Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Chromosomal Copy Number Variation Predicts EGFR-TKI Response and Prognosis for Patients with Non-Small Cell Lung Cancer
Authors He H, Ma H, Chen Z, Chen J, Wu D, Lv X, Zhu J
Received 22 April 2023
Accepted for publication 25 August 2023
Published 13 September 2023 Volume 2023:16 Pages 835—846
DOI https://doi.org/10.2147/PGPM.S418320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Haiyan He,1,* Hang Ma,1,* Zhuo Chen,2 Jingliang Chen,1 Dandan Wu,1 Xuedong Lv,1 Jie Zhu1
1Department of Respiratory Medicine, The Second Affiliated Hospital of Nantong University, Nantong First People’s Hospital, Nantong, Jiangsu, 226001, People’s Republic of China; 2Department of Invasive Technology, The Second Affiliated Hospital of Nantong University, Nantong First People’s Hospital, Nantong, Jiangsu, 226001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xuedong Lv; Jie Zhu, The Second Affiliated Hospital of Nantong University, Nantong First People’s Hospital, 6 North Road Hai’er Xiang, Nantong, 226001, Jiangsu, People’s Republic of China, Email [email protected]; [email protected]
Purpose: Chromosomal abnormalities represent genomic signatures linked to cancer prognosis and responses to chemotherapy, immunotherapy, and drug resistance. This study aimed to investigate the impact of chromosome copy number variants (CNVs) on the efficacy of tyrosine kinase inhibitors (TKIs) in EGFR-mutated non-small cell lung cancer (NSCLC) patients, as well as its prognostic implications for progression-free survival (PFS) and overall survival (OS) in EGFR wild-type patients.
Methods: A total of 110 patients with advanced NSCLC were enrolled in this study and categorized into EGFR-mutated and wild-type groups. Utilizing next-generation sequencing (NGS) technology, we assessed 24 genes and chromosome CNVs associated with lung cancer pathways in patients’ tissue samples.
Results: Within the EGFR-mutated group, patients with a gain in Chr 1p13.3-p13.1 exhibited poor TKI responses, a high relapse rate, and shortened PFS (P = 0.002). Conversely, EGFR-mutated patients with a gain in 14q31.1-q31.3 demonstrated favorable TKI responses and relatively extended PFS (P = 0.005). Among EGFR wild-type patients, the presence of 7q31.1-q31.31 CNV emerged as an independent factor influencing both PFS and OS (P = 0.013, P = 0.004). Notably, patients with a gain in 7q31.1-q31.31 exhibited prolonged PFS and OS. Additionally, independent prognostic significance for OS in EGFR wild-type patients was observed for CNVs in 9q21.31-q22.2 and 11p11.11-q12.1 regions (P = 0.001). Patients with gains in these regions experienced extended OS, while losses were predictive of poorer outcomes.
Conclusion: Our results suggested that chromosomal copy number variation is a practical indicator for predicting the response of EGFR-targeted therapy and prognosis for NSCLC patients.
Keywords: copy number variations, non-small cell lung cancer, EGFR-TKI response, prognosis
Introduction
Lung cancer stands as the most menacing malignant tumor, jeopardizing both human life and health. Its incidence and fatality rates take the forefront among all malignancies, with over 80% constituting non-small cell lung cancer (NSCLC).1 Despite ongoing enhancement of therapeutic techniques, radiotherapy often falls short in delivering satisfactory efficacy, resulting in a bleak prognosis.2 During the treatment of NSCLC patients, the emergence of activated epidermal growth factor receptor (EGFR) mutations holds the potential to heighten tumor sensitivity to EGFR-tyrosine kinase inhibitors (EGFR-TKIs) like gefitinib, afatinib, and erlotinib.3 Yet, despite the promising initial responses, a majority of EGFR-mutated (EGFR+) patients inevitably experience relapse after 9–13 months of treatment, likely attributed to the development of acquired resistance.4–6 In the case of EGFR+ patients, the evolution of TKI resistance primarily follows two distinct pathways. The first hinges on an EGFR-dependent route involving T790M and EGFR amplification, while the second takes a non-EGFR-dependent trajectory, potentially encompassing MET or HER2 amplification, as well as BRAF and KRAS mutations.7–9 Nevertheless, additional information is still needed to better overcome TKI resistance.
Most of the previous studies have focused on the alteration of single gene.7–9 However, the mechanisms underlying multiple TKI resistance are still needed to be further investigate.10 Currently, several approaches are being explored to address EGFR-TKI resistance. These include the application of next-generation TKIs to overcome resistance to prior-generation TKIs, the use of inhibitors targeting other molecules, and the utilization of immune checkpoint inhibitors. These strategies are applicable not only for assessing the prognosis of EGFR-mutated patients but also for detecting other genetic variants such as ALK, MET, and ROS1.11,12 While next-generation sequencing (NGS) and immune checkpoint inhibitor (ICI) tests can be performed in NSCLC patients, it is important to note that the benefits of targeted therapy and immunotherapy are not universally observed in all patients. Therefore, the selection of appropriate predictive indicators is crucial for accurately predicting the prognosis of a wider range of NSCLC patients before initiating treatment.13,14
Genomic DNA copy number variations (CNVs) can be detected in lung cancer and are valuable for assessing the progression of the disease. They provide insights into gene structure changes, gene expression, and tumor pathogenesis.15–18 Amplification and deletion of chromosomes can activate oncogenes or inactivate tumor suppressor genes.19 Genomic instability, which includes phases of tetraploidization, can lead to asymmetric cell division, chromosome loss, increased tumor heterogeneity, and multidrug resistance.20–22 Thus, CNV patterns are currently utilized as biomarkers for disease diagnosis and prognosis in clinical practice, as well as for drug development.23,24 Nevertheless, to our knowledge, there exists a dearth of pertinent research regarding CNV prediction of TKI responsiveness.25
Here, we conducted next-generation sequencing on tumor tissues obtained from 110 advanced NSCLC patients to assess the predictive value of CNVs for TKI responsiveness and prognosis. The patients were categorized into two groups: those with EGFR mutations (EGFR+) and those without (EGFR−). Our findings identified specific CNV patterns that could predict TKI response in EGFR-mutated patients and significant CNVs associated with the prognosis of EGFR wild-type patients. This study contributes novel insights and reliable biological indicators for predicting TKI response and prognosis in NSCLC patients. The following article adheres to the STARD reporting checklist.
Methods
Patients
Patients with NSCLCs who underwent surgical resection at Nantong First People’s Hospital between October of 2017 and July of 2020 were included in this study and were enrolled according to the following criteria: (i) availability of puncture tissues for obtaining samples and completion of imaging examination resulting in a specific pathological diagnosis, (ii) diagnosed with stage III and IV NSCLCs, (iii) presence of sufficient nucleic acid content in the tissue for NGS detection, and (iv) availability of complete prognostic information meeting the criteria for at least 6 months. Among the enrolled patients, all those with EGFR mutations received treatment with TKIs. Progression-free survival (PFS) was calculated from the surgery date to the date of disease progression or death (Due date), while overall survival (OS) was calculated from the surgery date to the date of death (Due date). The study protocol was approved by the Institutional Review Board of Nantong First People’s Hospital (No. 2017KY198), and all recruited patients provided written informed consent.
Next Generation Sequencing
Genomic DNA was extracted from formalin-fixed paraffin-embedded (FFPE) lung cancer samples using the QIAampDNA FFPE Tissue Kit (product number: 56404, QIAGEN) following the manufacturer’s instructions. The FFPE tissue sections were cut to a thickness of 10 μm, with the tumor cell content exceeding 50%. Qubit dsDNA High Sensitivity (HS) Assay Kit (product number: Q32854, Thermo Fisher Scientific) was used to detect the concentration of DNA (> 300 ng and 10 ng/l). For cases where DNA input was < 50 ng, an additional PCR cycle was incorporated during target enrichment. Subsequently, enzymatic shearing was applied to the extracted DNA, and sequencing libraries were prepared using the TIANSeq DirectFast DNA Library Prep Kit (product number: NG101, TIANGEN Biotech (Beijing) Co., Ltd.). Quantification of nucleic acid was performed using the VAHTS Library Quantification Kit for Illumina (product number: NG101, Nanjing Vazyme Biotech Co., Ltd.) when the Ct value of the library was below 10. The purified sequencing libraries underwent massively parallel sequencing using the Illumina HiSeq Xten platform (Illumina, Inc.), generating 4G of raw sequencing data per sample. 4G sequencing raw data per sample were filtered and aligned to the human reference genome. The NSCLC-panel contained 23 genes of the Wnt pathway and hot-spot regions out of the EGFR, BRAF, MET, HER2, KRAS, and TP53 genes.
Analyses of Gene Copy Number Variation
The Ultrasensitive Chromosomal Aneuploidy Detector (UCAD) pipeline was utilized to determine CNVs. The sequencing reads were aligned to the reference genome (UCSC hg19 assembly) using BWA-MEM (version 0.7.17-r1188). Sequencing coverage for each 200 K bin was computed and subsequently subjected to GC normalization. The median of the sample data was taken as the control samples. The sequencing coverage was further normalized by control samples. Twice Z-tests were employed to normalize both internal and external parameters. The total count of qualified reads within the sample was divided by the count of reads within each specified region. This division aimed to establish the relative proportion of reads within each region. Additionally, the data from normal tissue samples was juxtaposed with data from samples taken from patients with identified tumors. This comparison yielded relative values signifying either an increase or loss within each region of the identified samples. Subsequently, chromosome characteristic values were extracted and summarized.
In this study, all raw sequencing reads were mapped to human reference genome hg19, and genomic coverage was counted by using software samtools mpileup. Data from a total of 110 tumor samples were aggregated. Median counts for chromosome 1–18 were tabulated and then averaged. The cutoff > 1.1 indicated amplification, while the cutoff < 0.9 indicated deletion.
Statistical Analysis
The statistical analysis was conducted using SPSS software (version 18.0, SPSS Inc., Chicago, IL, USA). To assess the significance of associations between variables, appropriate tests were employed: the chi-squared test or Fisher’s exact test, and the Kruskal–Wallis test or Mann–Whitney U-test. The correlation between variables was explored using the Pearson correlation coefficient (R). Survival curves for PFS and OS were plotted using the Kaplan-Meier method, and differences between curves were estimated using the Log rank test. Furthermore, a multivariate analysis was performed using a Cox proportional hazards model to identify independent predictors of survival. The inclusion of proven prognostic factors from the univariate analysis was accomplished using a forced entry method. For each factor, the hazard ratio (HR) and its corresponding 95% confidence interval (CI) were calculated. All statistical tests were two-tailed, and statistical significance was defined as P < 0.05.
Results
Patient Characterization
Detailed characterization of the patient population is depicted in Table 1. Among the 110 advanced NSCLC patients enrolled in this study, most were male (59.1%), and their median age was 67.6 years. Most of the tumors were in the right lung (59.1%) and were adenocarcinoma (87.3%). The initial metastases were intrathoracic metastases (78.2%) and extracranial metastases (83.6%). The CEA was divided into four levels, and 42.7% of CEA levels were less than 10. Overall, EGFR mutations were identified in 43.6% of patients. According to the mutation of EGFR, 110 patients were divided into two groups: EGFR-mutated (EGFR+) group and EGFR wild-type (EGFR-) group. As shown in Table 1, the clinical and pathological factors associated with EGFR mutations were gender (female, P < 0.001), pathological pattern (adenocarcinoma, P < 0.001), metastatic sites (extrathoracic metastasis, P = 0.035), high CEA levels (P = 0.011) and non-smoking status (none, P = 0.001).
 |
Table 1 The Association Between Clinic Pathologic Features and EGFR Mutations |
At the time of analysis, the mean follow-up time for all patients was 17.5 ± 10.5 months (range, 1–34 months), during which 56 patients (50.9%) died, and 92 patients (83.6%) experienced recurrence. Kaplan-Meier survival analysis showed that the EGFR mutation was significantly associated with long PFS (P < 0.0001) and OS (P < 0.0001) (Figure 1).
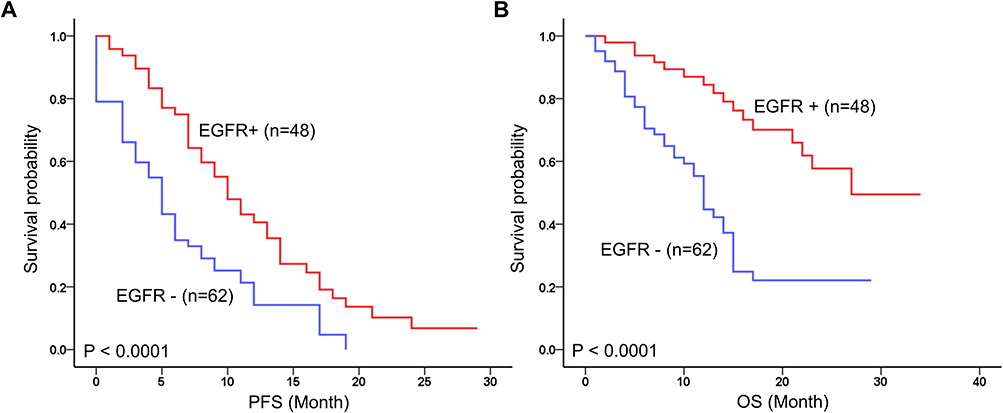 |
Figure 1 Prognostic value of EGFR mutations status in 110 advanced NSCLC. Kaplan-Meier survival curves of EGFR+ for (A) progression-free survival (PFS) and (B) Overall survival (OS). |
CNV Patterns Predicted the TKI Response of EGFR-Mutated NSCLC Patients
As shown in Table 2, among the 48 EGFR-mutated NSCLC patients, 96% (46 patients) had EGFR mutations in either exons 19 (19del) or 21 (L858R). All of them received EGFR-TKI treatment, sometimes followed by chemo or radiotherapy. Among the patients who received EGFR-TKI, 38 patients (79%) were administered the first-generation TKIs (gefitinib/Icotinib), 1 of them (2%) received afatinib, and the other 9 patients (19%) were treated with both gefitinib and osimertinib. All enrolled patients were categorized into three groups based on the PFS (Table 2): short-term (0–6 months, n = 13), medium-term (7–12 months, n = 19), and long-term (> 13 months, n = 16). The analysis revealed no significant differences in age (P = 0.060), gender (P = 0.599), EGFR mutation type (P = 0.286), and EGFR-TKI drugs (P = 0.085) among the different PFS groups.
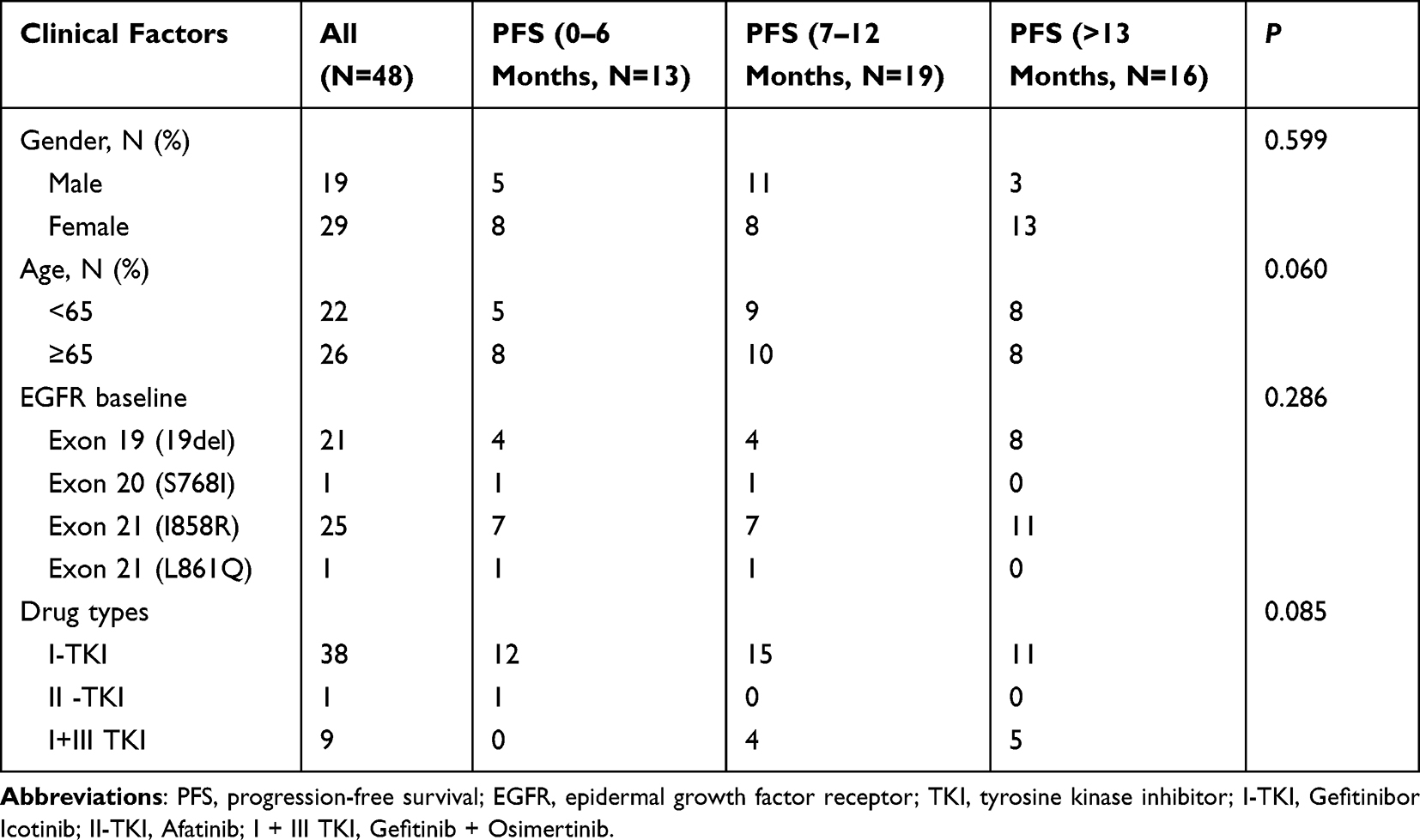 |
Table 2 The Clinical and Pathological Information of EGFR+ Patients |
The respective medians of chromosome 1–18 in EGFR+/EGFR- samples were counted and indicated by a blue line. Our results showed that amplification and deletion occurred in all the chromosomes from 1 to 18. Among all patients, chromosomes 7, 14, and 17 exhibited coverage higher than the normalized average, while chromosome 1 showed normal coverage (Figure 2A). In EGFR wild-type patients, chromosomes 12 and 17 exhibited coverage higher than the normalized average, while chromosome 1 remained normal (Figure 2B). Our observations indicate that patients with EGFR mutations exhibit higher coverage of chromosomes 1 and 4 compared to the wild-type patients. Focal amplifications were detected on Chr7 and Chr14 (Figure 2C). Additionally, the results demonstrate elevated amplification frequencies of chromosomes 1, 7, and 14 in EGFR mutation-positive patients compared to those with wild-type EGFR.
 |
Figure 2 DNA somatic copy numbers of (A) all advanced NSCLC patients; (B) EGFR+ patients; (C) EGFR- patients. Chromosome 1 to 18 is layout from left to right with red and green colors. |
The comprehensive analysis further demonstrated significant changes in chromosome loci between tumors with EGFR mutations and EGFR wild-type tumors. Figure 3 presents a heat-map illustrating the most significant CNVs associated with EGFR mutation. The results showed that the amplification of chromosomes 7p (45.6% vs 19%) and 14q (36.17% vs 15%) were significantly identified in EGFR-mutated patients. Moreover, the amplification of chromosome 9q (8.25% vs 30%) and 12p (10.83% vs 31%) were frequently found in EGFR wild-type patients. Notably, certain CNV regions were significantly observed in EGFR-mutated patients, including 1p13.3-p13.1 gain (P =0.01), 3q27.1-q27.3 loss (P =0.002), 15q15.1-q21.1 loss (P =0.031), 18p11.32-p11.22 loss (P =0.005). Conversely, we also found that 5p14.4-p13.3 loss (P =0.011) and 15q15.1-q21.1 gain (P =0.031) were more frequent in wild-type patients.
 |
Figure 3 Heat-map view of chromosomal changes in EGFR mut and EGFR wild-type patients. Significant copy-gain segments with Z-score larger than 1.5 are marked in red color, Z-score between 1.1–1.5 in yellow, 0.9–1.1 in white, Significant copy-loss with Z-score between 0.5–0.9 in light blue, less than 0.5 in dark blue. |
The univariate analysis of the Cox proportional hazard model demonstrated that 1p13.3-p13.1, 14q31.1-q31.3, and 14q32.11-q32.2 were significantly associated with PFS of EGFR-mutated patients (P = 0.002, P = 0.005, P = 0.022) (Figure 4). Additionally, 1p13.3-p13.1 and 14q13.1-q31.3 were identified as independent risk factors for TKI treatment. Patients with chromosome 1p 13.3-p13.1 amplification exhibited a poor response to TKI (P = 0.005, HR, 3.066, 95% CI, 1.391–6.759). Conversely, patients with chromosome 14q13.1-q31.3 gain displayed a good response to TKI (P = 0.024, HR, 0.400, 95% CI, 0.180–0.888) (Figure 4).
 |
Figure 4 Univariate and multivariate survival analysis for PFS in EGFR-mutated patients. |
CNV Patterns Predicted the PFS and OS for EGFR Wild-Type NSCLC Patients
Among the 62 EGFR wild-type NSCLC patients, 27% (17 patients) had KRAS mutations. Kaplan-Meier survival analysis showed that KRAS mutation was significantly associated with poor PFS (P = 0.020), and the Chr 8p23.3-p23.1 loss was associated with longer PFS (P = 0.007; Figure 5). Chr 7q31.1-q31.31 gain was found to be associated with longer PFS (P = 0.001) and OS (P = 0.016). In multivariate analysis, the Chr 7q31.1-q31.31 and Chr 8p23.3-p23.1 were independent risk factors for PFS of EGFR wild-type patients (Figure 6A). In addition, patients with chromosome Chr 7q31.1-q31.31 gain (P=0.013, HR, 0.113, 95% CI, 0.02–0.625) and Chr 8p23.3-p23.1 loss (P=0.006, HR, 0.131, 95% CI, 0.031–0.563) were found to have longer PFS.
 |
Figure 5 Kaplan-Meier survival curves of (A) KRAS mutation for PFS; (B) Chr8.1 variation for PFS; (C) Chr 7.12 variation for PFS and (D) Chr 7.12 variation for OS. |
 |
Figure 6 Multivariate survival analysis for (A) PFS and (B) OS in EGFR wild-type patients. |
Furthermore, the association of CNV with OS of EGFR wild-type patients was analyzed. Results of cox multivariate regression analysis revealed that the 4p15.1-p14, 7q31.1-q31.31, 9q21.31-q22.2, 10q21.2-q22.1, and 11p11.11-q12.1 were independent risk factors for OS. EGFR wild-type patients with 4p15.1-p14 gain (P=0.001, HR, 20.1, 95% CI, 3.382–119.457) had a poor OS (Figure 6B). Besides, patients with 7q31.1-q31.31 gain (P=0.004, HR, 0.133, 95% CI, 0.034–0.521) and 10q21.2-q22.1 loss (P=0.036, HR, 0.273, 95% CI, 0.081–0.916) had longer OS. Notably, amplifications in 9q21.31-q22.2 and 11p11.11-q12.1 predicted longer OS, while the deletion of these regions predicted poor OS (Figure 6B).
Discussion
In this study, we proposed the application of CNV patterns as a predictor of prognosis in advanced NSCLC patients. Specifically, the 1p and 14q gain assessing TKI efficacy for EGFR+ patients and the 7q and others for EGFR- patients. These findings can help to predict the response of EGFR-TKI treatment and the risk of recurrence for advanced NSCLC patients, potentially enhancing the current therapeutic landscape for NSCLC.
Previous studies have reported that 20–30% of advanced lung cancer patients with EGFR mutations exhibit intrinsic resistance to TKIs, while another subset of patients maintains TKI efficacy for over 2 years.26 Acquired resistance refers to cancer cells developing the ability to withstand the effects of TKIs following initial treatment, often involving new mutations or alterations in genes within the targeted pathways. These changes can lead to reduced drug binding affinity, increased drug efflux, or activation of alternative signaling pathways bypassing the target. Additionally, certain cancer cells may possess inherent genetic alterations or mechanisms that confer innate resistance to TKIs from the outset, rendering them less susceptible to the inhibitory effects of the drugs. Therefore, comprehending the mechanisms underlying EGFR-TKI resistance and identifying early predictive indicators of TKI responsiveness are of paramount importance for enhancing clinical practice. Growing evidence showed that chromosomal abnormalities represent genomic features related to cancer prognosis and response to chemotherapy and immunotherapy.27 It has been previously shown that Chr1 and Chr7 gain are common in patients with lung adenocarcinoma and 8p23.1 loss is common in NSCLC.28–34 Our findings are in congruence with prior research: (i) distinct CNVs exist among patients with EGFR mutation and EGFR wild-type in advanced lung cancer, and (ii) significant focal amplifications were also found on chromosome 1, 7 and 14 in EGFR-mutated patients compared to wild-type patients. Our results also showed that the 1p and 14q are two factors that independently affect PFS, and the 1p13.3-p13.1 gain patients were prone to drug-resistant relapse and poor TKI efficacy, while the 14q31.1-q31.3 gain patients had longer PFS and good TKI efficacy. It has been suggested that the miRNA located at 14q32.31 could potentially serve as a prognostic indicator across various cancers. In a prior investigation of chromosomal variants in lung cancer, Patrick’s study established a correlation between the loss of 14q31.1–32.33 and favorable outcomes in patients with squamous lung cancer across different stages.35 Our results, which showed that the 14q had an impact on prognosis and drug resistance in lung cancer patients, appear do not match that finding, and demonstrate that a number of altered chromosome 1 genes may have subtype- and stage- specificities in lung cancers and could be considered as a biomarker for indicating diagnosis and prognosis.36
Numerous driver oncogenes associated with resistance to EGFR-TKI, chemotherapy or immunotherapy, were previously found to locate in CNV regions.7–9 In our research, the 1p13.3-p13.1 gain was found to affect TKI response and predisposes to drug resistance, while the oncogene NRAS located at 1p13.2, 7q31.3-q31.31gain had better PFS and OS. The MET gene on 7q31.2 and the BRAF gene on 7q34, 9q21.31-q22.2 and 10q21.2-q22.1 are independent factors affecting OS of EGFR wild-type patients. Consistent with this, an acquired sequence variant in the NRAS gene on chromosome 1 was identified in an in vitro TKI-resistant model involving imatinib and nilotinib.37 The emergence of TKI resistance may involve MET or HER2 amplification on chromosome 7 and BRAF mutation.7 Refer to Table 3 for a subset of chromosome 1 and 7 genes associated with TKI resistance.38–40 Besides, other regions were also found to be involved in tumorigenesis and drug resistance, such as NTRK2 located on 9q21.33, PTEN located on 10q23.31, and RET located on 10q11.21. Furthermore, other regions, such as NTRK2 on 9q21.33, PTEN on 10q23.31, and RET on 10q11.21, were implicated in tumorigenesis and drug resistance. Notably, chromosome 7 appears pivotal in NSCLC development and progression. Aberrations involving EGFR, cMET, and BRAF, coupled with chromosomal anomalies, constitute cytogenetic events contributing to distinct molecular signatures in NSCLC patients.32 Genetic targets MET, RET, BRAF, and NTRK2 in lung cancers affect patient prognosis and can be addressed through targeted drugs. NRAS and PTEN mutations represent adverse prognostic indicators, necessitating further investigation into the impact of abnormalities in their chromosomal loci on subsequent clinical treatment.
 |
Table 3 Chromosome 1 and 7 Related TKI Resistance Genes |
NGS serves as a critical adjunct to conventional cytogenetics for comprehensive chromosome profiling in cancers, enabling direct testing of various chromosomal aberrations.36,41,42 In this study, NGS was employed to determine chromosome CNVs in advanced lung cancer tissue samples. Our results not only unveiled chromosomes linked to EGFR variants but also identified chromosomes predictive of TKI response in EGFR-mutated patients. Moreover, we found that specific chromosomal somatic copy number variants (7q, 9q, etc.) could forecast survival in EGFR wild-type patients. Mounting evidence underscores the strong association between CNV abnormalities and tumorigenesis, chemotherapy response, and immunotherapy outcomes. Chromosomal abnormalities, alone or in conjunction with TMB, could potentially serve as a more effective and prevalent immunotherapy biomarker compared to TMB alone.43–46 This study delved into the impact of CNV patterns on predicting response to EGFR-TKI targeted therapy and survival rates in EGFR wild-type patients receiving chemotherapy and immunotherapy. The predictive significance of CNV pattern in EGFR-TKI targeted therapy response holds crucial implications for the development of prospective prognostic indicators for survival assessment and offers valuable insights for prognostic evaluation in NSCLC patients. However, a major limitation of this study is the absence of empirical data to directly support the proposed conclusions. Additionally, the analytical approach is overly simplistic, and the sample size is inadequate. In forthcoming endeavors, we intend to validate the value of CNV markers in predicting EGFR-TKI targeted therapy response and patient prognosis by conducting in vitro or in vivo experiments, increasing the sample size, and incorporating non-malignant control tissues.
Abbreviation
EGFR, Epidermal Growth Factor Receptor; TKI, Tyrosine Kinase Inhibitor; PFS, Progression-Free Survival; OS, Overall Survival; NGS, Next Generation Sequencing; NSCLC, Non-Small Cell Lung Cancer; CNV, Copy Number Variation; UCAD, Ultrasensitive Chromosomal Aneuploidy Detector; TMB, Tumor Mutant Burden.
Data Sharing Statement
All data that can prove the conclusion of this article are included in the article.
Ethical Approval
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The trial was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The protocol was approved by the institutional Review Board of Nantong First People’s Hospital (NO.2017KY198).
Consent to Participate
All included patients got approved information materials and have provided a written consent.
Consent for Publication
All authors have agreed to publish the current document.
Acknowledgments
Haiyan He and Hang Ma are co-first authors for this study. The authors acknowledge the Suzhou Biomedical Research & Development Center to provide excellent NGS detection technology.
Funding
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20180267, BK20191207).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376(22):2109–2121. doi:10.1056/NEJMoa1616288
3. Reck M, Rabe KF. Precision diagnosis and treatment for advanced non-small-cell lung cancer. N Engl J Med. 2017;377(9):849–861. doi:10.1056/NEJMra1703413
4. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi:10.1056/NEJMra0802714
5. Yu D, Zhao W, Vallega KA, Sun SY. Managing acquired resistance to third-generation EGFR tyrosine kinase inhibitors through co-targeting MEK/ERK signaling. Lung Cancer. 2021;12:1–10. doi:10.2147/LCTT.S293902
6. Shah R, Lester JF. Tyrosine kinase inhibitors for the treatment of EGFR mutation-positive non-small-cell lung cancer: a clash of the generations. Clin Lung Cancer. 2020;21(3):e216–e228. doi:10.1016/j.cllc.2019.12.003
7. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17(11):637–658. doi:10.1038/nrc.2017.84
8. Murtuza A, Bulbul A, Shen JP, et al. Novel third-generation EGFR tyrosine kinase inhibitors and strategies to overcome therapeutic resistance in lung cancer. Cancer Res. 2019;79(4):689–698. doi:10.1158/0008-5472.CAN-18-1281
9. Piper-Vallillo AJ, Sequist LV, Piotrowska Z. Emerging treatment paradigms for EGFR-mutant lung cancers progressing on osimertinib: a review. J Clin Oncol. 2020;38(25):2926–2936. doi:10.1200/JCO.19.03123
10. Le X, Puri S, Negrao MV, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin Cancer Res. 2018;24(24):6195–6203. doi:10.1158/1078-0432.CCR-18-1542
11. Proto C, Lo Russo G, Corrao G, et al. Treatment in EGFR-mutated non-small cell lung cancer: how to block the receptor and overcome resistance mechanisms. Tumori. 2017;103(4):325–337. doi:10.5301/tj.5000663
12. Fang W, Ma Y, Yin JC, et al. Comprehensive genomic profiling identifies novel genetic predictors of response to anti-PD-(L)1 therapies in non-small cell lung cancer. Clin Cancer Res. 2019;25(16):5015–5026. doi:10.1158/1078-0432.CCR-19-0585
13. Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–269. doi:10.1038/nm.4040
14. Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. doi:10.1056/NEJMoa1411817
15. Sequist LV, Rolfe L, Allen AR. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;373(6):578–579. doi:10.1056/NEJMc1506831
16. Sequist LV, Soria JC, Camidge DR. Update to rociletinib data with the recist confirmed response rate. N Engl J Med. 2016;374(23):2296–2297. doi:10.1056/NEJMc1602688
17. Bowcock AM. Invited review DNA copy number changes as diagnostic tools for lung cancer. Thorax. 2014;69(5):495–496. doi:10.1136/thoraxjnl-2013-204681
18. Liu L, Bai X, Wang J, et al. Combination of TMB and CNA stratifies prognostic and predictive responses to immunotherapy across Metastatic Cancer. Clin Cancer Res. 2019;25(24):7413–7423. doi:10.1158/1078-0432.CCR-19-0558
19. Cosenza M, Krämer A. Centrosome amplification, chromosomal instability and cancer: mechanistic, clinical and therapeutic issues. Chromosome Res. 2016;24(1):105–126. doi:10.1007/s10577-015-9505-5
20. Xiang L, Fu X, Wang X, et al. A potential biomarker of combination of tumor mutation burden and copy number alteration for efficacy of Immunotherapy in KRAS-mutant advanced lung adenocarcinoma. Front Oncol. 2020;10:559896. doi:10.3389/fonc.2020.559896
21. Kachouie NN, Shutaywi M, Christiani DC. Discriminant analysis of lung cancer using nonlinear clustering of copy numbers. Cancer Invest. 2020;38(2):102–112. doi:10.1080/07357907.2020.1719501
22. Kuznetsova AY, Seget K, Moeller GK, et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle. 2015;14(17):2810–2820. doi:10.1080/15384101.2015.1068482
23. Qiu M, Xia W, Chen R, et al. The circular RNA circPRKCI promotes tumor growth in lung adenocarcinoma. Cancer Res. 2018;78(11):2839–2851. doi:10.1158/0008-5472.CAN-17-2808
24. Dou T, Wang J, Han C, Shao X, Zhang J, Lu W. Cellular uptake and transport characteristics of chitosan modified nanoparticles in Caco-2 cell monolayers. Int J Biol Macromol. 2019;138(10):791–799. doi:10.1016/j.ijbiomac.2019.07.168
25. Cheng Y, Ma L, Liu Y, et al. Comprehensive characterization and clinical impact of concomitant genomic alterations in EGFR-mutant NSCLCs treated with EGFR kinase inhibitors. Lung Cancer. 2020;145:63–70. doi:10.1016/j.lungcan.2020.04.004
26. Santoni-Rugiu E, Melchior L, Urbanska E, et al. EGFRIntrinsic resistance to EGFR-tyrosine kinase inhibitors in -mutant non-small cell lung cancer: differences and similarities with acquired resistance. Cancers. 2019;11(7):923. doi:10.3390/cancers11070923
27. Kou F, Wu L, Ren X, Yang L. Chromosome abnormalities: new insights into their clinical significance in cancer. Mol Ther Oncolytics. 2020;17:562–570. doi:10.1016/j.omto.2020.05.010
28. Zhang VC, Wang J, Shao GG, et al. Comprehensive genomic and immunological characterization of Chinese non-small cell lung cancer patients. Nat Commun. 2019;10(1):1772. doi:10.1038/s41467-019-09762-1
29. Chen H, Carrot-Zhang J, Zhao Y, et al. Genomic and immune profiling of pre-invasive lung adenocarcinoma. Nat Commun. 2019;10(1):5472. doi:10.1038/s41467-019-13460-3
30. Bao L, Zhang Y, Wang J, et al. Variations of chromosome 2 gene expressions among patients with lung cancer or non-cancer. Cell Biol Toxicol. 2016;32(5):419–435. doi:10.1007/s10565-016-9343-z
31. Yuan S, Yu SL, Chen HY, et al. Clustered genomic alterations in chromosome 7p dictate outcomes and targeted treatment responses of lung adenocarcinoma with EGFR-activating mutations. J Clin Oncol. 2011;29(25):3435–3442. doi:10.1200/JCO.2011.35.3979
32. Zhu L, Liang J, Xia B, et al. Identification of somatic copy number variations in plasma cell free DNA correlating with intrinsic resistances to EGFR targeted therapy in T790M negative non-small cell lung cancer. J Thorac Dis. 2020;12(3):883–892. doi:10.21037/jtd.2019.12.97
33. Taylor AM, Shih J, Ha G, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33(4):676–689 e673. doi:10.1016/j.ccell.2018.03.007
34. Tan X, Banerjee P, Pham EA, et al. PI4KIIIbeta is a therapeutic target in chromosome 1q-amplified lung adenocarcinoma. Sci Transl Med. 2020;12(527). doi:10.1126/scitranslmed.aax3772
35. Micke P, Edlund K, Holmberg L, et al. Gene copy number aberrations are associated with survival in histologic subgroups of non-small cell lung cancer. J Thorac Oncol. 2011;6(11):1833–1840. doi:10.1097/JTO.0b013e3182295917
36. Hata T, Suenaga M, Marchionni L, et al. Genome-wide somatic copy number alterations and mutations in high-grade pancreatic intraepithelial neoplasia. Am J Pathol. 2018;188(7):1723–1733. doi:10.1016/j.ajpath.2018.03.012
37. Kaehler M, Osteresch P, Kunstner A, et al. Clonal evolution in tyrosine kinase inhibitor-resistance: lessons from in vitro-models. Front Oncol. 2023;13:1200897. doi:10.3389/fonc.2023.1200897
38. Wang HL, Liu YC, Long MP, Zheng C, Yang JH. Blocking ROR1 enhances the roles of erlotinib in lung adenocarcinoma cell lines. Oncol Lett. 2019;18(3):2977–2984. doi:10.3892/ol.2019.10643
39. Koba H, Kimura H, Yoneda T, et al. NOTCH alteration in EGFR-mutated lung adenocarcinoma leads to histological small-cell carcinoma transformation under EGFR-TKI treatment. Transl Lung Cancer Res. 2021;10(11):4161–4173. doi:10.21037/tlcr-21-536
40. Morgillo F, Amendola G, Della Corte CM, et al. Dual MET and SMO negative modulators overcome resistance to EGFR inhibitors in human nonsmall cell lung cancer. J Med Chem. 2017;60(17):7447–7458. doi:10.1021/acs.jmedchem.7b00794
41. Zhang Y, Wang H, Wang J, et al. Global analysis of chromosome 1 genes among patients with lung adenocarcinoma, squamous carcinoma, large-cell carcinoma, small-cell carcinoma, or non-cancer. Cancer Metastasis Rev. 2015;34(2):249–264. doi:10.1007/s10555-015-9558-0
42. Li G, Li W-R, Jin Y-G, Jie -Q-Q, Wang C-Y, Wu L. Tetrandrine attenuated doxorubicin-induced acute cardiac injury in mice. Biomed Res Int. 2020;2020(1):1–10.
43. Consortium ITP-CAoWG. Pan-cancer analysis of whole genomes. Nature. 2020;578(7793):82–93. doi:10.1038/s41586-020-1969-6
44. Hieronymus H, Schultz N, Gopalan A, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci USA. 2014;111(30):11139–11144. doi:10.1073/pnas.1411446111
45. Gerstung M, Jolly C, Leshchiner I, et al. The evolutionary history of 2658 cancers. Nature. 2020;578(7793):122–128. doi:10.1038/s41586-019-1907-7
46. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355(6322). doi:10.1126/science.aaf8399
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.