Back to Journals » Infection and Drug Resistance » Volume 8
Chronic Lyme disease: misconceptions and challenges for patient management
Authors Halperin J
Received 23 February 2015
Accepted for publication 24 March 2015
Published 15 May 2015 Volume 2015:8 Pages 119—128
DOI https://doi.org/10.2147/IDR.S66739
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Suresh Antony
John J Halperin
Department of Neurosciences, Overlook Medical Center, Summit, NJ, USA
Abstract: Lyme disease, infection with the tick-borne spirochete Borrelia burgdorferi, causes both specific and nonspecific symptoms. In untreated chronic infection, specific manifestations such as a relapsing large-joint oligoarthritis can persist for years, yet subside with appropriate antimicrobial therapy. Nervous system involvement occurs in 10%–15% of untreated patients and typically involves lymphocytic meningitis, cranial neuritis, and/or mononeuritis multiplex; in some rare cases, patients have parenchymal inflammation in the brain or spinal cord. Nervous system infection is similarly highly responsive to antimicrobial therapy, including oral doxycycline. Nonspecific symptoms such as fatigue, perceived cognitive slowing, headache, and others occur in patients with Lyme disease and are indistinguishable from comparable symptoms occurring in innumerable other inflammatory states. There is no evidence that these nonspecific symptoms reflect nervous system infection or damage, or that they are in any way specific to or diagnostic of this or other tick-borne infections. When these symptoms occur in patients with Lyme disease, they typically also subside after antimicrobial treatment, although this may take time. Chronic fatigue states have been reported to occur following any number of infections, including Lyme disease. The mechanism underlying this association is unclear, although there is no evidence in any of these infections that these chronic posttreatment symptoms are attributable to ongoing infection with B. burgdorferi or any other identified organism. Available appropriately controlled studies indicate that additional or prolonged courses of antimicrobial therapy do not benefit patients with a chronic fatigue-like state after appropriately treated Lyme disease.
Keywords: Lyme disease, Borrelia burgdorferi, chronic, diagnosis, treatment, chronic fatigue, neuroborreliosis
Background
The debate about “chronic Lyme disease” provides a remarkable example of how heated a conversation can become when people use words differently. Contested issues largely stem from very different understandings of what terms mean. The broad medical and scientific communities use the term “Lyme disease” to refer specifically to infections with Borrelia burgdorferi and closely related European Borrelia spp. Proponents of the concept of chronic Lyme disease, who typically refer to themselves as “Lyme literate”, use the term to refer to a constellation of disabling symptoms that may or may not be related to this or other infections they believe to be tick transmitted. This clinical syndrome largely overlaps with the disorder commonly known as “myalgic encephalomyelitis/chronic fatigue syndrome”, or as an Institute of Medicine committee recently recommended, “systemic exertion intolerance disease” or SEID1 – a disorder that is real, disabling, and may appear to develop following an infectious illness.
The medical/scientific community uses the term “nervous system Lyme disease” to refer to disorders in which there is objective evidence that this organism has physically invaded the nervous system and the infection, or the host response to it, is having a specific impact on neurologic function. The “Lyme literate” use the term to include a broad array of neurobehavioral phenomena, with no requirement of objective evidence of actual nervous system infection. The medical/scientific community uses the term “chronic Lyme disease” to describe individuals with objective evidence of longstanding ongoing infection, while the “Lyme literate” use this term to describe individuals with chronic, life-altering symptomatology without necessarily having biologic evidence of persisting infection. This might be considered little more than a semantic debate (as Humpty Dumpty famously said,2 “A word shall mean exactly what I choose it to mean, neither more nor less, it’s merely a question of who’s to be the master”). However, since the “Lyme literate” construct is used to justify prolonged courses of antimicrobial therapy with significant potential for complications, impact on community antimicrobial resistance, and consumption of health care resources, it is essential that the terms be defined with clarity.
Introduction – can Lyme disease cause chronic infection?
In 1977, Steere et al3 described a syndrome including tick bites, a rash termed at that time erythema chronicum migrans (now erythema migrans, EM), nonspecific symptoms including headache, malaise, fatigue, myalgias, and fever, and recurrent episodes of frank arthritis, with disease duration of up to 22 weeks. In these authors’ subsequent description of effective treatment of Lyme meningitis,4 meningeal symptoms developed on an average of 5 weeks (but up to 12 weeks) after initial evidence of the infection; patients were initially evaluated by the authors at a mean of 6 weeks (up to 12) after initial neurological abnormalities, and then were treated successfully. In a longitudinal assessment of individuals frequenting a Massachusetts island highly endemic for Lyme disease,5 untreated patients were identified with relapsing arthritis and fatigue lasting up to 15 years. Importantly, as many as half the individuals identified in that study as infected, based on seroconversion, remained asymptomatic. Subsequent work6 described untreated patients, symptomatic for an average of 2 years, and emphasized the cognitive difficulties experienced by patients otherwise symptomatic with this chronic infection. This work – and more – illustrates three undisputed facts. First, B. burgdorferi, the tick-borne spirochete responsible for Lyme disease, is quite capable of establishing a chronic (ie, many months in duration) infection. Second, this chronic infection, as in many other ongoing inflammatory states, can cause nonspecific symptoms such as malaise, fatigue, and perceived cognitive slowing in addition to more specific clinical manifestations. Third, individuals can be seropositive but asymptomatic following infection.
These observations, combined with misunderstandings about laboratory testing for the diagnosis of this infection, provide the underpinnings of the “debate” about “chronic Lyme disease”. Understanding the evolution of this “debate” requires an understanding of the biology of this infection, of the nature of nervous system infection, and of the ways in which nervous system function can be altered by non-neurologic disease.
History and ecology
EM, recognized as a common manifestation of Lyme disease, was first described over a century ago by the Swedish dermatologist, Afzelius,7,8 who postulated that this was related to the bites of hard-shelled Ixodes ticks. Two years after the publication of his observations, two French clinicians9 published a description of a 58-year-old man who, 3 weeks after a tick bite on the left buttock, developed an enlarging erythroderm at the site of the bite, accompanied by severe sciatic pain. Neuropathic pain subsequently affected both lower and the right upper extremities. Pain persisted for months, and he developed right shoulder weakness. Based on a cerebrospinal fluid (CSF) pleocytosis with elevated protein, and a slightly positive Wasserman test, the authors concluded that he had a non-syphilis spirochetal infection and treated him with neoarsphenamine (preferred treatment at that time for syphilis), and he recovered. This disorder, recognized as including painful radiculitis, lymphocytic meningitis, and subsequently cranial neuritis, came to be known as Garin–Bujadoux–Bannwarth syndrome.10 European clinicians have been well aware of this tick-bite-associated syndrome for many years, and by the 1950s, were treating it with penicillin.11
In the early 1980s, groups in the US12,13 and Europe14 established that North American Lyme disease and European EM/Garin–Bujadoux–Bannwarth syndrome were caused by closely related tick-borne spirochetes – B. burgdorferi in the US and Borrelia afzelii and Borrelia garinii in Europe. B. burgdorferi is responsible for all Lyme disease acquired in the US. All three strains, as well as several lesser ones such as Borrelia spielmanii, occur in Europe. Most European infections are attributable to B. garinii, responsible for the majority of neuroborreliosis, and B. afzelii, commonly associated with primarily cutaneous involvement. European and North American borreliosis share many clinical similarities (EM, radiculoneuritis) but have some differences. Once the causative organism was identified, North American Lyme disease became defined as infection with B. burgdorferi. Europeans have preferred the terms neuroborreliosis or Lyme borreliosis, referring to the cutaneous manifestations as EM and acrodermatitis chronica atrophicans, or ACA.
Zoonoses such as Lyme disease require specific conditions both to infect humans and to persist in an ecosystem.15–17 The first requirement is a competent reservoir host, a species that can sustain prolonged, preferably nonlethal, infection. For these Borrelia spp., this host consists primarily of field mice (although numerous other small mammals and occasionally birds or even reptiles can serve this purpose). These hosts can maintain a prolonged infection while apparently asymptomatic. While any blood-sucking arthropod could, in theory, ingest spirochete-containing blood, and if it feeds again while spirochetes are still viable, inject spirochetes into another host, the specific interactions of Borrelia with Ixodes ticks make this the primary, if not sole, competent vector – Ixodes scapularis and to a lesser extent Ixodes pacificus in the US, Ixodes ricinus in Europe, and Ixodes persulcatus elsewhere.
Ixodes ticks are born uninfected; there is no transovarial transmission to the egg or larva. Over the course of its typically 2-year life cycle, the tick will ingest a total of three blood meals, one at each life stage. Over the months that follow the larva’s ingestion of blood, the tick matures into a nymph, and will then have its second blood meal. The tick will then overwinter, often on a large furry mammal such as a sheep, deer, or bear. Following its final meal on this animal, the adult female tick lays its eggs and dies. Although deer (and corresponding large hosts elsewhere) are often blamed for the transmission of Lyme disease, they are actually only marginally relevant. Without them, the population of ticks will decline. However, if the tick feeds on a deer, this will be its final meal – even if the deer were infected, the tick will never bite another host, so from a Lyme disease perspective, this is a biologic “dead end”.
If any of the blood meals contains viable Borrelia, these can survive in the tick gut until the tick’s next meal. The presence of newly ingested blood at the next meal triggers proliferation of these spirochetes, which then migrate throughout the tick, including reaching its salivary glands. Tick feeding involves days of attachment, during which time tick saliva is injected into the host – injecting anticoagulants, local anesthetics, and other substances required for sustained attachment and feeding. Once the spirochetes migrate to the salivary glands, they can similarly be injected into the host as well. Since the multiplication and migration of Borrelia in the tick requires at least 24–48 hours following the initiation of feeding, attachment for periods shorter than this carries very little risk of transmitting infection.18–20
Nymphal ticks are the most common cause of human infection. Larvae are uninfected, so even if they were to bite a person, there could be no transmission. Nymphs can be infected and are quite small – about the size of a period on a printed page – so they can be difficult to see. They also substantially outnumber adults – every adult had to be a nymph, but only some nymphs survive to adulthood.
The bite provides the first and best opportunity to interrupt the transmission of Lyme disease. Since the tick must be attached for days to transmit infection, a daily tick check following potential exposure – careful inspection of the skin for attached ticks – with timely removal of any that are found – markedly reduces the risk of infection. Tick removal is best accomplished by insertion of a fine pair of tweezers between the tick mouthparts and the skin, applying slow backward traction. Notably, as the tick feeds, it becomes bloated and engorged. If still tiny and black, it is highly unlikely to have fed sufficiently to have transmitted infection.
Transmission of this infection occurs in many temperate parts of the world where the requisite vectors, infected permissive reservoir hosts, and humans coexist. In the US, it occurs primarily along the east coast, from Maine to Virginia, with small foci of infection in the upper Midwest (Wisconsin, MN) and northern California.21 The endemic areas have gradually enlarged over the years, but this has been a slow process. In the US, the Centers for Disease Control and Prevention reports about 30,000 cases per year meeting the strict epidemiologic case definition.21 The number of actual cases probably exceeds that, though by how much is difficult to ascertain.22
Laboratory-based diagnosis
In deciding the extent to which a clinical diagnosis of Lyme disease should rely on laboratory confirmation, it is essential first to understand the accuracy of the laboratory techniques, to permit appropriate balancing of clinical vs laboratory data.
Historically, diagnosis of most bacterial infections has relied on in vitro culture and identification of the responsible organism. This is challenging for some organisms, which are either impossible (Treponema pallidum) or difficult (B. burgdorferi) to grow in culture. Culture of B. burgdorferi requires special medium not generally available in clinical microbiology laboratories. More importantly, other than in EM, the number of organisms present in readily sampled fluids (blood, CSF) appears to be quite low. As a result, even in ideal laboratory circumstances, cultures of CSF obtained from individuals known to have Lyme meningitis are only positive about 10% of the time.23 Even using the remarkable technical sensitivity of polymerase chain reaction-based techniques does not substantially increase the rate of true positives.
As a result, laboratory support for the diagnosis relies on testing the host immune response to the infecting organism. In most infections, serodiagnosis relies on assessment of acute and convalescent specimens, reflecting the fact that early in any infection, there is little or no measurable antibody, but as infection persists, the host response reflected in the antibody concentration will substantially increase. For reasons probably related to the unfortunate historic comparison to syphilis (where any amount of measurable nonspecific reaginic antibody measured in screening tests is considered to be relevant), Lyme serodiagnosis has often relied on assessment of a single sample. We know that, very early in the course of the illness, such as during the acute rash, as many as 50% of patients will be seronegative.24 Even during early disseminated infection, occasional patients with Lyme disease-associated facial nerve palsy will only seroconvert weeks after initial clinical presentation.25 On the other hand, in individuals with symptoms of more than 1- to 2-months duration, essentially every patient is seropositive.20
Some studies performed in the 1980s suggested that early but incomplete treatment with antibiotics might permanently abrogate the antibody response.26 These studies relied in large part on diagnosing patients based on measures of the T-cell response to B. burgdorferi. Subsequent work showed this T-cell assay to be quite nonspecific,20,27 rendering this conclusion incorrect – only if very early treatment eradicates the infection, eliminating any ongoing immune stimulation, would treatment blunt the antibody response. Some have interpreted these early studies as indicating that simply ingesting antibiotics would render a patient seronegative, while the antibiotics were present in the patient’s system. There has never been any evidence to support this conclusion, nor is there any biologically plausible basis for making such an assertion.
Consequently, if the data indicate that immunocompetent patients with B. burgdorferi infection of more than a few months duration are virtually always seropositive, and if the definition of “chronic Lyme disease” requires symptoms of more than several months duration to be deemed chronic, all patients with “chronic Lyme disease” should be seropositive.
As these conclusions have become more and more firmly rooted in clinical experience, it has become commonplace for the “Lyme literate” to ascribe the symptomatology formerly attributed to “chronic Lyme disease” to chronic infections due to other organisms known to be found occasionally in the same ticks, broadening the definition of “chronic Lyme disease” to include these co-infections.28 Although laboratory tests confirming the presence of these infections are available, proponents appear either to not rely on the results of the most specific tests or to apply lax interpretive criteria for others, rendering conclusions suspect, or to rely on other tests that have not been subject to rigorous validation. Importantly, there is little if any evidence that these other organisms cause chronic infection, or any of the symptoms attributed to “chronic Lyme disease”.
Serologic testing has evolved over the years with most efforts aiming to improve specificity. Initial work used enzyme-linked immunosorbent assays (ELISAs) using sonicated whole organisms as the target antigens; a number of interpretive criteria were chosen to try to balance sensitivity and specificity. In the early 1990s, extensive studies in large populations of patients with and without Lyme disease led to the currently recommended two-tier approach,29,30 using a highly sensitive ELISA as a screening test, and then a Western blot to provide specificity. It is important to understand that Western blot criteria (Table 1) were not selected based on the uniqueness of any Borrelia epitopes but rather on statistical analyses of findings to identify those combinations with the greatest positive and negative predictive values. As a result of these studies, a set of three IgM and ten IgG bands were selected such that individuals with early disease typically have at least two of the three IgM bands, while patients with longstanding disease typically have at least five of the ten IgG bands.29 Two important facts must be borne in mind. First, the Western blot criteria were developed in individuals with positive or borderline ELISAs. Interpretation in patients with negative ELISAs is quite problematic and should only be attempted with great caution. Second, IgM tests are inherently quite cross-reactive, so false positives are commonplace. Patients with disease of more than 1-month or 2-month duration should be IgG seropositive, so only IgG blots provide reliable information. Any IgM findings in this setting should be considered, at best, uninterpretable, and more correctly as spurious.
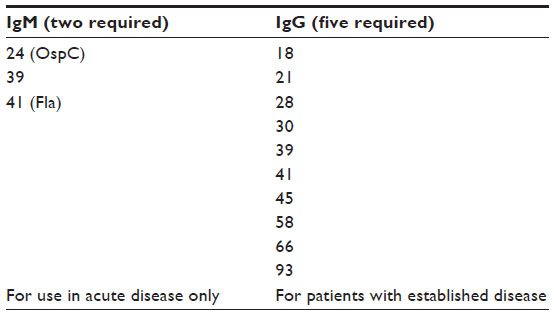 | Table 1 Western blot interpretation criteria |
Laboratory findings in central nervous system infection
In a significant number of patients with B. burgdorferi infection, the spirochete invades the central nervous system (CNS) quite early in the course of the disease.31 As with any CNS infection, this triggers a local inflammatory response, which can be used to support or refute the conclusion that the CNS is infected. Invasion appears to trigger local production of CXCL13,32,33 a chemokine that serves to attract circulating B-cells to the site of infection. B-cells that then enter the CNS remain there, producing specific antibody targeting B. burgdorferi. Since a small amount of circulating immunoglobulin normally crosses the blood–brain barrier, determining the relative concentrations of B. burgdorferi-specific IgG, after normalizing for the relative concentrations of nonspecific IgG, allows for the determination of intrathecal production of specific antibody.6,34–36 This measure turns out to be highly specific for CNS neuroborreliosis. The major drawback is that the derived index may remain elevated for a decade or more after effective treatment.37 However, since active infection elicits an inflammatory response, combining CSF serologic information with CSF cell counts and protein concentration provides invaluable diagnostic information. Just as in neurosyphilis, the best measure of resolution of the infection is the normalization of the CSF pleocytosis and elevated protein that are invariably found in active CNS infection.
Clinical phenomenology
Since EM, Garin–Bujadoux–Bannwarth syndrome, and Lyme disease were all clinically defined long before the causative organisms were identified, these disorders were originally described syndromically. Not surprisingly, the replacement of the syndromic definition with diagnosis based on a defined pathophysiology can result in confusion when lab tests and clinical phenomenology do not align. Many assert that the diagnosis of Lyme disease is a “clinical diagnosis” – a statement that is as true for Lyme disease as it is for anything else in medicine. A clinical diagnosis is one made by an informed clinician incorporating all available data. It makes no more sense to ignore relevant laboratory data in a patient clinically suspected to have Lyme disease than it would be to diagnose a lethal brain tumor in a patient with normal brain magnetic resonance imaging (MRI). In any patient, the diagnosis must be deduced based on the balance of the sensitivities and specificities of each of the clinical elements under consideration. In the appropriate context, EM is highly sensitive and specific. In the setting of very early infection, sensitivity of serologic testing is only about 50%, so clinical diagnosis should be based on the rash, not the serology. In patients with Lyme arthritis, sensitivity of serologic testing is for all intents and purposes 100%, so diagnosis requires a positive serology. In patients whose only symptoms are both commonplace and nonspecific – headache, fatigue, and perceived cognitive slowing – it is highly unlikely that even in a highly endemic area would more than 5% of patients with these symptoms have them attributable to this infection. Hence, the specificity of these symptoms is probably no more than 5%. In contrast, in individuals with Lyme disease of more than a month or two duration, sensitivity of serologic testing is over 95%. In this setting, attributing these symptoms to Lyme disease in seronegative patients would be inappropriate.38
Cutaneous manifestations
EM, as described by Afzelius, Garin and Bujadoux, and Scrimenti,39 is almost pathognomonic. Beginning as a small erythroderm at the site of the bite, this gradually expands as spirochetes migrate centrifugally from the initial focus of inoculation. For case definition purposes, it must be at least 5 cm in diameter. Obviously, it will start smaller than this, and if treated rapidly, may not attain this threshold. The erythroderm expands day by day and can become huge – the one described by Garin and Bujadoux involved both buttocks, the abdomen, and thigh of an adult male. The rash can be homogeneous and round but often takes on a target-like appearance as the leading edge becomes erythematous, while more central areas return to their more normal hue. The rash need not be round, its shape dictated by the anatomic areas involved. It is usually surprisingly asymptomatic – not necessarily pruritic or painful, despite its inflamed appearance. Biopsies generally reveal large numbers of spirochetes, much like in the painless chancre of syphilis. However, the clinical appearance and evolution are so typical that biopsy is rarely needed. The frequency with which EM occurs has been debated. In children, who presumably have parents attentively inspecting them at bath time, about 90% of infected children develop EM.40 In adults, where the rash might occur in areas not easily or routinely inspected, estimates are generally in the range of a half to two-thirds.
Spirochetes can disseminate hematogenously from this original nidus. In the US, about 25% or more of patients will develop multifocal EM as a result of this,41 each of the secondary EMs evolving in a manner similar to the original one. This only occurs in about 5% of European patients. In contrast, European patients may develop a Borrelia lymphocytoma – a dense lymphocytic infiltrate of the earlobe or areola of the breast, something seen very rarely in the US. Europeans with chronic untreated infection can also develop acrodermatitis atrophicans – a tissue paper-like thinning with purplish discoloration of the skin of a leg. This has never been reported in a US patient.
Non-neurologic extracutaneous symptoms
Early descriptions of Lyme disease included the occurrence of otherwise unexplained heart block in about 5% of patients. For unclear reasons, this has been less evident in more recent series. It has been described occasionally in European patients, but the incidence is certainly no more than a single-digit percent of infected individuals.
The original defining phenomenology in the US patients was Lyme arthritis.3 This is a relapsing remitting large-joint oligoarthritis – affecting a knee, elbow, shoulder, or hip. Typically, one joint is involved at a time, spontaneously becoming red, painful, and swollen, and then resolving after a few weeks. At different times, different joints can be involved. Small joints such as fingers, toes, or spine facets are involved infrequently. Arthritis too seems to be becoming less frequent with increased early recognition and treatment of Lyme disease.
Neuroborreliosis
The range of different disorders considered to be manifestations of nervous system involvement with B. burgdorferi, B. afzelii, and B. garinii can be considered to fall into three distinct groups.42 The “classic triad” occurs in early acute infection, presenting as acute inflammation in one or a few nerves or of the meninges.4,9,43 Occasional patients will have infection and inflammation involving the spinal cord or brain. Patients with more indolent, longstanding infection may have subacute-to-chronic infection of the peripheral nervous system (PNS)44 or CNS.6 Other patients – with evidence of active infection outside the nervous system – may have what has been termed “Lyme encephalopathy”45,46 – symptoms of cognitive and memory difficulty, fatigue, and malaise, identical to those seen in patients with other active infectious or inflammatory states.
“Classic triad”
Acute nervous system involvement occurs in 10%–15% of infected individuals – in both Europe and the US47 – and takes quite similar forms in both regions. The classic triad includes lymphocytic meningitis and painful radiculoneuritis, as described years ago by Garin and Bujadoux, as well as cranial neuritis.
Radiculitis, a term used generally to describe inflammation of spinal nerve roots, causes sciatica-like neuropathic pain, typically involving one or a few dermatomes, most commonly in a limb but occasionally the trunk. Patients often will have weakness or diminished deep tendon reflexes in muscles innervated by the affected dermatome. Non-Lyme disease radiculopathy is almost always caused by mechanical compression; patients with neuroborreliosis can develop remarkably similar symptoms, attributed to inflammation of the symptomatic nerve root or roots. Precisely the syndrome described by Garin and Bujadoux in 1922, it is now clear that this is just one clinical presentation of PNS involvement in Lyme disease.44 It appears likely that these clinically varied manifestations are all various forms of what is known as a mononeuropathy multiplex, a common type of peripheral nerve involvement in inflammatory diseases (vasculitides, infections such as leprosy) or other vasculopathic disorders, such as diabetes mellitus.
Although all rooted in the same pathophysiologic process, clinical manifestations of PNS Lyme will vary with the nerve(s) involved. If a single nerve is involved, the patient may develop a mononeuropathy. A single nerve root can produce signs and symptoms indistinguishable from a mechanical radiculopathy associated with disk herniation – with severe radicular neuropathic pain and segmental muscle denervation and reflex loss. Other patients may develop a plexopathy, involving either the brachial or lumbosacral plexus.
Cranial nerve involvement is also quite common, with the facial nerve, either unilaterally or occasionally bilaterally, involved in the majority of patients with Lyme disease-associated cranial neuropathies. It is important to emphasize that facial palsy is not a subtle finding – patients have severe drooping of the side of the face, and are typically unable to seal the lips, close the eye, or wrinkle the forehead on one side. Recovery is to be expected in this disorder, but the initial presentation is typically quite dramatic – and distressing to the patient. Other cranial nerves can be involved – the trigeminal nerve causing numbness and pain, the nerves to the extraocular muscles causing paralysis of innervated eye muscles and diplopia, or the acousticovestibular nerve affecting hearing and balance. These are all distinctly uncommon; involvement of the optic or olfactory nerves occurs rarely if ever.48 Similarly involvement of cranial nerves 9–12 occurs very infrequently.49 As with peripheral nerve involvement, facial palsy and these other cranial neuropathies appear to be differing manifestations of the same process – a mononeuropathy multiplex.
Each of these disorders – cranial neuritis, radiculoneuritis, and lymphocytic meningitis – can be considered typical of nervous system Lyme disease. It is therefore worth considering the specificity of these disorders for the diagnosis. In an observational study from an area highly endemic for Lyme disease, approximately 25% of adult cases of facial nerve palsy were attributable to Lyme disease.25 Cross-sectional studies of patients with “aseptic meningitis”50–53 suggest that in summer months, in areas highly endemic for Lyme disease, between one in two and one in five patients with lymphocytic meningitis might have this infection. Lumbar radiculopathy is estimated to affect 3%–5% of the adult population;54 cervical radiculopathy is slightly less prevalent. Since radiculopathy affects no more than 5% of the 30,000 patients with Lyme disease reported to the Centers for Disease Control and Prevention annually, this would translate to 1,500 cases per year, a very small fraction of the number of mechanically caused cases. From this, it should be clear that the specificity of even these very specific neurologic manifestations of Lyme disease is quite low – no more than 25% for facial nerve palsy, perhaps one in three for lymphocytic meningitis, and much less than 1% for radiculopathy – in areas highly endemic for Lyme disease. In areas where the incidence of Lyme disease is much lower, the potential positive predictive value for one of these presentations for the diagnosis of Lyme disease would drop precipitously. Even in endemic areas, a finding with no better than a 50:50 chance of predicting the diagnosis would never be considered a useful basis for making a clinical diagnosis.
Patients with more longstanding and indolent infection may have less acute forms of nervous system involvement. Originally described in European patients with acrodermatitis,55 there may be more diffuse, widespread involvement of peripheral nerves, clinically mimicking the much more common stocking–glove type of peripheral neuropathy. A very similar disorder was described in patients with chronic, untreated Lyme arthritis.56,57 In both, more detailed analysis suggests that this too is pathophysiologically a form of mononeuropathy – in this case, what is referred to as a confluent mononeuropathy multiplex – with mild diffuse involvement of multiple small nerves.
Lyme encephalitis vs Lyme encephalopathy
The issue that has caused the greatest confusion about nervous system Lyme disease relates to possible CNS involvement. Other than lymphocytic meningitis – a disorder that may cause severe headache but does not damage the brain itself – parenchymal CNS infection is remarkably rare. In European patients with Lyme radiculitis, the nerve root inflammation may extend proximally into the adjacent spinal cord, causing myelopathic changes – a disorder reported only anecdotally in the US (unpublished observations). Brain involvement was described primarily in the European literature in the 1980s.58,59 More recent reports are extraordinarily difficult to find, perhaps reflecting more aggressive early recognition and treatment of this infection. Similarly, in some rare cases, patients with apparent parenchymal brain involvement have been reported from the US,6,57 but this too is now remarkably infrequent. What little evidence exists about these patients indicates that it is due to active brain infection. Imaging studies appear inflammatory, PET scans appear hypermetabolic,60 CSF is inflammatory, and most patients have evidence of localized production of specific anti-Borrelia antibody within the CNS.6,61,62
In contrast to these rare occurrences in patients, in the days when patients with longstanding Lyme arthritis were frequently seen, it was almost the norm for them to describe cognitive slowing, fatigue, and memory problems. With extensive testing, including brain MRI, CSF examinations, neurophysiologic testing, and more, virtually none had evidence of CNS inflammation or infection.6 Some work suggested that this might be related to elevated concentrations of cytokines or other potential neuromodulators63,64 produced in the periphery and then entering the brain, but the mechanism remains unclear. What is clear is that the clinical phenomenology is indistinguishable from the “toxic metabolic” encephalopathy seen in active rheumatoid arthritis, sepsis, other serious infections, or even in patients receiving therapeutic interferon. It is equally clear that in virtually none of these individuals is there any evidence of CNS infection with B. burgdorferi, or of any potentially related inflammatory state. Rather, this seems to be the ubiquitous encephalopathy or delirium seen in innumerable medical patients with active systemic infection or inflammation. Just like that state, this alteration of cognitive function is reversible when the infection or inflammation resolves (although often not immediately) and does not require any specific alteration of treatment. Specifically, this does not require the selection of antimicrobial regimens based on their ability to penetrate the blood–brain barrier.
Treatment
The Borrelia spp. responsible for Lyme disease remain highly sensitive to readily available antibiotics, both in vivo and in vitro.20 Early studies in patients infected for extended periods of time demonstrated microbiologic cures in the vast majority of such individuals.20,65 Patients with significant end organ damage – those with severe arthritis or with longstanding brain or spinal cord inflammation – might have some residua from already established damage, but progressive or recurrent disease is distinctly uncommon. Although meningeal dose penicillin and ceftriaxone were both introduced to assure adequate treatment of CNS infection, there are now multiple studies demonstrating that oral doxycycline attains sufficient concentrations in the CSF to be as effective as intravenously administered ceftriaxone, at least in European patients66,67 (Table 2).
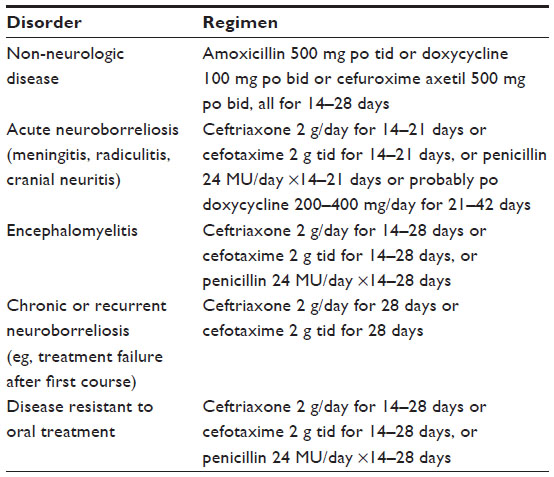 | Table 2 Commonly used antimicrobial regimens |
In contrast to the overwhelming evidence that Borrelia infections are readily cured with antimicrobial therapy, there is abundant evidence that antibiotic treatment is ineffective in patients with persistent fatigue and cognitive symptoms following appropriately diagnosed and treated Lyme disease.68–71
Conclusion
Lyme disease, defined as infection with B. burgdorferi or the two closely related European spirochete pathogens B. afzelii and B. garinii, infects the nervous system in up to 15% of patients. Untreated infection, particularly with joint involvement, can be chronic, lasting years. That said, it is essential to understand that the fatigue and cognitive difficulty seen in many individuals with Lyme disease are neither caused by or evidence of nervous system infection nor in any way specific to this disease.72 Patients without Lyme disease may develop the same disabling symptoms including severe fatigue and cognitive difficulty, a disorder formerly referred to as chronic fatigue syndrome, and for which the label “systemic exertion intolerance disease” has recently been suggested.1 Although the latter is both real and disabling, and appears to occur following any number of infections – including possibly Lyme disease – there is no evidence that it is caused by persisting infection with B. burgdorferi, other tick-borne pathogens, or any other as yet identified pathogen. As such, treatment of this symptom complex with antibiotics is unlikely to be helpful to patients but does incur substantial risk. This, combined with the impact of excessive antibiotic usage on the development of widespread antibiotic resistance among more potentially lethal pathogens, and the significant health care resource utilization and cost associated with prolonged administration of parenteral antibiotics, makes such treatment ill advised.
Disclosure
Expert witness, defending physicians in medical malpractice cases in which they have been accused of failure to diagnose or treat Lyme disease. The author owns equity in several pharmaceutical companies none of which is relevant to this topic. He has also received royalties from UpToDate and for “Lyme Disease, an evidence based approach” published by CABI in 2011. The author reports no other conflicts of interest in this work.
References
Committee on the Diagnostic Criteria for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome; Board on the Health of Select Populations; Institute of Medicine. Beyond Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: Redefining an Illness. Washington DC: The National Academies Press; 2015. | |
Dodgson C. Through the Looking-Glass. Gutenberg: Project Gutenberg; 2008. | |
Steere AC, Malawista SE, Hardin JA, Ruddy S, Askenase W, Andiman WA. Erythema chronicum migrans and Lyme arthritis. The enlarging clinical spectrum. Ann Intern Med. 1977;86(6):685–698. | |
Steere AC, Pachner AR, Malawista SE. Neurologic abnormalities of Lyme disease: successful treatment with high-dose intravenous penicillin. Ann Intern Med. 1983;99:767–772. | |
Steere AC, Taylor E, Wilson ML, Levine JF, Spielman A. Longitudinal assessment of the clinical and epidemiologic features of Lyme disease in a defined population. J Infect Dis. 1986;154:295–300. | |
Halperin JJ, Luft BJ, Anand AK, et al. Lyme neuroborreliosis: central nervous system manifestations. Neurology. 1989;39(6):753–759. | |
Afzelius A. Verhandlugen der dermatorischen Gesellshaft zu Stockholm. Arch Derm Syphiligr. [Proceedings of the Stockholm Dermatologic Society]. 1910;101:404. | |
Afzelius A. Erythema chronicum migrans. Acta Derm Venereol Suppl (Stockh). 1921;2:120–125. | |
Garin C, Bujadoux A. Paralysie par les tiques. [tick paralysis]. J Med Lyon. 1922;71: 765–767. | |
Bannwarth A. Chronische lymphocytare meningitis, entzundliche polyneuritis und “rheumatismus”. [Chronic lymphocytic meningitis, inflammatory polyneuritis and ‘rheumatism’]. Arch Psychiatr Nervenkr. 1941;113: 284–376. | |
Hollstrom E. Successful treatment of erythema migrans Afzelius. Acta Derm Venereol. 1951;31(2):235–243. | |
Benach JL, Bosler EM, Hanrahan JP, et al. Spirochetes isolated from the blood of two patients with Lyme Disease. N Engl J Med. 1983;308: 740–742. | |
Steere AC, Grodzicki RL, Kornblatt AN, et al. The spirochetal etiology of Lyme Disease. N Engl J Med. 1983;308:733–740. | |
Asbrink E, Hederstedt B, Hovmark A. The spirochetal etiology of acrodermatitis chronica atrophicans Herxheimer. Acta Derm Venereol. 1984;64:506–512. | |
Anderson JF. Epizootiology of Borrelia in Ixodes tick vectors and reservoir hosts. Rev Infect Dis. 1989;11(Suppl 6):S1451–S1459. | |
Smith RP. Ticks, the vectors of Lyme disease. In: Halperin JJ, editor. Lyme Disease, An Evidence-Based Approach. Wallingford: CABI; 2011:1–28 | |
Richer LM, Brisson D, Melo R, Ostfeld RS, Zeidner N, Gomes-Solecki M. Reservoir targeted vaccine against Borrelia burgdorferi: a new strategy to prevent Lyme disease transmission. J Infect Dis. 2014;209(12):1972–1980. | |
des Vignes F, Piesman J, Heffernan R, Schulze TL, Stafford KC, Fish D. Effect of tick removal on transmission of Borrelia burgdorferi and Ehrlichia phagocytophila by Ixodes scapularis nymphs. J Infect Dis. 2001;183(5):773–778. | |
Nadelman RB, Nowakowski J, Fish D, et al. Prophylaxis with single-dose doxycycline for the prevention of Lyme disease after an Ixodes scapularis tick bite. N Engl J Med. 2001;345(2):79–84. | |
Wormser GP, Dattwyler RJ, Shapiro ED, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the infectious diseases society of America. Clin Infect Dis. 2006;43:1089–1134. | |
Adams DA, Jajosky RA, Ajani U, et al; Centers for Disease Control and Prevention (CDC). Summary of notifiable diseases – United States, 2012. MMWR Morb Mortal Wkly Rep. 2014;61(53):1–121. | |
Hinckley AF, Connally NP, Meek JI, et al. Lyme disease testing by large commercial laboratories in the United States. Clin Infect Dis. 2014;59(5):676–681. | |
Karlsson M, Hovind HK, Svenungsson B, Stiernstedt G. Cultivation and characterization of spirochetes from cerebrospinal fluid of patients with Lyme borreliosis. J Clin Microbiol. 1990;28(3):473–479. | |
Aguero-Rosenfeld ME, Nowakowski J, Bittker S, Cooper D, Nadelman RB, Wormser GP. Evolution of the serologic response to Borrelia burgdorferi in treated patients with culture-confirmed erythema migrans. J Clin Microbiol. 1996;34(1):1–9. | |
Halperin JJ, Golightly M. Lyme borreliosis in Bell’s palsy. Long island neuroborreliosis collaborative study group. Neurology. 1992;42(7):1268–1270. | |
Dattwyler RJ, Volkman DJ, Luft BJ, Halperin JJ, Thomas J, Golightly MG. Seronegative Lyme disease. Dissociation of specific T- and B-lymphocyte responses to Borrelia burgdorferi. N Engl J Med. 1988; 319(22):1441–1446. | |
Dressler F, Yoshinari NH, Steere AC. The T-cell proliferative assay in the diagnosis of Lyme disease [see comments]. Ann Intern Med. 1991; 115(7):533–539. | |
Cameron DJ, Johnson LB, Maloney EL. Evidence assessments and guideline recommendations in Lyme disease: the clinical management of known tick bites, erythema migrans rashes and persistent disease. Expert Rev Anti Infect Ther. 2014;12(9):1103–1135. | |
Dressler F, Whalen JA, Reinhardt BN, Steere AC. Western blotting in the serodiagnosis of Lyme disease. J Infect Dis. 1993;167(2):392–400. | |
Centers for Disease Control and Prevention (CDC). Recommendations for test performance and interpretation from the second national conference on serologic diagnosis of Lyme disease. MMWR Morb Mortal Wkly Rep. 1995;44(31):590–591. | |
Luft BJ, Steinman CR, Neimark HC, et al. Invasion of the central nervous system by Borrelia burgdorferi in acute disseminated infection. JAMA. 1992;267(10):1364–1367. | |
Rupprecht TA, Plate A, Adam M, et al. The chemokine CXCL13 is a key regulator of B cell recruitment to the cerebrospinal fluid in acute Lyme neuroborreliosis. J Neuroinflammation. 2009;6:42. | |
Rupprecht TA, Pfister HW, Angele B, Kastenbauer S, Wilske B, Koedel U. The chemokine CXCL13 (BLC): a putative diagnostic marker for neuroborreliosis. Neurology. 2005;65(3):448–450. | |
Stiernstedt GT, Granstrom M, Hederstedt B, Skoldenberg B. Diagnosis of spirochetal meningitis by enzyme linked immunosorbent assay and indirect immunofluorescence assay in serum and cerebrospinal fluid. J Clin Microbiol. 1985;21:819–825. | |
Hansen K, Lebech AM. Lyme neuroborreliosis: a new sensitive diagnostic assay for intrathecal synthesis of Borrelia burgdorferi – specific immunoglobulin G, A, and M. Ann Neurol. 1991;30(2):197–205. | |
Steere AC, Berardi VP, Weeks KE, Logigian EL, Ackermann R. Evaluation of the intrathecal antibody response to Borrelia burgdorferi as a diagnostic test for Lyme neuroborreliosis. J Infect Dis. 1990;161(6):1203–1209. | |
Hammers Berggren S, Hansen K, Lebech AM, Karlsson M. Borrelia burgdorferi-specific intrathecal antibody production in neuroborreliosis: a follow-up study. Neurology. 1993;43(1):169–175. | |
Halperin JJ, Baker P, Wormser GP. Common misconceptions about Lyme disease. Am J Med. 2013;126(3):264. e261–e267. | |
Scrimenti RJ. Erythema chronicum migrans. Arch Dermatol. 1970; 102(1):104–105. | |
Gerber MA, Shapiro ED, Burke GS, Parcells VJ, Bell GL; Pediatric Lyme Disease Study Group. Lyme disease in children in southeastern Connecticut. N Engl J Med. 1996;335(17):1270–1274. | |
Steere AC, Bartenhagen NH, Craft JE, et al. The early clinical manifestations of Lyme disease. Ann Intern Med. 1983;99(1):76–82. | |
Halperin J. Spirochetal infections of the nervous system. In: Aminoff M, editor. Neurology and General Medicine. 4th ed. Amsterdam: Elsevier; 2014:817–832. | |
Reik L, Steere AC, Bartenhagen NH, Shope RE, Malawista SE. Neurologic abnormalities of Lyme disease. Medicine. 1979;58(4):281–294. | |
Halperin JJ, Luft BJ, Volkman DJ, Dattwyler RJ. Lyme neuroborreliosis – peripheral nervous system manifestations. Brain. 1990;113: 1207–1221. | |
Halperin JJ, Krupp LB, Golightly MG, Volkman DJ. Lyme borreliosis-associated encephalopathy. Neurology. 1990;40:1340–1343. | |
Logigian EL, Kaplan RF, Steere AC. Successful treatment of Lyme encephalopathy with intravenous ceftriaxone. J Infect Dis. 1999;180(2):377–383. | |
Bacon RM, Kugeler KJ, Mead PS. Surveillance for Lyme Disease – United States, 1992–2006. MMWR Morb Mortal Wkly Rep. 2008; 57(SS10):1–9. | |
Sibony P, Halperin J, Coyle P, Patel K. Reactive Lyme serology in patients with optic neuritis and papilledema. J Neuro Ophthal. 2005;25(2):71–82. | |
Ishaq S, Quinet R, Saba J. Phrenic nerve paralysis secondary to Lyme neuroborreliosis. Neurology. 2002;59(11):1810–1811. | |
Tuerlinckx D, Bodart E, Jamart J, Glupczynski Y. Prediction of Lyme meningitis based on a logistic regression model using clinical and cerebrospinal fluid analysis: a European study. Pediatr Infect Dis J. 2009;28(5):394–397. | |
Garro AC, Rutman M, Simonsen K, Jaeger JL, Chapin K, Lockhart G. Prospective validation of a clinical prediction model for Lyme meningitis in children. Pediatrics. 2009;123(5):e829–e834. | |
Shah SS, Zaoutis TE, Turnquist J, Hodinka RL, Coffin SE. Early differentiation of Lyme from enteroviral meningitis. Pediatr Infect Dis J. 2005;24(6):542–545. | |
Tuerlinckx D, Bodart E, Garrino MG, de Bilderling G. Clinical data and cerebrospinal fluid findings in Lyme meningitis versus aseptic meningitis. Eur J Pediatr. 2003;162(3):150–153. | |
Tarulli AW, Raynor EM. Lumbosacral radiculopathy. Neurol Clin. 2007;25(2):387–405. | |
Hopf HC. Peripheral neuropathy in acrodermatitis chronica atrophicans. J Neurol Neurosurg Psychiatry. 1975;38:452–458. | |
Halperin JJ, Little BW, Coyle PK, Dattwyler RJ. Lyme disease – a treatable cause of peripheral neuropathy. Neurology. 1987;37: 1700–1706. | |
Logigian EL, Kaplan RF, Steere AC. Chronic neurologic manifestations of Lyme disease. N Engl J Med. 1990;323(21):1438–1444. | |
Ackermann R, Rehse KB, Gollmer E, Schmidt R. Chronic neurologic manifestations of erythema migrans borreliosis. Ann N Y Acad Sci. 1988;539:16–23. | |
Ackermann R, Gollmer E, Rehse KB. Progressiv Borrelia enzephalomyelitis. Chronische manifestation erythem chronicum migrans krankheit des nervensystems. [Progressive Borrelia encephalomyelitis. Chronic manifestation of erythema chronicum migrans disease of the nervous system]. Dtsch Med Wochenschr. 1985;110(26):1039–1042. | |
Kalina P, Decker A, Kornel E, Halperin JJ. Lyme disease of the brainstem. Neuroradiology. 2005;47(12):903–907. | |
Henriksson A, Link H, Cruz M, Stiernstedt G. Immunoglobulin abnormalities in cerebrospinal fluid and blood over the course of lymphocytic meningoradiculitis (Bannwarth’s syndrome). Ann Neurol. 1986;20: 337–345. | |
Baig S, Olsson T, Hansen K, Link H. Anti-Borrelia burgdorferi antibody response over the course of Lyme neuroborreliosis. Infect Immun. 1991;59(3):1050–1056. | |
Halperin JJ, Heyes MP. Neuroactive kynurenines in Lyme borreliosis. Neurology. 1992;42(1):43–50. | |
Jacek E, Fallon BA, Chandra A, Crow MK, Wormser GP, Alaedini A. Increased IFNalpha activity and differential antibody response in patients with a history of Lyme disease and persistent cognitive deficits. J Neuroimmunol. 2013;255(1–2):85–91. | |
Dattwyler RJ, Halperin JJ, Volkman DJ, Luft BJ. Treatment of late Lyme disease. Lancet. 1988;1:1191–1193. | |
Halperin JJ, Shapiro ED, Logigian EL, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: treatment of nervous system Lyme disease. Neurology. 2007;69(1):91–102. | |
Bremell D, Dotevall L. Oral doxycycline for Lyme neuroborreliosis with symptoms of encephalitis, myelitis, vasculitis or intracranial hypertension. Eur J Neurol. 2014;21(9):1162–1167. | |
Klempner MS, Baker PJ, Shapiro ED, et al. Treatment trials for post-lyme disease symptoms revisited. Am J Med. 2013;126(8):665–669. | |
Krupp LB, Hyman LG, Grimson R, et al. Study and treatment of post Lyme disease (STOP-LD): a randomized double masked clinical trial. Neurology. 2003;60(12):1923–1930. | |
Fallon BA, Keilp JG, Corbera KM, et al. A randomized, placebo-controlled trial of repeated IV antibiotic therapy for Lyme encephalopathy. Neurology. 2008;70:992–1003. | |
Klempner MS, Hu LT, Evans J, et al. Two controlled trials of antibiotic treatment in patients with persistent symptoms and a history of Lyme disease. N Engl J Med. 2001;345(2):85–92. | |
Luo N, Johnson J, Shaw J, Feeny D, Coons S. Self-reported health status of the general adult US population as assessed by the EQ-5D and health utilities index. Med Care. 2005;43(11):1078–1086. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.