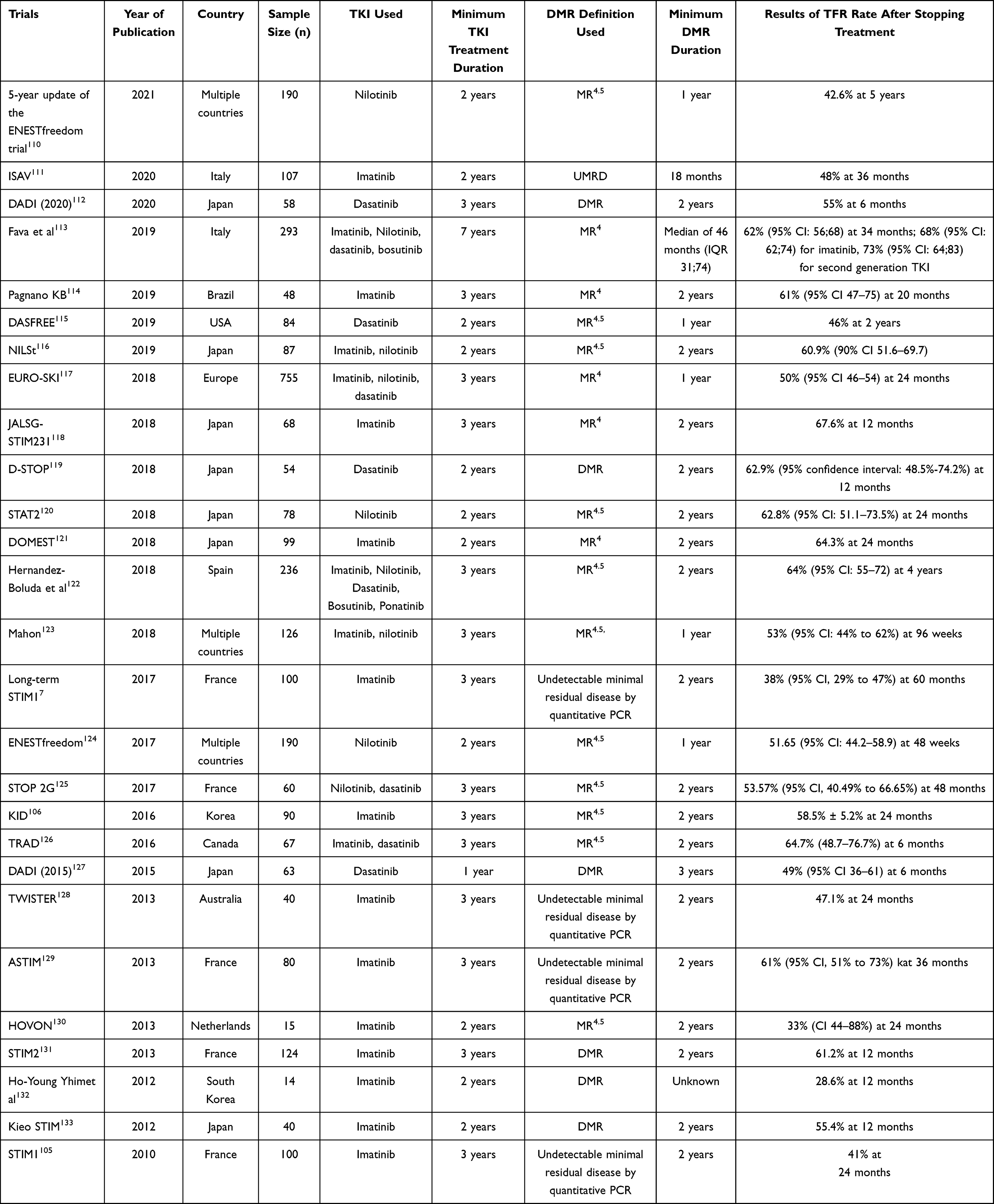 |
Table 4 Summary of TFR Studies |
Predictors and Biomarkers for TFR
Clinical parameters and biomarkers that can be used to predict TFR in CML patients are still under investigation and this remains a topic of interest (Figure 2). Vigón et al identified several immunological parameters that predict relapse.134 These parameters are low levels of natural killer cells, natural killer T cells, and CD8±TCRγβ+ T cells.134 Similarly, Rea et al found that higher number of natural killer (NK) cells were observed in CML patients that did not relapse.135 Both CD26 and CD93 which are circulating CML stem cell markers appear to be correlated with TFR as well.51,136,137 Other possible markers include regulatory T cells, cytotoxic CD8+ T lymphocyte (CTL), and somatic mutations.134,138–140 Type of BCR-ABL1 transcript also has a role; in a study by D’Adda et al, TFR maintenance was observed to be higher in CML patients with e14a2 transcript when compared with e13a2 transcripts.141 However, all of these biomarkers are not readily available for routine clinical use, and thus, clinical parameters remain the most widely used or accessible parameters for TFR.142
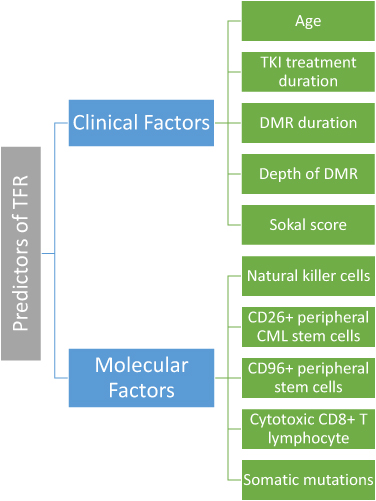 |
Figure 2 Known predictors of treatment-free remission. |
Many studies have data that can be used to identify clinical parameters that can be used for TFR predictions.115,143–145 In general, longer duration of TKI therapy, longer DMR duration, older age, undetectable BCR-ABL1 3 months after TKI discontinuation, and deeper DMR status are observed as predictors for TFR.115,143,144,146,147 Recent study by Kim et al proposed six years imatinib treatment and 4.5 years of MR4 response as the optimal duration for starting TFR.147
It is speculated that adding concomitant ruxolitinib or pembrolizumab to patients’ TKI therapy may produce a beneficial outcome.148 In the future, it is hoped that treatment-free remission can become more achievable for CML patients and can also become a treatment outcome for other cancers as well.
Side Effects of TKI Discontinuation
Several long-term CML patients who have been treated with a TKI have had side effects when discontinuing their TKI. Side effects of TKI discontinuation include mostly muscle and joint pain. These side effects can last up to a few months and can be treated with supportive care such as NSAIDs. Around 30% of patients may experience musculoskeletal pain after TKI discontinuation.143,149,150 Musculoskeletal pain may be experienced in different regions of the body such as the shoulders, extremities, hips, hands, and feet.150 A study by Hernandez-Boluda et al revealed that patients that stopped TKI had elevated cholesterol level after discontinuing with the exception of nilotinib.122
Currently, the mechanisms of TKI withdrawal symptoms are unclear.106 It is speculated that TKIs inhibit c-kit and platelet-derived growth factor receptor (PDGFR), which results in an increased number of osteoclasts and osteoblasts.149,150 A small study by Katagiri et al found that patients with symptoms of musculoskeletal pain after discontinuing TKI treatment had a lower body weight and lower BMI, but other factors that can predict whether patients will develop musculoskeletal pain are still unknown.149
Generally, the side effects of TKI discontinuation are not a major concern, as most patients’ symptoms resolve within 3 months and most of the symptoms are mild.151 However, clinicians should be aware of the side effects and educate patients as needed to prepare them for this possibility.
Conclusion
Many advances have been made in improving our understanding of the pathophysiological processes of CML. However, the etiology of the Philadelphia chromosome and mechanisms of LSC persistence are still under investigation. Somatic mutations and additional chromosomal abnormalities are associated with CML progression into blast phase. The frontline treatment of CML currently is TKI as it is more effective than previous therapies. The current aim of CML treatment is to achieve TFR, and CML patients who achieve a DMR and receive long-term TKI therapy have a higher likelihood of achieving this. In the future, it is hoped that more patients can achieve treatment-free remission.
Acknowledgement
The authors would like to express their gratitude to Yehezkiel Alexander Eduard George, MD, for his professional assistance in proofreading the galley proof of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am J Hematol. 2020;95(6):691–709. doi:10.1002/ajh.25792
2. Nicholson E, Holyoake T. The chronic myeloid leukemia stem cell. Clin Lymphoma Myeloma. 2009;9(Suppl 4):S376–381. doi:10.3816/CLM.2009.s.037
3. Frazer R, Irvine AE, McMullin MF. Chronic myeloid leukaemia in the 21st century. Ulster Med J. 2007;76(1):8–17.
4. García-Gutiérrez V, Hernández-Boluda JC. Tyrosine kinase inhibitors available for chronic myeloid leukemia: efficacy and safety. Front Oncol. 2019;9. doi:10.3389/fonc.2019.00603
5. Sasaki K, Strom SS, O’Brien S, et al. Relative survival in patients with chronic-phase chronic myeloid leukaemia in the tyrosine-kinase inhibitor era: analysis of patient data from six prospective clinical trials. Lancet Haematol. 2015;2(5):e186–193. doi:10.1016/S2352-3026(15)00048-4
6. Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917–927. doi:10.1056/NEJMoa1609324
7. Etienne G, Guilhot J, Rea D, et al. Long-term follow-up of the French stop imatinib (STIM1) study in patients with chronic myeloid leukemia. J Clin Oncol off J Am Soc Clin Oncol. 2017;35(3):298–305. doi:10.1200/JCO.2016.68.2914
8. Oka S, Muroi K, Mori M, et al. Prediction of response to imatinib in patients with chronic myelogenous leukemia by flow cytometric analysis of bone marrow blastic cell phenotypes. Leuk Lymphoma. 2009;50(2):290–293. doi:10.1080/10428190802627598
9. Kang ZJ, Liu YF, Xu LZ, et al. The Philadelphia chromosome in leukemogenesis. Chin J Cancer. 2022;35:48. doi:10.1186/s40880-016-0108-0
10. Holmberg M. Is the primary event in radiation-induced chronic myelogenous leukemia the induction of the t(9;22) translocation? Leuk Res. 1992;16(4):333–336. doi:10.1016/0145-2126(92)90134-S
11. Ismail SI, Naffa RG, Yousef AMF, Ghanim MT. Incidence of bcr‑abl fusion transcripts in healthy individuals. Mol Med Rep. 2014;9(4):1271–1276. doi:10.3892/mmr.2014.1951
12. Aplan PD. Causes of oncogenic chromosomal translocation. Trends Genet TIG. 2006;22(1):46–55. doi:10.1016/j.tig.2005.10.002
13. Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood. 2022;113(8):1619–1630. doi:10.1182/blood-2008-03-144790
14. Verma D, Kantarjian HM, Jones D, et al. Chronic myeloid leukemia (CML) with P190 BCR-ABL: analysis of characteristics, outcomes, and prognostic significance. Blood. 2009;114(11):2232–2235. doi:10.1182/blood-2009-02-204693
15. Arun AK, Senthamizhselvi A, Mani S, et al. Frequency of rare BCR-ABL1 fusion transcripts in chronic myeloid leukemia patients. Int J Lab Hematol. 2017;39(3):235–242. doi:10.1111/ijlh.12616
16. Cortes JE, Talpaz M, Beran M, et al. Philadelphia chromosome-negative chronic myelogenous leukemia with rearrangement of the breakpoint cluster region. Long-term follow-up results. Cancer. 1995;75(2):464–470. doi:10.1002/1097-0142(19950115)75:2<464::AID-CNCR2820750209>3.0.CO;2-E
17. Seong D, Kantarjian HM, Albitar M, et al. Analysis of Philadelphia chromosome-negative BCR-ABL-positive chronic myelogenous leukemia by hypermetaphase fluorescence in situ hybridization. Ann Oncol off J Eur Soc Med Oncol. 1999;10(8):955–959. doi:10.1023/A:1008349405763
18. Onida F, Ball G, Kantarjian HM, et al. Characteristics and outcome of patients with Philadelphia chromosome negative, bcr/abl negative chronic myelogenous leukemia. Cancer. 2002;95(8):1673–1684. doi:10.1002/cncr.10832
19. Ohyashiki JH, Ohyashiki K, Ito H, Toyama K. Molecular and clinical investigations in Philadelphia chromosome-negative chronic myelogenous leukemia. Cancer Genet Cytogenet. 1988;33(1):119–126. doi:10.1016/0165-4608(88)90057-X
20. Wang SA, Hasserjian RP, Fox PS, et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood. 2014;123(17):2645–2651. doi:10.1182/blood-2014-02-553800
21. Diamantopoulos PT, Viniou NA. Atypical chronic myelogenous leukemia, BCR-ABL1 negative: diagnostic criteria and treatment approaches. Front Oncol. 2021;11:722507. doi:10.3389/fonc.2021.722507
22. Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–2264. doi:10.1172/JCI41246
23. Yohannan B, George B. B-lymphoid blast phase–chronic myeloid leukemia: current therapeutics. Int J Mol Sci. 2022;23(19):11836. doi:10.3390/ijms231911836
24. Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104(12):3746–3753. doi:10.1182/blood-2004-05-1941
25. Koptyra M, Falinski R, Nowicki MO, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108(1):319–327. doi:10.1182/blood-2005-07-2815
26. Skorski T. Oncogenic tyrosine kinases and the DNA-damage response. Nat Rev Cancer. 2002;2(5):351–360. doi:10.1038/nrc799
27. Chereda B, Melo JV. Natural course and biology of CML. Ann Hematol. 2015;94(Suppl 2):107–121. doi:10.1007/s00277-015-2325-z
28. Quintás-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood.2009;113(8):1619–1630. doi:10.1182/blood-2008-03-144790
29. How HR. I treat CML blast crisis. Blood. 2012;120(4):737–747. doi:10.1182/blood-2012-03-380147
30. Adnan Awad S, Dufva O, Ianevski A, et al. RUNX1 mutations in blast-phase chronic myeloid leukemia associate with distinct phenotypes, transcriptional profiles, and drug responses. Leukemia. 2021;35(4):1087–1099. doi:10.1038/s41375-020-01011-5
31. Ochi Y, Yoshida K, Huang YJ, et al. Clonal evolution and clinical implications of genetic abnormalities in blastic transformation of chronic myeloid leukaemia. Nat Commun. 2021;12(1):2833. doi:10.1038/s41467-021-23097-w
32. Soliman DS, Amer AM, Mudawi D, et al. Chronic myeloid leukemia with cryptic Philadelphia translocation and extramedullary B-lymphoid blast phase as an initial presentation. Acta Bio. 2018;89(3–S):38–44.
33. Zagaria A, Anelli L, Albano F, et al. A fluorescence in situ hybridization study of complex t(9;22) in two chronic myelocytic leukemia cases with a masked Philadelphia chromosome. Cancer Genet Cytogenet. 2004;150(1):81–85. doi:10.1016/j.cancergencyto.2003.08.018
34. Alhuraiji A, Kantarjian H, Boddu P, et al. Prognostic significance of additional chromosomal abnormalities at the time of diagnosis in patients with chronic myeloid leukemia treated with frontline tyrosine kinase inhibitors. Am J Hematol. 2018;93(1):84–90. doi:10.1002/ajh.24943
35. Krishna Chandran R, Geetha N, Sakthivel KM, Suresh Kumar R, Jagathnath Krishna KMN, Sreedharan H. Impact of additional chromosomal aberrations on the disease progression of chronic myelogenous leukemia. Front Oncol. 2019;5(9):88. doi:10.3389/fonc.2019.00088
36. Chen Z, Shao C, Wang W, et al. Cytogenetic landscape and impact in blast phase of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Leukemia. 2017;31(3):585–592. doi:10.1038/leu.2016.231
37. Clark RE, Apperley JF, Copland M, Cicconi S. Additional chromosomal abnormalities at chronic myeloid leukemia diagnosis predict an increased risk of progression. Blood Adv. 2021;5(4):1102–1109. doi:10.1182/bloodadvances.2020003570
38. Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–984. doi:10.1038/s41375-020-0776-2
39. Meggyesi N, Kozma A, Halm G, Nahajevszky S, Bátai A, Fekete S, et al. Additional chromosome abnormalities, BCR-ABL tyrosine kinase domain mutations and clinical outcome in Hungarian tyrosine kinase inhibitor-resistant chronic myelogenous leukemia patients. Acta Haematol. 2012;127(1):34–42.
40. Siti Mariam I, Norhidayah R, Zulaikha AB, et al. Differential prognostic impact of stratified additional chromosome abnormalities on disease progression among Malaysian chronic myeloid leukemia patients undergoing treatment with imatinib mesylate. Front Oncol. 2022;12. doi:10.3389/fonc.2022.720845
41. Houshmand M, Simonetti G, Circosta P, et al. Chronic myeloid leukemia stem cells. Leukemia. 2019;33(7):1543–1556. doi:10.1038/s41375-019-0490-0
42. Jiang X, Zhao Y, Smith C, et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia. 2007;21(5):926–935. doi:10.1038/sj.leu.2404609
43. Kavalerchik E, Goff D, Jamieson CHM. Chronic myeloid leukemia stem cells. J Clin Oncol off J Am Soc Clin Oncol. 2008;26(17):2911–2915. doi:10.1200/JCO.2008.17.5745
44. Herrmann H, Cerny-Reiterer S, Gleixner KV, et al. CD34(+)/CD38(-) stem cells in chronic myeloid leukemia express Siglec-3 (CD33) and are responsive to the CD33-targeting drug gemtuzumab/ozogamicin. Haematologica. 2012;97(2):219–226. doi:10.3324/haematol.2010.035006
45. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. doi:10.1038/nrd4253
46. Minami Y, Kono T, Miyazaki T, Taniguchi T. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol. 1993;11:245–268. doi:10.1146/annurev.iy.11.040193.001333
47. Sadovnik I, Herrmann H, Eisenwort G, et al. Expression of CD25 on leukemic stem cells in BCR-ABL1+ CML: potential diagnostic value and functional implications. Exp Hematol. 2022;51:17–24. doi:10.1016/j.exphem.2017.04.003
48. Herrmann H, Sadovnik I, Cerny-Reiterer S, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood. 2014;123(25):3951–3962. doi:10.1182/blood-2013-10-536078
49. Gschwandtner M, Paulitschke V, Mildner M, et al. Proteome analysis identifies L1CAM/CD171 and DPP4/CD26 as novel markers of human skin mast cells. Allergy. 2017;72(1):85–97. doi:10.1111/all.12919
50. Ali S, Huber M, Kollewe C, Bischoff SC, Falk W, Martin MU. IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci U S A. 2007;104(47):18660–18665. doi:10.1073/pnas.0705939104
51. Kinstrie R, Horne GA, Morrison H, et al. CD93 is expressed on chronic myeloid leukemia stem cells and identifies a quiescent population which persists after tyrosine kinase inhibitor therapy. Leukemia. 2020;34(6):1613–1625. doi:10.1038/s41375-019-0684-5
52. Chu S, McDonald T, Lin A, et al. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood. 2022;118(20):5565–5572. doi:10.1182/blood-2010-12-327437
53. Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–779. doi:10.1038/nature07737
54. Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet. 2009;41(7):783–792. doi:10.1038/ng.389
55. Kuepper MK, Bütow M, Herrmann O, et al. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia. 2019;33(8):1964–1977. doi:10.1038/s41375-019-0427-7
56. Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood. 2017;129(12):1595–1606. doi:10.1182/blood-2016-09-696013
57. Skorski T. BCR/ABL, DNA damage and DNA repair: implications for new treatment concepts. Leuk Lymphoma. 2008;49(4):610–614. doi:10.1080/03093640701859089
58. Stoklosa T, Poplawski T, Koptyra M, et al. BCR/ABL inhibits mismatch repair to protect from apoptosis and induce point mutations. Cancer Res. 2008;68(8):2576–2580. doi:10.1158/0008-5472.CAN-07-6858
59. Cramer K, Nieborowska-Skorska M, Koptyra M, et al. BCR/ABL and other kinases from chronic myeloproliferative disorders stimulate single-strand annealing, an unfaithful DNA double-strand break repair. Cancer Res. 2008;68(17):6884–6888. doi:10.1158/0008-5472.CAN-08-1101
60. Zhang B, Chu S, Agarwal P, et al. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood. 2016;128(23):2671–2682. doi:10.1182/blood-2015-11-679928
61. Bhatia R. TARGETING LEUKEMIA STEM CELL RESISTANCE IN CHRONIC MYELOGENOUS LEUKEMIA. Trans Am Clin Climatol Assoc. 2019;130:246–254.
62. Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol OncolJ Hematol Oncol. 2020;13(1):104. doi:10.1186/s13045-020-00937-8
63. Gallipoli P, Cook A, Rhodes S, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood. 2014;124(9):1492–1501. doi:10.1182/blood-2013-12-545640
64. Hochhaus A, Saussele S, Rosti G, et al. Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol. 2017;1(28):iv41–51. doi:10.1093/annonc/mdx219
65. Deininger MW, Shah NP, Altman JK, et al. Chronic myeloid leukemia, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw JNCCN. 2020;18(10):1385–1415. doi:10.6004/jnccn.2020.0047
66. Asnafi AA, Deris Zayeri Z, Shahrabi S, Zibara K, Vosughi T. Chronic myeloid leukemia with complex karyotypes: prognosis and therapeutic approaches. J Cell Physiol. 2019;234(5):5798–5806. doi:10.1002/jcp.27505
67. ClinicalKey. Chronic myeloid leukemia; 2022. Available from: https://www.clinicalkey.com/#!/content/book/3-s2.0-B9780323357623000676?scrollTo=%23hl0000264.
68. Arunachalam AK, Jain M, Kumar A, Kushwaha R, Singh US, Tripathi AK. Megakaryocytes in chronic phase of chronic myeloid leukemia: a descriptive case series. Ann Pathol Lab Med. 2016;3(3):A176–182.
69. Mughal Z, Babar H, Ashraf S, Hamid A, Khokhar A, Qamar S. Megakaryocytic clustering in chronic myeloid leukemia: can it be a predictor of clinical outcome? J Coll Physicians Surg. 2021;31(1):34–38. doi:10.29271/jcpsp.2021.01.34
70. Dekmezian R, Kantarjian HM, Keating MJ, Talpaz M, McCredie KB, Freireich EJ. The relevance of reticulin stain-measured fibrosis at diagnosis in chronic myelogenous leukemia. Cancer. 1987;59(10):1739–1743. doi:10.1002/1097-0142(19870515)59:10<1739::AID-CNCR2820591011>3.0.CO;2-2
71. Haznedaroğlu İC, Kuzu I, Ilhan O. WHO 2016 definition of chronic myeloid leukemia and tyrosine kinase inhibitors. Turk J Hematol. 2020;37(1):42–47.
72. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
73. Ivanov S, Sharma P, Jobanputra Y, Zhang Y. Transformation of chronic myeloid leukemia to acute biphenotypic leukemia. J Med Cases. 2020;11(8):239–242. doi:10.14740/jmc3511
74. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi:10.1182/blood-2016-01-643569
75. Padhi P, Topalovski M, El Behery R, Cantu ES, Medavarapu R, Rare A. Case of chronic myelogenous leukemia presenting as T-cell lymphoblastic crisis. Case Rep Oncol Med. 2018;2018:7276128. doi:10.1155/2018/7276128
76. Andretta E, Costa C, Longobardi C, et al. Potential approaches versus approved or developing chronic myeloid leukemia therapy. Front Oncol. 2022;11:801779. doi:10.3389/fonc.2021.801779
77. Simonsson B, Gedde-Dahl T, Markevärn B, et al. Combination of pegylated IFN-α2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood. 2011;118(12):3228–3235. doi:10.1182/blood-2011-02-336685
78. Preudhomme C, Guilhot J, Nicolini FE, et al. Imatinib plus peginterferon alfa-2a in chronic myeloid leukemia. N Engl J Med. 2010;363(26):2511–2521. doi:10.1056/NEJMoa1004095
79. Gratwohl A, Pfirrmann M, Zander A, et al. Long-term outcome of patients with newly diagnosed chronic myeloid leukemia: a randomized comparison of stem cell transplantation with drug treatment. Leukemia. 2016;30(3):562–569. doi:10.1038/leu.2015.281
80. O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi:10.1056/NEJMoa022457
81. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract. 2022;2014:357027.
82. Uzer E, Ünal A, Köker MY, Doğan SA. The Side Effects of Imatinib. Turk J Hematol. 2022;30(3):341. doi:10.4274/TJH-2011.0018
83. Mughal TI, Schrieber A. Principal long-term adverse effects of imatinib in patients with chronic myeloid leukemia in chronic phase. Biol Targets Ther. 2010;2(4):315–323. doi:10.2147/BTT.S5775
84. Prost S, Le Dantec M, Augé S, et al. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signaling pathway in macaques. J Clin Invest. 2008;118(5):1765–1775. doi:10.1172/JCI33037
85. Breccia M, Alimena G. Second-Generation Tyrosine Kinase Inhibitors (Tki) as salvage therapy for resistant or intolerant patients to prior TKIs. Mediterr J Hematol Infect Dis. 2014;6(1):e2014003. doi:10.4084/mjhid.2014.003
86. Cortes JE, Saglio G, Kantarjian HM, et al. Final 5-year study results of DASISION: the dasatinib versus imatinib study in treatment-naïve chronic myeloid leukemia patients trial. J Clin Oncol off J Am Soc Clin Oncol. 2016;34(20):2333–2340. doi:10.1200/JCO.2015.64.8899
87. Cortes JE, Jiang Q, Wang J, et al. Dasatinib vs. imatinib in patients with chronic myeloid leukemia in chronic phase (CML-CP) who have not achieved an optimal response to 3 months of imatinib therapy: the DASCERN randomized study. Leukemia. 2020;34(8):2064–2073. doi:10.1038/s41375-020-0805-1
88. Hochhaus A, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30(5):1044–1054. doi:10.1038/leu.2016.5
89. Cortes JE, Gambacorti-Passerini C, Deininger MW, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J Clin Oncol off J Am Soc Clin Oncol. 2018;36(3):231–237. doi:10.1200/JCO.2017.74.7162
90. Do YR, Kwak JY, Kim JA, et al. Long-term data from a Phase 3 study of radotinib versus imatinib in patients with newly diagnosed, chronic myeloid leukaemia in the chronic phase (RERISE). Br J Haematol. 2020;189(2):303–312. doi:10.1111/bjh.16381
91. O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16(5):401–412. doi:10.1016/j.ccr.2009.09.028
92. Zhou T, Commodore L, Huang WS, et al. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem Biol Drug Des. 2011;77(1):1–11. doi:10.1111/j.1747-0285.2010.01054.x
93. Chan O, Talati C, Isenalumhe L, et al. Side-effects profile and outcomes of ponatinib in the treatment of chronic myeloid leukemia. Blood Adv. 2020;4(3):530–538. doi:10.1182/bloodadvances.2019000268
94. Alves R, Gonçalves AC, Rutella S, et al. Resistance to tyrosine kinase inhibitors in chronic myeloid leukemia-from molecular mechanisms to clinical relevance. Cancers. 2021;13(19):4820. doi:10.3390/cancers13194820
95. Meenakshi Sundaram DN, Jiang X, Brandwein JM, Valencia-Serna J, Remant KC, Uludağ H. Current outlook on drug resistance in chronic myeloid leukemia (CML) and potential therapeutic options. Drug Discov Today. 2019;24(7):1355–1369. doi:10.1016/j.drudis.2019.05.007
96. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A Phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–1796. doi:10.1056/NEJMoa1306494
97. Galinsky I, Buchanan S. Guide to interpreting disease responses in chronic myeloid leukemia. J Adv Pract Oncol. 2022;3(4):225–236.
98. Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108(6):1809–1820. doi:10.1182/blood-2006-02-005686
99. Branford S. Why is it critical to achieve a deep molecular response in chronic myeloid leukemia? Haematologica. 2022;105(12):2730–2737. doi:10.3324/haematol.2019.240739
100. Ebian HF, Abdelnabi ASM, Abdelazem AS, Khamis T, Fawzy HM, Hussein S. Peripheral blood CD26 positive leukemic stem cells as a possible diagnostic and prognostic marker in chronic myeloid leukemia. Leuk Res Rep. 2022;17:100321. doi:10.1016/j.lrr.2022.100321
101. Kuntegowdanahalli LC, Kanakasetty GB, Thanky AH, et al. Prognostic and predictive implications of Sokal, Euro and EUTOS scores in chronic myeloid leukaemia in the imatinib era—experience from a tertiary oncology centre in Southern India. Ecancermedicalscience. 2022;10:679.
102. Pfirrmann M, Clark RE, Prejzner W, et al. The EUTOS long-term survival (ELTS) score is superior to the Sokal score for predicting survival in chronic myeloid leukemia. Leukemia. 2022;34(8).
103. Munje C, Copland M. Exploring stem cell heterogeneity in chronic myeloid leukemia. Trends Cancer. 2022;4(3):167–169. doi:10.1016/j.trecan.2017.12.001
104. Jeanpierre S, Arizkane K, Thongjuea S, et al. The quiescent fraction of chronic myeloid leukemic stem cells depends on BMPR1B, Stat3 and BMP4-niche signals to persist in patients in remission. Haematologica. 2022;106(1):111–122. doi:10.3324/haematol.2019.232793
105. Mahon FX, Réa D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035. doi:10.1016/S1470-2045(10)70233-3
106. Lee SE, Choi SY, Song HY, et al. Imatinib withdrawal syndrome and longer duration of imatinib have a close association with a lower molecular relapse after treatment discontinuation: the KID study. Haematologica. 2016;101(6):717–723. doi:10.3324/haematol.2015.139899
107. Campiotti L, Suter MB, Guasti L, et al. Imatinib discontinuation in chronic myeloid leukaemia patients with undetectable BCR-ABL transcript level: a systematic review and a meta-analysis. Eur J Cancer Oxf Engl. 2017;77:48–56.
108. Irani YD, Hughes A, Clarson J, et al. Successful treatment-free remission in chronic myeloid leukaemia and its association with reduced immune suppressors and increased natural killer cells. Br J Haematol. 2020;191(3):433–441. doi:10.1111/bjh.16718
109. Ilander M, Olsson-Strömberg U, Schlums H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia. 2017;31(5):1108–1116. doi:10.1038/leu.2016.360
110. Radich JP, Hochhaus A, Masszi T, et al. Treatment-free remission following frontline nilotinib in patients with chronic phase chronic myeloid leukemia: 5-year update of the ENESTfreedom trial. Leukemia. 2021;35(5):1344–1355. doi:10.1038/s41375-021-01205-5
111. Diral E, Mori S, Antolini L, et al. Increased tumor burden in patients with chronic myeloid leukemia after 36 months of imatinib discontinuation. Blood. 2020;136(19):2237–2240. doi:10.1182/blood.2019004371
112. Kimura S, Imagawa J, Murai K, et al. Treatment-free remission after first-line dasatinib discontinuation in patients with chronic myeloid leukaemia (first-line DADI trial): a single-arm, multicentre, phase 2 trial. Lancet Haematol. 2020;7(3):e218–25. doi:10.1016/S2352-3026(19)30235-2
113. Fava C, Rege-Cambrin G, Dogliotti I, et al. Observational study of chronic myeloid leukemia Italian patients who discontinued tyrosine kinase inhibitors in clinical practice. Haematologica. 2019;104(8):1589–1596. doi:10.3324/haematol.2018.205054
114. Pagnano KB, Seguro FS, Miranda EC, et al. Duration of major molecular response and discontinuation in deep molecular response (MR4.5) were associated with longer treatment-free survival after imatinib discontinuation - results from two prospective Brazilian trials. Blood. 2019;134:1655. doi:10.1182/blood-2019-125368
115. Shah NP, García-Gutiérrez V, Jiménez-Velasco A, et al. Dasatinib discontinuation in patients with chronic-phase chronic myeloid leukemia and stable deep molecular response: the DASFREE study. Leuk Lymphoma. 2020;61(3):650–659. doi:10.1080/10428194.2019.1675879
116. Nagafuji K, Matsumura I, Shimose T, et al. Cessation of nilotinib in patients with chronic myelogenous leukemia who have maintained deep molecular responses for 2 years: a multicenter phase 2 trial, stop nilotinib (NILSt). Int J Hematol. 2019;110(6):675–682. doi:10.1007/s12185-019-02736-5
117. Saussele S, Richter J, Guilhot J, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): a prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018;19(6):747–757. doi:10.1016/S1470-2045(18)30192-X
118. Takahashi N, Tauchi T, Kitamura K, et al. Deeper molecular response is a predictive factor for treatment-free remission after imatinib discontinuation in patients with chronic phase chronic myeloid leukemia: the JALSG-STIM213 study. Int J Hematol. 2018;107(2):185–193. doi:10.1007/s12185-017-2334-x
119. Kumagai T, Nakaseko C, Nishiwaki K, et al. Dasatinib cessation after deep molecular response exceeding 2 years and natural killer cell transition during dasatinib consolidation. Cancer Sci. 2018;109(1):182–192. doi:10.1111/cas.13430
120. Takahashi N, Nishiwaki K, Nakaseko C, et al. Treatment-free remission after two-year consolidation therapy with nilotinib in patients with chronic myeloid leukemia: STAT2 trial in Japan. Haematologica. 2018;103(11):1835–1842. doi:10.3324/haematol.2018.194894
121. Fujisawa S, Ueda Y, Usuki K, et al. Feasibility of the imatinib stop study in the Japanese clinical setting: delightedly overcome CML expert stop TKI trial (DOMEST Trial). Int J Clin Oncol. 2019;24(4):445–453. doi:10.1007/s10147-018-1368-2
122. Hernández-Boluda JC, Pereira A, Pastor-Galán I, et al. Feasibility of treatment discontinuation in chronic myeloid leukemia in clinical practice: results from a nationwide series of 236 patients. Blood Cancer J. 2018;8(10):91. doi:10.1038/s41408-018-0125-0
123. Mahon FX, Boquimpani C, Kim DW, et al. Treatment-free remission after second-line nilotinib treatment in patients with chronic myeloid leukemia in chronic phase: results from a single-group, phase 2, open-label study. Ann Intern Med. 2018;168(7):461–470. doi:10.7326/M17-1094
124. Hochhaus A, Masszi T, Giles FJ, et al. Treatment-free remission following frontline nilotinib in patients with chronic myeloid leukemia in chronic phase: results from the ENESTfreedom study. Leukemia. 2017;31(7):1525–1531. doi:10.1038/leu.2017.63
125. Rea D, Nicolini FE, Tulliez M, et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: interim analysis of the STOP 2G-TKI study. Blood. 2017;129(7):846–854. doi:10.1182/blood-2016-09-742205
126. Kim DDH, Bence-Bruckler I, Forrest DL, et al. Treatment-Free Remission Accomplished By Dasatinib (TRAD): preliminary results of the pan-Canadian tyrosine kinase inhibitor discontinuation trial. Blood. 2016;128(22):1922. doi:10.1182/blood.V128.22.1922.1922
127. Imagawa J, Tanaka H, Okada M, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. 2015;2(12):e528–535. doi:10.1016/S2352-3026(15)00196-9
128. Ross DM, Branford S, Seymour JF, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122(4):515–522. doi:10.1182/blood-2013-02-483750
129. Rousselot P, Charbonnier A, Cony-Makhoul P, et al. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J Clin Oncol off J Am Soc Clin Oncol. 2014;32(5):424–430. doi:10.1200/JCO.2012.48.5797
130. Thielen N, van der Holt B, Cornelissen JJ, et al. Imatinib discontinuation in chronic phase myeloid leukaemia patients in sustained complete molecular response: a randomised trial of the Dutch-Belgian cooperative trial for haemato-oncology (HOVON). Eur J Cancer Oxf Engl. 2013;49(15):3242–3246.
131. Mahon FX, Nicolini FE, Noël MP, et al. Preliminary report of the STIM2 study: a multicenter stop imatinib trial for chronic phase chronic myeloid leukemia de novo patients on imatinib. Blood. 2013;122(21):654. doi:10.1182/blood.V122.21.654.654
132. Yhim HY, Lee NR, Song EK, et al. Imatinib mesylate discontinuation in patients with chronic myeloid leukemia who have received front-line imatinib mesylate therapy and achieved complete molecular response. Leuk Res. 2012;36(6):689–693. doi:10.1016/j.leukres.2012.02.011
133. Matsuki E, Ono Y, Tonegawa K, et al. Detailed Investigation on characteristics of Japanese patients with chronic phase CML who achieved a durable CMR after discontinuation of imatinib – an updated result of the keio STIM study. Blood. 2012;120(21):2788. doi:10.1182/blood.V120.21.2788.2788
134. Vigón L, Luna A, Galán M, et al. Identification of immunological parameters as predictive biomarkers of relapse in patients with chronic myeloid leukemia on treatment-free remission. J Clin Med. 2021;10(1):42. doi:10.3390/jcm10010042
135. Rea D, Henry G, Khaznadar Z, et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. 2017;102(8):1368–1377. doi:10.3324/haematol.2017.165001
136. Raspadori D, Pacelli P, Sicuranza A, et al. Flow cytometry assessment of CD26+ leukemic stem cells in peripheral blood: a simple and rapid new diagnostic tool for chronic myeloid leukemia. Cytometry B Clin Cytom. 2019;96(4):294–299. doi:10.1002/cyto.b.21764
137. Bocchia M, Sicuranza A, Abruzzese E, et al. Residual peripheral blood CD26+ leukemic stem cells in chronic myeloid leukemia patients during TKI Therapy and during treatment-free remission. Front Oncol. 2018;8:194. doi:10.3389/fonc.2018.00194
138. Stuckey R, López Rodríguez JF, Gómez-Casares MT. Discontinuation of tyrosine kinase inhibitors in patients with chronic myeloid leukemia: a review of the biological factors associated with treatment-free remission. Curr Oncol Rep. 2022;24(4):415–426. doi:10.1007/s11912-022-01228-w
139. Cayssials E, Jacomet F, Piccirilli N, et al. Sustained treatment-free remission in chronic myeloid leukaemia is associated with an increased frequency of innate CD8(+) T-cells. Br J Haematol. 2019;186(1):54–59. doi:10.1111/bjh.15858
140. Claudiani S, Apperley JF, Khan A, Khorashad J, Milojkovic D. Prolonged treatment-free remission in chronic myeloid leukemia patients with previous BCR-ABL1 kinase domain mutations. Haematologica. 2020;105(5):e225–7. doi:10.3324/haematol.2019.234179
141. D’Adda M, Farina M, Schieppati F, et al. The e13a2 BCR-ABL transcript negatively affects sustained deep molecular response and the achievement of treatment-free remission in patients with chronic myeloid leukemia who receive tyrosine kinase inhibitors. Cancer. 2019;125(10):1674–1682. doi:10.1002/cncr.31977
142. Castagnetti F, Binotto G, Capodanno I, et al. Making Treatment-Free Remission (TFR) easier in chronic myeloid leukemia: fact-checking and practical management tools. Target Oncol. 2021;16(6):823–838. doi:10.1007/s11523-021-00831-4
143. Mori S, Vagge E, le Coutre P, et al. Age and dPCR can predict relapse in CML patients who discontinued imatinib: the ISAV study. Am J Hematol. 2015;90(10):910–914. doi:10.1002/ajh.24120
144. Atallah E, Schiffer CA, Radich JP, et al. Assessment of outcomes after stopping tyrosine kinase inhibitors among patients with chronic myeloid leukemia: a nonrandomized clinical trial. JAMA Oncol. 2021;7(1):42–50. doi:10.1001/jamaoncol.2020.5774
145. Clark RE, Polydoros F, Apperley JF, et al. De-escalation of tyrosine kinase inhibitor therapy before complete treatment discontinuation in patients with chronic myeloid leukaemia (DESTINY): a non-randomised, phase 2 trial. Lancet Haematol. 2019;6(7):e375–83. doi:10.1016/S2352-3026(19)30094-8
146. Atallah E, Treatment-Free Remission: SK. the New Goal in CML Therapy. Curr Hematol Malig Rep. 2021;16(5):433–439. doi:10.1007/s11899-021-00653-1
147. Kim DDH, Novitzky-Basso I, Kim TS, et al. Optimal duration of imatinib treatment/deep molecular response for treatment-free remission after imatinib discontinuation from a Canadian tyrosine kinase inhibitor discontinuation trial. Br J Haematol. 2021;193(4):779–791. doi:10.1111/bjh.17447
148. ECOG-ACRIN Cancer Research Group. Phase II study of adding the anti-PD-1 pembrolizumab to tyrosine kinase inhibitors in patients with chronic myeloid leukemia and persistently detectable minimal residual disease; 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT03516279.
149. Katagiri S, Tauchi T, Ando K, Okabe S, Gotoh M, Ohyashiki K. Low body weight and body mass index may be associated with musculoskeletal pain following imatinib discontinuation in chronic myeloid leukemia. Leuk Res Rep. 2017;7:33–35. doi:10.1016/j.lrr.2017.04.002
150. Richter J, Söderlund S, Lübking A, et al. Musculoskeletal Pain in Patients With Chronic Myeloid Leukemia After Discontinuation of Imatinib: a Tyrosine Kinase Inhibitor Withdrawal Syndrome? J Clin Oncol. 2014;32(25):2821–2823. doi:10.1200/JCO.2014.55.6910
151. Flynn KE, Atallah E, Lin L, et al. Patient- and physician-reported pain after tyrosine kinase inhibitor discontinuation among patients with chronic myeloid leukemia. Haematologica. 2022;107(11):2641–2649. doi:10.3324/haematol.2021.280377
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.