Back to Archived Journals » Metalloproteinases In Medicine » Volume 3
Chondrocyte responses to neurovascular peptides, cytokines, and a 3D environment: focus on ADAMs
Authors Bevan D, Baker N, Goldring M, Donell S, Gavrilovic J ![]()
Received 8 March 2016
Accepted for publication 11 May 2016
Published 1 September 2016 Volume 2016:3 Pages 63—74
DOI https://doi.org/10.2147/MNM.S108003
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yoshifumi Itoh
Damon Bevan,1 Natasha Baker,1 Mary B Goldring,2 Simon T Donell,3 Jelena Gavrilovic1
1School of Biological Sciences, University of East Anglia, Norwich, UK; 2Hospital for Special Surgery, Research Division, Weill Cornell Medical College, New York, NY, USA; 3Department of Orthopaedics, Norfolk and Norwich University Hospital, Norwich, UK
Abstract: Chondrocyte exposure to inflammatory stimuli in several arthritic conditions, including osteoarthritis, results in the well-characterized induction of extracellular matrix degrading proteinases, notably members of a disintegrin and metalloproteinase (ADAM) with thrombospondin domains and matrix metalloproteinase families. Here we briefly review the less-studied ADAM family of proteinases in chondrocyte and cartilage biology. Following damage, cartilage is exposed to neurovascular peptides, and in this study we hypothesized that substance P and bradykinin, alongside inflammatory cytokines, may modulate chondrocyte steady-state messenger RNA levels for the proteolytic ADAM family members as well as for key cytokines and neuropeptides. We compared chondrocytes cultured in both two-dimensional and three-dimensional (3D) environments and found that 3D culture generally resulted in repression of expression of the genes under investigation, with the exception of anti-inflammatory interleukin 10 which was markedly upregulated in a 3D environment. Substance P and bradykinin had little effect on ADAM family expression, but further investigation revealed that a combination of bradykinin and cytokines led to enhanced expression of ADAM28 and a synergistic upregulation of interleukin 6, also observed under hypoxic conditions. Overall these data reveal wider chondrocyte responses to neurovascular peptides which may have an impact in an osteoarthritis context.
Keywords: chondrocyte, ADAM, metalloproteinase, cytokine; bradykinin
Introduction
The cartilage environment
Chondrocytes are surrounded by an extensive extracellular matrix comprising type II collagen and aggrecan as well as a large number of other collagens, proteoglycans, and proteins together providing a remarkable, unique environment. Chondrocytes lack the capacity to drive efficient repair and thus in osteoarthritis (OA) articular cartilage is irreversibly damaged. While earlier studies viewed OA as a wear-and-tear condition, there is now a consensus, reviewed by Konitten et al,1 that chondrocytes both respond to and may themselves produce proinflammatory cytokines which can result in cartilage breakdown through modulating the expression of metalloproteinases including a disintegrin and metalloproteinase with thrombospondin domains (ADAMTS) enzymes, matrix metalloproteinases (MMPs), and serine proteinases.2
Interplay of pain-related neurovascular peptides with cartilage biology
OA is increasingly viewed as a disease of the whole joint, including cartilage, synovium, bone, and, in knee OA, the infrapatellar fat pad.3,4 Adult cartilage is avascular and the resulting hypoxic environment promotes chondrocyte differentiation,5–7 in addition to maintaining the chondrocyte phenotype.8 Cartilage is also aneural, but damaged cartilage is recognized as a potentially permissive environment for neurovascular infiltration.9–11 Sensory nerve fibers thus accompany new blood microvessels that grow into damaged cartilage in OA.
While effects of several cytokines and chemokines on neuronal excitation have been explored,12 as yet the effects of neurovascular factors on chondrocyte gene expression and function remain less studied. Several neuropeptides, which play key roles in transmission of pain signals from the periphery to the central nervous system, are upregulated in joint disease. For example, substance P is elevated in the synovial fluid in both OA and rheumatoid arthritis (RA) joints, and the expression of substance P is particularly associated with more active OA.13 Im et al13 also demonstrated that substance P and its receptor NK1-R are expressed at higher levels in human articular cartilage from OA patients than controls. Substance P is produced in nerve fibers of the intracellular fat pad where a role in inflammation and cartilage destruction has been suggested.4
Kinins, including bradykinin, are often associated with acute and chronic inflammation, and, more recently, attention has turned to potential roles in OA. Bradykinin is a vasodilator with established roles in inflammation and a number of studies show that it is elevated in OA synovial fluid.14 An antagonist of bradykinin has been shown to reduce pain levels in a small study of patients with very painful OA.15 Functional bradykinin receptors are expressed in human chondrocytes with evidence of release of inflammatory cytokines interleukin (IL)6 and IL8 following bradykinin stimulation.16 The synovial compartment, which is itself innervated, may also respond to neuropeptide release and bradykinin is important in synovitis in OA.17 A more recent study showed that an antagonist of bradykinin receptor B (BDKRB)1 provides protection against joint injury in the rat anterior cruciate ligament model.18 In addition, a polymorphism in BDKRB2 is associated with both higher risk and higher severity of OA in one study.19
As well as roles in regulation of gene expression, neuropeptides may also modify aspects of chondrocyte biology including cell proliferation and migration. Chondrocytes can migrate in vitro and in vivo in development and possibly in OA.20 Opolka et al21 demonstrated that substance P can induce proliferation of murine newborn costal chondrocytes and elevate cell adhesion and focal contact formation, indicating novel roles in cell-matrix interactions.
ADAMs in chondrocytes and cartilage
A disintegrin and metalloproteinase (ADAM) family members with proteolytic activity may play roles in chondrocyte biology and OA. While regulation of both MMPs and ADAMTS proteinases has been studied extensively in chondrocytes, less attention has been given to the ADAM family. Several ADAMTS proteinases can degrade proteoglycans22 and ADAMTS4 and 5 are known to play key roles in proteoglycan degradation in cartilage with human ADAMTS5 showing 1,000-fold greater aggrecan degradative capacity than ADAMTS4 in biochemical analyses.23 Both enzymes can degrade aggrecan in human cartilage explants22 and recently developed antibodies specifically block ADAMTS5 aggrecan fragment release,24 in one case to a far greater extent than possible through blockade of ADAMTS4.25 In addition adamts5 (but not adamts4) null mice are protected from experimentally induced OA.26,27
ADAMs have a structure comprising a propeptide, catalytic, disintegrin, cysteine-rich, transmembrane, and cytoplasmic domains and are furin-activated (furin cleavage site located between pro- and catalytic domains).28 As transmembrane enzymes, ADAMs are well placed for their extensive shedding functions, where cell adhesion molecules, growth factors, and their receptors among many other proteins are cleaved and thus shed from the cell surface.29 Certain MMPs share shedding functions (including, for example, MMP9), but this seems to be a function mainly restricted to ADAMs. Through their disintegrin domains, several ADAMs can bind to integrins, a function also observed with certain MMPs (even though in MMPs disintegrin domains are absent),30 but ADAMTS enzymes, despite the presence of a disintegrin domain, seem to lack this function.28 Several proteolytic ADAMs are expressed in chondrocytes or OA cartilage.31,32 Here, we briefly review findings regarding ADAMs with proteolytic activity where expression and/or function relating to chondrocyte biology and/or OA have been reported.
ADAM8
Expression of ADAM8 has been observed in cartilage during mouse development,33 and expression of an inactive ADAM8 mutant results in protection against murine experimental inflammatory arthritis.34 In addition, ADAM8 is expressed in OA cartilage, and when isolated from chondrocyte culture medium, this protease is able to cleave fibronectin.35 This is an important finding, since fibronectin fragments are found in damaged joint tissues and are able to induce MMP gene expression and aggrecan degradation in cartilage.36
ADAM9
Flannery et al37 showed the downregulation of ADAM9 expression in IL1-treated porcine chondrocytes in three-dimensional (3D) compared to two-dimensional (2D) culture,37 and this proteinase is expressed during human mesenchymal cell differentiation along the chondrocyte lineage.38 ADAM9 expression is upregulated in end-stage OA cartilage.23
ADAM10
Earlier studies revealed increased ADAM10 mRNA levels in IL1-treated bovine chondrocytes,37 and enhanced ADAM10 protein levels were observed in OA compared to healthy cartilage and in IL1-stimulated bovine nasal cartilage.39 More recent observations indicate that ADAM10 cleavage of the cell adhesion molecule, N-cadherin, has an important role in chondroprogenitor cell–cell interactions leading to condensation and differentiation.40
ADAM12
This ADAM has been associated with OA in genetic studies,41,42 and ADAM12 mRNA and protein (by immunocytochemistry) levels are upregulated in end-stage OA cartilage.22,23 ADAM12 plays a role in chondrocyte proliferation through cleavage of insulin-like growth factor binding protein (IGFBP)-5,22 which may be important in attempted repair by chondrocytes in OA.
ADAM15
IL1 was shown to enhance the expression of ADAM15 in monolayer cultures of porcine chondrocytes37 and ADAM15 (also known as metargidin) is upregulated in end-stage OA cartilage.43 Interestingly, ADAM15 null mice developed a more accelerated OA,44 suggesting that ADAM15 is protective against the development of OA. Later studies revealed that ADAM15 is anti-apoptotic in OA chondrocytes and promotes cell adhesion to type II collagen when overexpressed in chondrocytes.45 The anti-apoptotic mechanism involves ADAM15 cytoplasmic tail interaction with focal adhesion kinase (FAK) resulting in enhanced FAK phosphorylation and promotion of cell survival under genotoxic conditions.46
ADAM17
Due to its role in shedding of tumor necrosis factor alpha (TNFα), along with cleavage of other relevant molecules, ADAM17 has been highly associated with RA.47 Soluble IL6-receptor (sIL6-R) is often generated through ADAM17 cell surface cleavage, enabling sIL6-R to increase IL6-mediated cartilage damage. A very recent study has shown that ADAM17-mediated shedding of IL6-R is blocked by secreted frizzled-related protein 3 (sFRP3) and the rare OA double-mutant variant of sFRP3 loses this inhibitory capacity.48 This could result in greater release of sIL6-R and hence enhanced IL6 signaling leading to further cartilage destruction.
ADAM19
Expression of this ADAM is elevated during in vitro differentiation of human chondrocytes.38 ADAM19 can shed TNFα from the cell surface,49 though this has not as yet been demonstrated in chondrocytes.
ADAM28
ADAM28 is not observed in undamaged human cartilage; however, it is expressed in end-stage OA cartilage.23 More recently, a proteomic approach found ADAM28 in the pericellular matrix of chondrocytes in articular cartilage.50 Since ADAM28 can degrade IGFBP-3,51 it is possible that this proteinase plays a role in chondrocyte proliferation, as has been demonstrated for breast cancer cells.52 Another function of ADAM28 appears to be in the shedding of TNFα from the cell surface, demonstrated in monocytes and which may be of relevance in arthritic conditions.53 ADAM28 can be regulated by 9-cis retinoic acid and peroxisome proliferator-activated receptor gamma ligands in differentiated monocytes,54 as well as by 9-cis retinoic acid in chondrocytes,55 indicating the involvement of potentially similar pathways across cell types. ADAM28 also degrades human chondrocyte proteoglycans,55 such that co-culture of bovine nasal cartilage discs with ADAM28-expressing COS-7 cells results in greater proteoglycan release.55
Neurovascular peptides and metalloproteinase expression
Studies on neurovascular peptides substance P and bradykinin in an arthritic context have to date focused on MMP or ADAMTS expression. Miller et al12 showed that treatment of cartilage fragments with substance P in combination with fibroblast growth factor 2 results in cartilage degradation. In addition, exposure of human chondrocytes to increasing concentrations of substance P leads to enhanced expression of several MMPs.13 Taken together, these data suggest that substance P may be an important regulator of catabolism of articular cartilage in OA. Bradykinin levels correlate with glycosoaminoglycan fragments in synovial fluid of OA patients,56 which could suggest a link to matrix degradation. Bradykinin is also known for its ability to induce the activation of the ADAM17-EGFR transactivation pathway57 and to indirectly enhance MMP production in a number of other cell types, but this has not been explored in chondrocytes.
The potential interplay between neurovascular peptides released in the OA joint and ADAM proteinase expression remains little explored. We hypothesized that a 3D culture environment may regulate chondrocyte responses to cytokines and neurovascular peptides. Specifically in the experiments reported here, we have surveyed the expression of genes encoding proteolytic ADAM family members, tissue inhibitors of metalloproteinases (TIMPs) and selected cytokines/neuropeptides and receptors in chondrocytes in response to the cytokines IL1α and oncostatin M (OSM), substance P, and bradykinin in both 2D and 3D culture environments.
Materials and methods
Materials
Unless otherwise stated chemical reagents were purchased from Sigma Aldrich, Gillingham, UK, culture media from Gibco Thermo Fisher Scientific, UK, and tissue culture plastics from Nunc Thermo Scientific, UK.
Cell culture model
C28I/2 chondrocytes were cultured essentially as previously described58 in DMEM with 10% fetal calf serum (FCS) at a cell density of 190,000 cells/24 well in 2D monolayer culture or in a 3D model with culture of chondrocytes over polyHEMA,58 for a total of 96 hours. During the final 24 hours, the cells were stimulated with a combination of IL1α (10 ng/mL) and OSM (5 ng/mL), bradykinin (1 µM), or substance P (100 µM) in DMEM with 1% FCS. Where indicated cells were also exposed to a hypoxic environment (1% oxygen in a Billins chamber) for 24 hours prior to and during treatments.
RNA extraction, TaqMan low-density array, and qRT-PCR analysis
Following cell lysis, RNA was extracted using the RNeasy plus mini kit (Qiagen) according to the manufacturer’s instructions. Total RNA (1 μg) was reverse transcribed with 1 μg random primer oligonucleotides (Invitrogen, Paisley, UK) and 200 units of M-MLV reverse transcriptase (Promega, Southampton, UK). cDNA samples were analyzed on custom-designed TaqMan low-density array (TLDA) plates (Applied Biosystems, Warrington, UK) to detect the expression of the genes encoding ADAMs 8, 9, 10, 12, 15, 17, 19, 20, 21, 22, 28, 33, TIMPs 1–4, the cytokines IL6 and IL10, the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), neuropeptide Y (NPY), and bradykinin receptors and the housekeeping gene 18S. (ADAM22 was included in this initial TLDA screen as, while it lacks proteolytic activity, ADAM22 expression is highly upregulated in OA cartilage compared to healthy cartilage, at least at the mRNA level23). Prior to TLDA analysis, samples were analyzed on geNorm (Primer Design; Qiagen, Manchester, UK) where 18S ribosomal RNA was found to be the least-regulated housekeeping gene (of the 12 explored) for all treatment regimens (data not shown). TLDA analysis was performed in triplicate for each condition. A total of 150 ng of RNA was loaded into each port of the TLDA plates and analyzed in an ABI 7900HT Instrument (Applied Biosystems).
QRT-PCR using an ABI prism 7500 sequence detection system (Applied Biosystems) was performed as previously described59 with primer probe sets from Applied Biosystems. Genes were analyzed using predesigned Applied Biosystems inventorized primer probe sets (Table 1) or primer probe sets designed using Primer Express 1.0 Software (Applied Biosystems; Table 1). Primers were designed spanning exon–intron boundaries to avoid amplification of genomic DNA and were tested for specificity by BLAST analysis. 18S ribosomal RNA was used as an endogenous control to normalize for differences in the amount of total RNA in each sample (18S rRNA primers and probe were purchased from Applied Bioscience).
 | Table 1 Inventorized primer set codes for TLDA analysis and for further experimentation (ADAM28) Abbreviations: ADAM, a disintegrin and metalloproteinase; TLDA, TaqMan low-density array; TIMP, tissue inhibitors of metalloproteinase; IL, interleukin; BDNF, brain-derived neurotrophic factor; NGF, nerve growth factor; NPY, neuropeptide Y; BDKRB, bradykinin receptor B. |
Results
Effects of culture in 3D and of cytokine stimulation
Of the 12 ADAMs whose steady-state mRNA expression was determined, eleven showed modulation of expression in chondrocytes by TLDA analysis (Figure 1; Table 2). 3D culture resulted in the downregulation of steady-state mRNA levels of seven of the ADAMs expressed in chondrocytes, with ADAM9, ADAM20, and ADAM33 mRNAs showing at least a twofold suppression (Figure 1; Table 2). TIMP2 mRNA was also significantly repressed, but other TIMPs were unaffected by 3D culture. Cytokine stimulation resulted in the upregulation of expression of several ADAM mRNAs in 2D, but in 3D culture, this was restricted to ADAMs12 and 28 (Table 2). Interestingly, ADAM10 and ADAM15 mRNAs were no longer modulated by cytokines when cells were cultured in 3D and that of ADAM19 and ADAM21 was now repressed (Table 2). TIMP1 mRNA was increased by cytokines in both 2D and 3D cultures, whereas that of TIMP3 was repressed by cytokines in both culture environments (Table 2).
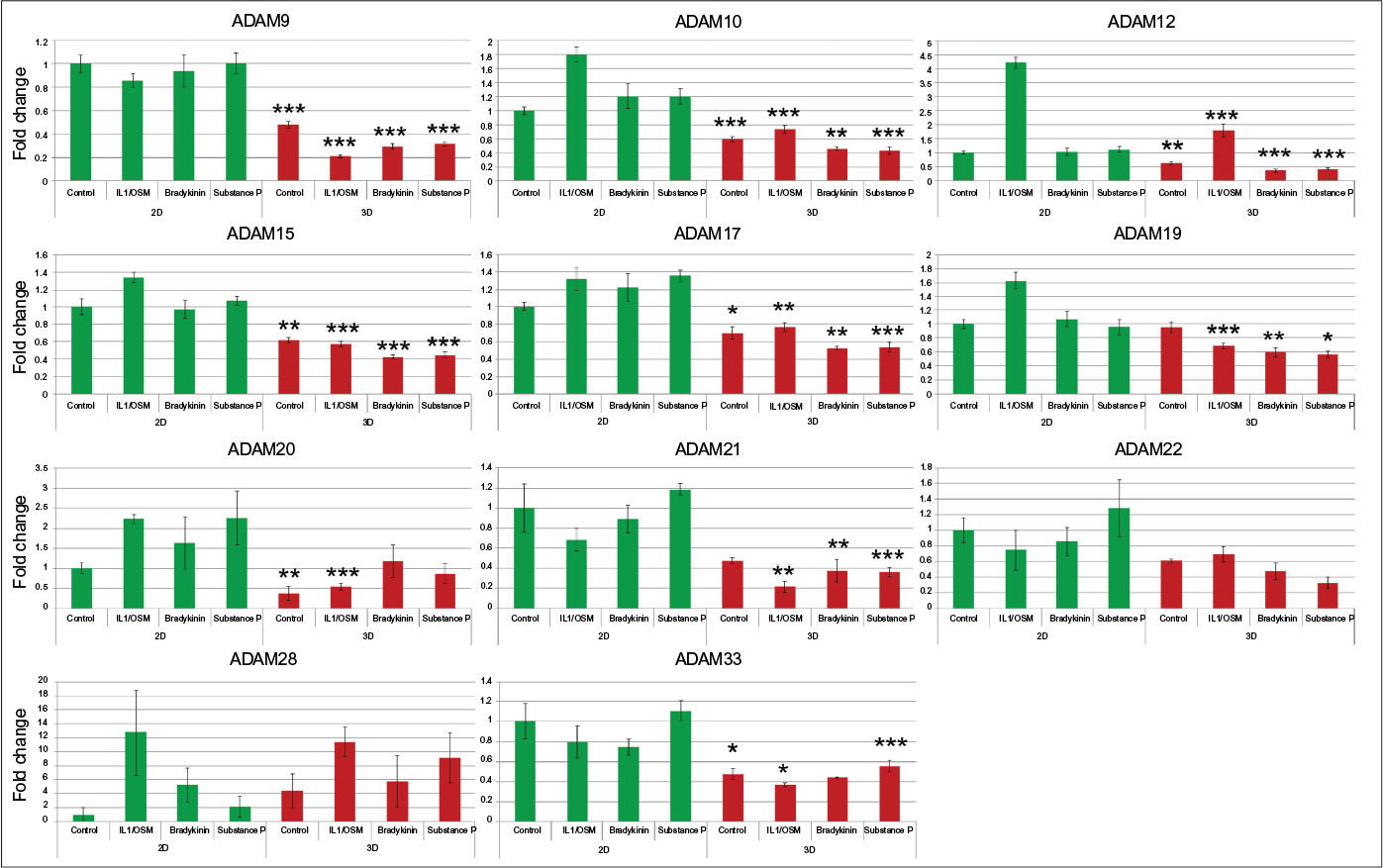 | Figure 1 Culture in 3D in general suppresses ADAM steady-state mRNA levels, irrespective of the stimulus added. Notes: C28I/2 chondrocytes were cultured either in 2D (green) or 3D (red) with the indicated stimuli, and following RNA extraction and reverse transcription, TLDA analysis was performed. Each bar represents the mean ± SEM for triplicate samples following normalization to 18S RNA. For this fold-change analysis, control (unstimulated) cells were set as 1. Comparisons between the levels in 2D and 3D for control conditions and for each stimulus are indicated. *P<0.05; **P<0.02; ***P<0.01. Abbreviations: 2D, two dimensional; 3D, three dimensional; TLDA, TaqMan low-density array; SEM, standard error of mean; ADAM, a disintegrin and metalloproteinase; IL, interleukin; OSM, oncostatin M. |
 | Table 2 Modulation of steady-state mRNA levels for ADAMs, TIMPs, cytokines, and neurovascular factors under the indicated conditions, determined by TLDA analysis Notes: Significant fold changes are indicated compared to unstimulated cells in either 2D or 3D culture (or comparison between 2D and 3D for unstimulated cells only). ↔ indicates no change. Abbreviations: IL1/OSM, interleukin 1/oncostatin M; subP, substance P; BK, bradykinin; ADAM, a disintegrin and metalloproteinase; TIMP, tissue inhibitor of metalloproteinase; IL6, interleukin 6; IL10, interleukin 10; BDNF, brain-derived neurotrophic factor; NGF, nerve growth factor; NPY, neuropeptide Y; BDKRB1, bradykinin receptor B1; BDKRB2, bradykinin receptor B2; TLDA, TaqMan low-density array. |
We surveyed a small number of receptors for neuropeptides as well as neurotrophins and cytokines. It is interesting to note that cytokines upregulated the expression of bradykinin receptors 1 and 2 both in 2D and in 3D cultures of chondrocytes. In addition IL6 and IL10 mRNAs were strongly upregulated by cytokines in both 2D and 3D (Table 2). In contrast, cytokines repressed the expression of BDNF, NPY, and tachykinin 3 in both culture environments.
Effects of substance P and bradykinin
Substance P treatment slightly upregulated ADAM17 mRNA (1.4-fold) but bradykinin did not alter ADAM expression in 2D culture. In chondrocytes in 3D culture, substance P and bradykinin both significantly repressed the gene expression of several ADAMs (Figure 1; Table 2). Substance P repressed TIMPs 2 and 3 whereas bradykinin repressed TIMPs 2 and 4, respectively (Figure 1; Table 2). Steady-state mRNA levels of few other genes were modulated by either substance P or bradykinin (Figure 1; Table 2).
Effects of co-stimulation with neurovascular peptides and cytokines under normoxic and hypoxic conditions
In general, little overlap of induction of gene expression was observed under different stimuli. The small number of genes regulated by substance P or bradykinin alone led us to explore the effects of co-stimulation with cytokines and these neurovascular peptides. We were particularly interested to explore further the regulation of ADAM28 since this proteinase has recently been found in the pericellular matrix of chondrocytes.50 We pursued its expression alongside that of IL6, since this cytokine was upregulated by several stimuli. We exposed cells in 3D culture to cytokines or neuropeptides both alone and in combination with cytokines under normoxic and hypoxic conditions.
In 3D culture under normoxic conditions, we confirmed the upregulation of expression of ADAM28 in the presence of cytokines. Interestingly expression was further enhanced by co-stimulation with bradykinin (Figure 2A). A further TLDA survey of the other proteolytic ADAMs did not reveal any effect of bradykinin on their expression in the presence of cytokines (results not shown). Preliminary investigations revealed that exposure of chondrocytes to hypoxic conditions significantly upregulated ADAM28 mRNA by 3.7-fold and in the presence of cytokines alone no further regulation was observed. However, under hypoxic conditions co-stimulation with bradykinin led to further upregulation of ADAM28 mRNA (Figure 2A). Cytokine induction of IL6 mRNA was confirmed in 3D culture under normoxic conditions and an apparently synergistic induction with bradykinin co-stimulation was observed (Figure 2B). In contrast to ADAM28, the cytokine-induced expression of IL6 mRNA was suppressed under hypoxic conditions (2.4-fold) but synergistic induction with co-stimulation with bradykinin was again observed (Figure 2B). Expression of TNFα was also explored but no synergy with bradykinin was observed (data not shown).
 | Figure 2 Confirmation of co-stimulatory effects of bradykinin and cytokines in chondrocytes cultured in 3D. Notes: (A) ADAM28 steady-state mRNA levels are elevated by a combination of cytokines and bradykinin under normoxic conditions (red bars) and are maintained under hypoxic conditions (blue). (B) IL6 expression is synergistically enhanced by the combination of cytokines and bradykinin under normoxic conditions (red) and overall responses are reduced under hypoxic conditions (blue). Steady-state mRNA levels for ADAM28 and IL6 were determined by qRT-PCR. For this fold-change analysis, control (unstimulated) cells were set as 1. Each bar represents the mean ± SEM for triplicate samples following normalization to 18S RNA. *P<0.05; **P<0.02; ***P<0.01. Abbreviations: ADAM, a disintegrin and metalloproteinase; IL, interleukin; OSM, oncostatin M |
Discussion
Neurovascular mediators including substance P and bradykinin are released into the synovial fluid in both OA and RA and are involved in pain perception. Here we show that the cellular environment has profound effects on constitutive ADAM expression as well as on the modulating effects of soluble mediators, either cytokines or neurovascular peptides.
3D suspension culture results in downregulation of mRNA levels for many genes studied
Overall, we observed that the culture of chondrocytes in 3D significantly repressed steady-state mRNA levels of seven ADAMs including ADAM9, as well as IL6, TIMP2, BDNF, and NGF. Downregulation of ADAM9 mRNA in chondrocytes has been reported previously in 3D cultures.37 Collagen I downregulation was observed in primary chondrocytes in a 3D culture model similar to that used here 60 (indicative of chondrocyte redifferentiation) and we observed similar results in our cultures (data not shown). Repression of IL6 mRNA in 3D collagen gel culture (compared to culture on a thin coating of collagen) was observed previously in ovarian tumor cells.61 Future studies will reveal whether similar mechanisms are involved in downregulation of steady-state mRNA levels for the 11 genes affected by 3D culture in our studies. Uniquely among the genes explored in our study, the anti-inflammatory cytokine, IL10, was upregulated in cells cultured in 3D. In relation to this finding, it is interesting to note that fibroblasts cultured in a 3D matrix of enhanced stiffness in a wound healing context express greater levels of IL10.62 Taken together, our data on IL6 and IL10 suggest that chondrocytes in a 3D environment have an overall anti-inflammatory phenotype.
Effects of cytokines and neurovascular peptides
We exposed chondrocytes to a standard combination of IL1α and OSM since these cytokines have previously been shown to be present in synovial fluid of OA patients63 and have long been known to act synergistically to induce complete breakdown of both bovine nasal cartilage64 and human OA cartilage.65
ADAMs
In 2D culture, cytokine treatment upregulated the steady-state mRNA levels for six ADAMs, several of which were previously reported to be expressed in chondrocytes or OA cartilage.31,32 In our 3D cultures, we no longer saw cytokine-mediated upregulation of ADAM10, ADAM15, and ADAM19, and this could reflect the overall suppressive nature of the 3D environment. ADAM12 mRNA was elevated by cytokines in both 2D and 3D culture, with overall expression levels approximately halved in 3D compared to monolayer culture. Recently an ADAM12 SNP associated with severity of knee OA was reported.42 ADAM12 (among other ADAMs) can shed the cell surface receptor LRP-1 which mediates endocytosis of TIMP3 and aggrecan-degrading ADAMTS5.66 Thus, alterations in ADAM12 levels could indirectly have consequences on cartilage degradation.
In our initial survey, ADAM28 showed a trend toward increased expression following cytokine treatment in 3D culture (confirmed in our later experiment) and overall expression levels were not repressed in 3D, in contrast to other ADAMs. Cytokines regulate ADAM28 in monocytic cells53 and ADAM28 expression is elevated in peripheral blood mononuclear cells in Kashin–Beck disease, characterized by chondrocyte necrosis.67 We saw a trend toward increase in expression with bradykinin treatment alone, but this did not reach significance.
TIMP3
TIMP3 expression was repressed following cytokine treatment, an effect seen in synovial cells68 and observed in endothelial cells by us in previous studies.69 Substance P also repressed TIMP3 mRNA levels in 3D culture, suggesting a potentially important role for this neuropeptide in cartilage homeostasis. TIMP3 deletion in mice results in the degradation of collagen and aggrecan over time and development of OA-like symptoms,70 and very recently, a TIMP3 polymorphism associated with the severity of knee OA was reported,71 although any functional consequences remain unknown. As well as inhibiting many MPs involved in cartilage degradation (for example, ADAMTS5 and MMP13), TIMP3 also inhibits ADAM17 and other sheddases. Thus the repression of TIMP3 mRNA observed here may also have profound consequences on the cell surface ecology of chondrocytes.
Cytokines
The increase in steady-state mRNA levels for anti-inflammatory IL10 seen in response to cytokines in both 2D and 3D may reflect an attempt to counteract the damaging effects of proinflammatory cytokine stimulation. Exogenous IL10 can protect against cartilage damage72 and IL10 levels are upregulated in OA and postulated to be a prorepair response.73 Cytokine upregulation of IL6 is in agreement with previous findings in OSM-treated mouse articular chondrocytes, for example, where a hierarchy of responses suggests that OSM may be a better therapeutic target than IL6.74 Cytokines repressed neurotrophin BDNF expression as well as that of NPY and tachykinin 3 (both in 2D and in 3D). Perhaps of relevance is the finding that levels of BDNF are enhanced after exercise in patients with knee OA and may have anti-inflammatory benefits with corresponding improved pain scores.75
Bradykinin and cytokine co-stimulation: effects on ADAM28 and IL6 expression
Enhanced expression of ADAM28 in the presence of cytokines and bradykinin may be due to the increased bradykinin receptor expression observed in the presence of cytokines alone. The synergistic increase in IL6 expression with cytokine and bradykinin co-treatment in normoxia is reminiscent of that seen in an early study in gingival fibroblasts where IL6 protein production was elevated by stimulation with bradykinin and IL1 or TNFα.76 Similar synergistic effects of bradykinin and IL1β on the expression of COX-2 and PGE-2 in synoviocytes have been observed.77 These authors demonstrated that this effect was mediated by bradykinin receptors, using the inhibitor fasitibant, and was dependent on the NFkB pathway. Future studies will determine mechanisms underpinning the upregulation of steady-state mRNA levels for ADAM28 and IL6 seen with bradykinin and cytokine co-stimulation in chondrocytes.
In our preliminary study, hypoxia exerted a positive effect on both constitutive and cytokine-induced ADAM28 mRNA levels but was suppressive for IL6 (and for the expression of several other genes; data not shown). Several studies have explored IL6 gene expression and/or protein secretion under various levels of hypoxia with 3% oxygen resulting in upregulation of IL6 secretion in human synovial cells,78 but 5% oxygen leading to downregulation in placental cells.79 Previous work showed that hypoxia (1% oxygen as used in our study) promotes chondrocyte differentiation7 and perhaps the repression of IL6 under hypoxic conditions is anti-inflammatory and prorepair.
Overall our experiments demonstrate interesting and novel regulation of steady-state mRNA levels for ADAMs and cytokines in C28/I2 chondrocytes, a readily available immortalized human culture model. Future experiments will reveal mechanisms underpinning our observations as well as any parallels in primary human chondrocytes and cartilage.
Limitations of the study
While we have demonstrated novel effects of neurovascular peptides and hypoxic conditions on ADAM and cytokine steady-state mRNA levels, we have not confirmed effects at the protein level nor have we explored functional consequences of any changes in expression. Future studies following confirmation of protein levels would include functional assessment of enzyme activity with relevant substrates and gene silencing approaches to determine, for example, ADAM28’s role in proteoglycan degradation/cell proliferation in chondrocytes under bradykinin and cytokine stimulation.
Conclusion
This study reveals the novel regulation of ADAM proteinases at the mRNA level by a 3D culture environment which also modulates cytokine responses in chondrocytes. In addition, steady-state mRNA levels of ADAM28 and IL6 are elevated by bradykinin, in the presence of cytokines, which may represent an important modulator of the chondrocyte milieu. These findings pave the way for future studies of the impact of bradykinin and downstream pathways on cartilage function under inflammatory stress.
Acknowledgments
This work was supported by Action Arthritis and by the Research Sustainability Fund (Norfolk and Norwich University Hospital).
Disclosure
The authors report no conflicts of interest in this work.
References
Konttinen YT, Sillat T, Barreto G, Ainola M, Nordstrom DC. Osteoarthritis as an autoinflammatory disease caused by chondrocyte-mediated inflammatory responses. Arthritis Rheum. 2012;64(3):613–616. | ||
Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta. 2012;1824(1):133–145. | ||
Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64(6):1697–1707. | ||
Clockaerts S, Bastiaansen-Jenniskens YM, Runhaar J, et al. The infrapatellar fat pad should be considered as an active osteoarthritic joint tissue: a narrative review. Osteoarthritis Cartilage. 2010;18(7):876–882. | ||
Thoms BL, Murphy CL. Inhibition of hypoxia-inducible factor-targeting prolyl hydroxylase domain-containing protein 2 (PHD2) enhances matrix synthesis by human chondrocytes. J Biol Chem. 2010;285(27):20472–20480. | ||
Strobel S, Loparic M, Wendt D, et al. Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Res Ther. 2010;12(2):R34. | ||
Lafont JE, Talma S, Hopfgarten C, Murphy CL. Hypoxia promotes the differentiated human articular chondrocyte phenotype through SOX9-dependent and -independent pathways. J Biol Chem. 2008;283(8):4778–4786. | ||
Murphy CL, Thoms BL, Vaghjiani RJ, Lafont JE. Hypoxia. HIF-mediated articular chondrocyte function: prospects for cartilage repair. Arthritis Res Ther. 2009;11(1):213. | ||
Suri S, Gill SE, Massena de Camin S, Wilson D, McWilliams DF, Walsh DA. Neurovascular invasion at the osteochondral junction and in osteophytes in osteoarthritis. Ann Rheum Dis. 2007;66(11):1423–1428. | ||
Walsh DA, McWilliams DF, Turley MJ, et al. Angiogenesis and nerve growth factor at the osteochondral junction in rheumatoid arthritis and osteoarthritis. Rheumatology (Oxford). 2010;49(10):1852–1861. | ||
Grassel SG. The role of peripheral nerve fibers and their neurotransmitters in cartilage and bone physiology and pathophysiology. Arthritis Res Ther. 2014;16(6):485. | ||
Miller RE, Miller RJ, Malfait AM. Osteoarthritis joint pain: the cytokine connection. Cytokine. 2014;70(2):185–193. | ||
Im HJ, Li X, Muddasani P, et al. Basic fibroblast growth factor accelerates matrix degradation via a neuro-endocrine pathway in human adult articular chondrocytes. J Cell Physiol. 2008;215(2):452–463. | ||
Meini S, Maggi CA. Knee osteoarthritis: a role for bradykinin? Inflamm Res. 2008;57(8):351–361. | ||
Song IH, Althoff CE, Hermann KG, et al. Contrast-enhanced ultrasound in monitoring the efficacy of a bradykinin receptor 2 antagonist in painful knee osteoarthritis compared with MRI. Ann Rheum Dis. 2009;68(1):75–83. | ||
Meini S, Cucchi P, Catalani C, Bellucci F, Giuliani S, Maggi CA. Bradykinin and B(2) receptor antagonism in rat and human articular chondrocytes. Br J Pharmacol. 2011;162(3):611–622. | ||
Nishimura M, Segami N, Kaneyama K, Suzuki T, Miyamaru M. Relationships between pain-related mediators and both synovitis and joint pain in patients with internal derangements and osteoarthritis of the temporomandibular joint. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;94(3):328–332. | ||
Kaufman GN, Zaouter C, Valteau B, Sirois P, Moldovan F. Nociceptive tolerance is improved by bradykinin receptor B1 antagonism and joint morphology is protected by both endothelin type A and bradykinin receptor B1 antagonism in a surgical model of osteoarthritis. Arthritis Res Ther. 2011;13(3):R76. | ||
Chen S, Zhou Y, Li J, Shan LQ, Fan QY. The effect of bradykinin B2 receptor polymorphisms on the susceptibility and severity of osteoarthritis in a Chinese cohort. J Biomed Biotechnol. 2012;2012:597637. | ||
Morales TI. Chondrocyte moves: clever strategies? Osteoarthritis Cartilage. 2007;15(8):861–871. | ||
Opolka A, Straub RH, Pasoldt A, Grifka J, Grassel S. Substance P and norepinephrine modulate murine chondrocyte proliferation and apoptosis. Arthritis Rheum. 2012;64(3):729–739. | ||
Stanton H, Melrose J, Little CB, Fosang AJ. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim Biophys Acta. 2011;1812(12):1616–1629. | ||
Gendron C, Kashiwagi M, Lim NH, et al. Proteolytic activities of human ADAMTS-5: comparative studies with ADAMTS-4. J Biol Chem. 2007;282(25):18294–18306. | ||
Santamaria S, Yamamoto K, Botkjaer K, et al. Antibody-based exosite inhibitors of ADAMTS-5 (aggrecanase-2). Biochem J. 2015;471(3):391–401. | ||
Larkin J, Lohr TA, Elefante L, et al. Translational development of an ADAMTS-5 antibody for osteoarthritis disease modification. Osteoarthritis Cartilage. 2015;23(8):1254–1266. | ||
Stanton H, Rogerson FM, East CJ, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434(7033):648–652. | ||
Glasson SS, Askew R, Sheppard B, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005;434(7033):644–648. | ||
Shiomi T, Lemaitre V, D’Armiento J, Okada Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol Int. 2010;60(7):477–496. | ||
Dreymueller D, Pruessmeyer J, Groth E, Ludwig A. The role of ADAM-mediated shedding in vascular biology. Eur J Cell Biol. 2012;91(6–7):472–485. | ||
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. | ||
Okada A, Mochizuki S, Yatabe T, et al. ADAM-12 (meltrin alpha) is involved in chondrocyte proliferation via cleavage of insulin-like growth factor binding protein 5 in osteoarthritic cartilage. Arthritis Rheum. 2008;58(3):778–789. | ||
Swingler TE, Waters JG, Davidson RK, et al. Degradome expression profiling in human articular cartilage. Arthritis Res Ther. 2009;11(3):R96. | ||
Kelly K, Hutchinson G, Nebenius-Oosthuizen D, et al. Metalloprotease-disintegrin ADAM8: expression analysis and targeted deletion in mice. Dev Dyn. 2005;232(1):221–231. | ||
Zack MD, Melton MA, Stock JL, et al. Reduced incidence and severity of experimental autoimmune arthritis in mice expressing catalytically inactive A disintegrin and metalloproteinase 8 (ADAM8). Clin Exp Immunol. 2009;158(2):246–256. | ||
Zack MD, Malfait AM, Skepner AP, et al. ADAM-8 isolated from human osteoarthritic chondrocytes cleaves fibronectin at Ala(271). Arthritis Rheum. 2009;60(9):2704–2713. | ||
Stanton H, Ung L, Fosang AJ. The 45 kDa collagen-binding fragment of fibronectin induces matrix metalloproteinase-13 synthesis by chondrocytes and aggrecan degradation by aggrecanases. Biochem J. 2002;364(Pt 1):181–190. | ||
Flannery CR, Little CB, Caterson B, Hughes CE. Effects of culture conditions and exposure to catabolic stimulators (IL-1 and retinoic acid) on the expression of matrix metalloproteinases (MMPs) and disintegrin metalloproteinases (ADAMs) by articular cartilage chondrocytes. Matrix Biol. 1999;18(3):225–237. | ||
Djouad F, Delorme B, Maurice M, et al. Microenvironmental changes during differentiation of mesenchymal stem cells towards chondrocytes. Arthritis Res Ther. 2007;9(2):R33. | ||
Chubinskaya S, Mikhail R, Deutsch A, Tindal MH. ADAM-10 protein is present in human articular cartilage primarily in the membrane-bound form and is upregulated in osteoarthritis and in response to IL-1alpha in bovine nasal cartilage. J Histochem Cytochem. 2001;49(9):1165–1176. | ||
Nakazora S, Matsumine A, Iino T, et al. The cleavage of N-cadherin is essential for chondrocyte differentiation. Biochem Biophys Res Commun. 2010;400(4):493–499. | ||
Valdes AM, Hart DJ, Jones KA, et al. Association study of candidate genes for the prevalence and progression of knee osteoarthritis. Arthritis Rheum. 2004;50(8):2497–2507. | ||
Wang L, Guo L, Tian F, Hao R, Yang T. Analysis of single nucleotide polymorphisms within ADAM12 and risk of knee osteoarthritis in a Chinese Han population. Biomed Res Int. 2015;2015:518643. | ||
Bohm BB, Aigner T, Gehrsitz A, Blobel CP, Kalden JR, Burkhardt H. Up-regulation of MDC15 (metargidin) messenger RNA in human osteoarthritic cartilage. Arthritis Rheum. 1999;42(9):1946–1950. | ||
Bohm BB, Aigner T, Roy B, Brodie TA, Blobel CP, Burkhardt H. Homeostatic effects of the metalloproteinase disintegrin ADAM15 in degenerative cartilage remodeling. Arthritis Rheum. 2005;52(4):1100–1109. | ||
Bohm B, Hess S, Krause K, et al. ADAM15 exerts an antiapoptotic effect on osteoarthritic chondrocytes via up-regulation of the X-linked inhibitor of apoptosis. Arthritis Rheum. 2010;62(5):1372–1382. | ||
Fried D, Bohm BB, Krause K, Burkhardt H. ADAM15 protein amplifies focal adhesion kinase phosphorylation under genotoxic stress conditions. J Biol Chem. 2012;287(25):21214–21223. | ||
Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45(2):146–169. | ||
Oldefest M, Dusterhoft S, Desel C, et al. Secreted Frizzled-related protein 3 (sFRP3)-mediated suppression of interleukin-6 receptor release by A disintegrin and metalloprotease 17 (ADAM17) is abrogated in the osteoarthritis-associated rare double variant of sFRP3. Biochem J. 2015;468(3):507–518. | ||
Zheng Y, Saftig P, Hartmann D, Blobel C. Evaluation of the contribution of different ADAMs to tumor necrosis factor alpha (TNFalpha) shedding and of the function of the TNFalpha ectodomain in ensuring selective stimulated shedding by the TNFalpha convertase (TACE/ADAM17). J Biol Chem. 2004;279(41):42898–42906. | ||
Zhang Z, Jin W, Beckett J, Otto T, Moed B. A proteomic approach for identification and localization of the pericellular components of chondrocytes. Histochem Cell Biol. 2011;136(2):153–162. | ||
Mochizuki S, Shimoda M, Shiomi T, Fujii Y, Okada Y. ADAM28 is activated by MMP-7 (matrilysin-1) and cleaves insulin-like growth factor binding protein-3. Biochem Biophys Res Commun. 2004;315(1):79–84. | ||
Mitsui Y, Mochizuki S, Kodama T, et al. ADAM28 is overexpressed in human breast carcinomas: implications for carcinoma cell proliferation through cleavage of insulin-like growth factor binding protein-3. Cancer Res. 2006;66(20):9913–9920. | ||
Jowett JB, Okada Y, Leedman PJ, et al. ADAM28 is elevated in humans with the metabolic syndrome and is a novel sheddase of human tumour necrosis factor-alpha. Immunol Cell Biol. 2012;90(10):966–973. | ||
Worley JR, Baugh MD, Hughes DA, et al. Metalloproteinase expression in PMA-stimulated THP-1 cells. Effects of peroxisome proliferator-activated receptor-gamma (PPAR gamma) agonists and 9-cis-retinoic acid. J Biol Chem. 2003;278(51):51340–51346. | ||
Hikichi Y, Yoshimura K, Takigawa M. All-trans retinoic acid-induced ADAM28 degrades proteoglycans in human chondrocytes. Biochem Biophys Res Commun. 2009;386(2):294–299. | ||
Bellucci F, Meini S, Cucchi P, et al. Synovial fluid levels of bradykinin correlate with biochemical markers for cartilage degradation and inflammation in knee osteoarthritis. Osteoarthritis Cartilage. 2013;21(11):1774–1780. | ||
Schafer B, Marg B, Gschwind A, Ullrich A. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J Biol Chem. 2004;279(46):47929–47938. | ||
Baker N, Sharpe P, Culley K, et al. Dual regulation of metalloproteinase expression in chondrocytes by Wnt-1-inducible signaling pathway protein 3/CCN6. Arthritis Rheum. 2012;64(7):2289–2299. | ||
Murray MY, Birkland TP, Howe JD, et al. Macrophage migration and invasion is regulated by MMP10 expression. PLoS One. 2013;8(5):e63555. | ||
Stokes DG, Liu G, Dharmavaram R, Hawkins D, Piera-Velazquez S, Jimenez SA. Regulation of type-II collagen gene expression during human chondrocyte de-differentiation and recovery of chondrocyte-specific phenotype in culture involves Sry-type high-mobility-group box (SOX) transcription factors. Biochem J. 2001;360(Pt 2):461–470. | ||
Barbolina MV, Adley BP, Kelly DL, et al. Downregulation of connective tissue growth factor by three-dimensional matrix enhances ovarian carcinoma cell invasion. Int J Cancer. 2009;125(4):816–825. | ||
Branco da Cunha C, Klumpers DD, Li WA, et al. Influence of the stiffness of three-dimensional alginate/collagen-I interpenetrating networks on fibroblast biology. Biomaterials. 2014;35(32):8927–8936. | ||
Tsuchida AI, Beekhuizen M, t Hart MC, et al. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res Ther. 2014;16(5):441. | ||
Cawston TE, Ellis AJ, Humm G, Lean E, Ward D, Curry V. Interleukin-1 and oncostatin M in combination promote the release of collagen fragments from bovine nasal cartilage in culture. Biochem Biophys Res Commun. 1995;215(1):377–385. | ||
Litherland GJ, Hui W, Elias MS, et al. Glycogen synthase kinase 3 inhibition stimulates human cartilage destruction and exacerbates murine osteoarthritis. Arthritis Rheumatol. 2014;66(8):2175–2187. | ||
Etique N, Verzeaux L, Dedieu S, Emonard H. LRP-1: a checkpoint for the extracellular matrix proteolysis. Biomed Res Int. 2013;2013:152163. | ||
Wang S, Guo X, Wu XM, Lammi MJ. Genome-wide gene expression analysis suggests an important role of suppressed immunity in pathogenesis of Kashin-Beck disease. PLoS One. 2012;7(1):e28439. | ||
Gatsios P, Haubeck HD, Van de Leur E, et al. Oncostatin M differentially regulates tissue inhibitors of metalloproteinases TIMP-1 and TIMP-3 gene expression in human synovial lining cells. Eur J Biochem. 1996;241(1):56–63. | ||
Singh RJ, Mason JC, Lidington EA, et al. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc Res. 2005;67(1):39–49. | ||
Sahebjam S, Khokha R, Mort JS. Increased collagen and aggrecan degradation with age in the joints of Timp3(-/-) mice. Arthritis Rheum. 2007;56(3):905–909. | ||
Su SL, Yang HY, Lee HS, et al. Gene-gene interactions between TGF-beta/Smad3 signalling pathway polymorphisms affect susceptibility to knee osteoarthritis. BMJ Open. 2015;5(6):e007931. | ||
van Meegeren ME, Roosendaal G, Jansen NW, et al. IL-4 alone and in combination with IL-10 protects against blood-induced cartilage damage. Osteoarthritis Cartilage. 2012;20(7):764–772. | ||
Wojdasiewicz P, Poniatowski LA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014;2014:561459. | ||
Liu X, Liu R, Croker BA, Lawlor KE, Smyth GK, Wicks IP. Distinctive pro-inflammatory gene signatures induced in articular chondrocytes by oncostatin M and IL-6 are regulated by Suppressor of Cytokine Signaling-3. Osteoarthritis Cartilage. 2015;23(10):1743–1754. | ||
Gomes WF, Lacerda AC, Mendonca VA, et al. Effect of exercise on the plasma BDNF levels in elderly women with knee osteoarthritis. Rheumatol Int. 2014;34(6):841–846. | ||
Modeer T, Anduren I, Yucel-Lindberg T. Bradykinin synergistically stimulates interleukin 6 production in human gingival fibroblasts challenged with interleukin 1 or tumour necrosis factor alpha. Cytokine. 1998;10(1):26–31. | ||
Meini S, Cucchi P, Tinti L, et al. Fasitibant prevents the bradykinin and interleukin 1beta synergism on prostaglandin E(2) release and cyclooxygenase 2 expression in human fibroblast-like synoviocytes. Naunyn Schmiedebergs Arch Pharmacol. 2012;385(8):777–786. | ||
Gao W, McCormick J, Connolly M, Balogh E, Veale DJ, Fearon U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann Rheum Dis. 2015;74(6):1275–1283. | ||
Shirasuna K, Shimamura N, Seno K, et al. Moderate hypoxia down-regulates interleukin-6 secretion and TLR4 expression in human Sw.71 placental cells. Cell Physiol Biochem. 2015;36(6):2149–2160. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.