Back to Journals » International Journal of Nanomedicine » Volume 14
Cholesterol-rich lipid-mediated nanoparticles boost of transfection efficiency, utilized for gene editing by CRISPR-Cas9
Authors Hosseini ES, Nikkhah M ![]() , Hosseinkhani S
, Hosseinkhani S
Received 21 December 2018
Accepted for publication 16 April 2019
Published 11 June 2019 Volume 2019:14 Pages 4353—4366
DOI https://doi.org/10.2147/IJN.S199104
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Elaheh Sadat Hosseini,1 Maryam Nikkhah,1 Saman Hosseinkhani2
1Department of Nanobiotechnology, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, Iran; 2Department of Biochemistry, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, Iran
Purpose: Gene therapy has become a promising remedy to treat disease by modifying the person’s genes. The therapeutic potential of related tools such as CRISPR-Cas9 depends on the efficiency of delivery to the targeted cells. Numerous transfection reagents have been designed and lots of efforts have been devoted to develop carriers for this purpose. Therefore, the aim of the present study was to develop novel cholesterol-rich lipid-based nanoparticles to enhance transfection efficiency and serum stability.
Materials and methods: We constructed two-, three- and four-component cationic liposomes (CLs) to evaluate the combined effect of cholesterol domain and DOPE (dioleoyl phosphatidylethanolamine), a fusogenic lipid, and the PEG (polyethylene glycol) moiety location inside or outside of the cholesterol domain on transfection efficiency and other properties of the particle. Lipoplex formation and pDNA (plasmid DNA) entrapment were assessed by gel retardation assay at different N/P ratios (3, 5, 7). Physicochemical characteristics, cytotoxicity, serum stability and endosomal escape capability of the lipoplexes were studied and transfection potential was measured by firefly luciferase assay. Next, HEK293 cell line stably expressing GFP was utilized to demonstrate the editing of a reporter through Cas9 and sgRNA plasmids delivery by the selected CL formula, which showed the highest transfection efficiency.
Results: Among the designed CLs, the four-component formula [DOTAP (1,2-dioleoyl-3-trimethylammoniumpropane)/DOPE/cholesterol/Chol-PEG (cholesterol-polyethylene glycol)] showed the highest rate of transfection at N/P 3. Finally, transfection of Cas9/sgRNA by this formulation at N/P 3 resulted in 39% gene-editing efficiency to knockout GFP reporter. The results also show that this CL with no cytotoxicity effect can totally protect the plasmids from enzymatic degradation in serum.
Conclusion: The novel PEGylated cholesterol domain lipoplex providing serum stability, higher transfection efficiency and endosomal release can be used for in vivo Cas9/sgRNA delivery and other future gene-therapy applications.
Keywords: cationic liposomes, cholesterol domains, PEGylation, Cas9/sgRNA delivery
Introduction
Gene-based therapy largely depends on efficient transfection into the target cells with maximum efficacy and minimal toxicity which originates from the vector essence. In spite of the importance of modified viruses in efficient and precise gene delivery, these vectors can cause severe immunological responses and are potentially carcinogenic. Non-viral vectors including cationic lipids, polymers, dendrimers, and peptides are promising vectors of overcoming this limitation which offer potential routes for compacting DNA for targeted delivery.1–5 Although, the non-viral vectors show reduced transfection efficiency compared to their viral counterpart some of their characteristics like biocompatibility, non-immunogenicity and potential for large-scale production make these compounds increasingly attractive for modern therapy.
Clustered regularly interspaced short palindromic repeat-CRISPR-associated protein (CRISPR-Cas9) systems, found in nature as microbial adaptive immune system, have been repurposed into a programmable platform for precision gene editing, giving rise to unprecedented opportunities for basic research in life sciences as well as great perspectives in treatment of disease.6–8 CRISPR-Cas9 functions as a ribonucleoprotein (RNP) endonuclease formed between a Cas9 protein and a guide RNA molecule. The guide RNA (gRNA) contains a constant sequence, which interacts with the Cas9 protein, and a variable sequence, which anneals to a complementary region on the target DNA molecule. When the guide RNA anneals to the target sequence, Cas9 introduces a double-stranded break (DSB) in the bound DNA.9 Once double strand break happens, either the non-homologous end joining (NHEJ) or homology directed repair (HDR) pathway will occur. The NHEJ repair pathway is often associated with the insertion/deletion (InDel) generation at the break site leading to premature stop codons within the open reading frame (ORF) and frameshift in the target gene, typically causing a “knock out.” In contrast to NHEJ, HDR as an error-free repair mechanism leading to the correct sequence insertion precisely which causes a “knock in” in the target site. Generally, the rate of HDR occurrence in the cells is much less than NHEJ.10 Compared to previously used tools for gene editing including ZFN and TALEN, CRISPR-Cas9 is a newly developed one but holds high promise for human gene therapy because of the encouraging results due to complex diseases such as cancer, blood disorders, blindness, AIDS, cystic fibrosis, muscular dystrophy and Huntington’s.11 In spite of applicable attributations of CRISPR-Cas9 complex, some challenges hamper its potential in the clinic such as a lack of safe, efficient and specific delivery carrier. Gene therapy with adeno-associated viruses (AAVs) is currently the most advanced methodology for delivering Cas9 in vivo. However, developing Cas9 therapeutics based on AAV delivery is challenging because a large fraction of the human population has pre-existing immunity towards AAV, making them ineligible for AAV-based therapies. Moreover, AAV-based Cas9 delivery has the potential to cause significant off-target genomic damage due to the sustained expression of Cas9.12 Nanoparticles are currently being developed for many therapeutic applications including gene delivery.13 Cationic liposomes (CLs) have always been among the most encouraging candidates with a broad utilization in pDNA, siRNA and mRNA delivery.14–16 As stated by our previous studies, multicomponent (MC) lipoplexes have emerged as encouraging transfection agents. We depicted that pDNA was transfected by three-component lipoplexes including DDAB/DPPC/Cholesterol with 10% cationic lipid more efficient than binary complexes. However, it was still low compared to lipofectamine 2000.17 Afterwards, in another study, we demonstrated a high number of long-chain helper lipid components incorporation in the DDAB/Cholesterol resulted in pDNA entrapment increase. But the highest transfection efficiency was acquired when DSPC or DPPC integrated to the DDAB/DOPE/Cholesterol.18
In this stuy, we prepared two-, three- and four-component CLs to assess their gene transfer abiliy to the HEK293 cells. Since serum instability is the problem associated with the CLs, two types of PEG-linked lipids, DSPE-PEG2000 and PEG linked Cholesterol (Chol-PEG), were used for making the particles PEGylated.19 In fact, the surface of the liposomes was designed by integration of Chol-PEG in cholestrol domains and DSPE-PEG in non-cholestrol domains. Furthermore, DOPE role in endosomal escape and rapid endosomal trafficking was evaluated. Thus, we hypothesized that the impact of Chol-PEG integration in the cholesterol domain coupled with DOPE maximizes transfection efficiency and serum stability. First, a luciferase-expressing plasmid (pGL3) was deliverd to the HEK293 cells by (PEGylated) liposomes. Next, the formulation with the highest transfection was selected for Cas9 and sgRNA plasmid delivery to a stably expressing EGFP cell line to knock out the integrated EGFP in AAVS1 locus (Figure 1).
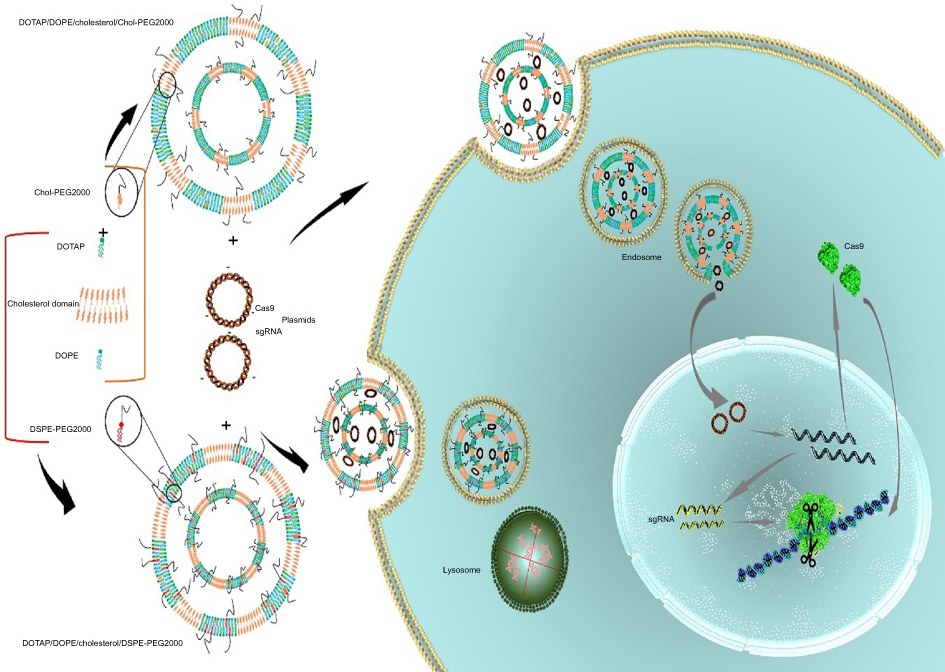 | Figure 1 Schematic illustration of the Chol-rich lipid-mediated nanoparticle carrying Cas9/sgRNA plasmids.Notes: Encapsulation of Cas9 and sgRNA plasmids in DOTAP/DOPE/Cholesterol/Chol-PEG and DOTAP/DOPE/Cholesterol/DSPE-PEG liposomes has happened via electrostatic interactions between the positively charged cationic lipid (DOTAP) and the negatively charged pDNA. By the lipoplex formation, transfection was performed by which the complexes entered the cells through endocytosis. Entrapped pDNAs in DOTAP/DOPE/Cholesterol/Chol-PEG escaped from the endosome while the ones in DOTAP/DOPE/Cholesterol/DSPE-PEG were kept in the endosome and degraded by lysosomal nucleases. Released pDNAs were transferred to the nucleus; after transcription and translation, the ribonucleoprotein complex of Cas9/sgRNA was formed and the chromosome was scanned for the target locus to introduce the double-strand cut for editing. |
Materials and methods
Materials
All chemicals were purchased from Sigma-Aldrich Inc. (St Louis, MO, USA) unless otherwise stated. DOTAP (1,2-dioleoyl-3-trimethylammoniumpropane) and DOPE (dioleoylphosphatidylethanolamine) have been kindly provided by Professor Mahmoud Reza Jafari (Mashhad University of Medical Sciences, Mashhad, Iran). D-luciferin potassium salt was purchased from Resem (Resem, the Netherlands) and adenosine-5´triphosphate (ATP) were obtained from Sigma Aldrich. 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) was purchased from Avanti Polar lipids Inc. (Sigma Aldrich Inc.) MeO-PEG2000-Cholesterol was synthesized using the modified procedure reported by Zhao et al.20 All other chemicals were commercial analytical grade which were used without further purification. Dulbecco’s modified eagle medium (DMEM), fetal bovine serum (FBS), sodium pyruvate, penicillin streptomycin (PenStrep®), Trypsine-DTA and Opti-MEM were purchased from Gibco (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA).
Liposome preparation
Two and three components of CLs including DOTAP/DOPE (1:1), DOTAP/Cholesterol (1:1), DOTAP/DOPE/Cholesterol (1:1:2.1) were prepared. To obtain PEGylated liposomes, 2 mol% of DSPE-PEG and 0.4 mol% of Chol-PEG were added to the mixture. According to the mentioned molar ratios, suitable amounts of lipid powders were dissolved and mixed in chloroform (obtaining liposomes at a final concentration of 5 mM). The chloroform was evaporated using a rotary evaporator (temperature 37°C, under vacuum and 40 rpm) and the lipid thin film was dried by high vacuum for several hours. At the next step, the dried film was hydrated by addition of distilled water at 45°C with repeated rotatory movement to ensure complete hydration of the lipids and vortexed vigorously for 10 minutes to obtain dispersed liposomes. Primary homogenization was performed by sonicator bath (Soltec, Milan, Italy) for 5 min. Thereafter, the liposomes were downsized 10 times through two stacked 100 nm polycarbonate membrane filters (400 nm, 200 nm, and 100 nm) at room temperature using an Avanti Mini-Extruder (Avanti Polar, Sigma Aldrich).
Preparation and characterization of (PEGylated) lipoplexes
Appropriate amounts of CLs were added to pGL3 (1 µg/µl) in N/P ratios (3, 5, 7) and incubated at room temperature for 20 minutes. PEGylated lipoplexes were prepared according to the post PEGylation method. Briefly, incubation of pGL3 with different amounts of CLs at pointed N/P ratios was carried out and then 2 mol% of DSPE-PEG or 0.4 mol% Chol-PEG was added. The average particle size of the (PEGylated) liposomes and (PEGylated) lipoplexes were determined by dynamic light scattering using photon correlation spectroscopy. The measurements were performed at 25°C using a Zetasizer Nano ZS instrument (Malvern Instruments Ltd, Malvern, Worcestershire, UK) equipped with a helium-neon laser and a scattering angle of 173°. The zeta potential of liposomes indirectly reflects vesicle surface net charge and therefore can be used to evaluate the extent of interaction of the liposomal surface cationic charges with the anionic charges of DNA. Hence, the zeta potential of the (PEGylated) liposome dispersion was measured with the same instrument at 25°C by the electrophoretic mobility. All samples were measured after a 20-fold dilution in water for each particle size and zeta potential measurement.
Gel retardation analysis of (PEGylated) lipoplexes
To examine the (PEGylated) liposomes ability to entrap and release the pDNA, agarose gel electrophoresis was carried out. Lipoplexes at different N/P ratios (3, 5, 7) were prepared in two groups: 20 µl of one group was loaded directly on the agarose gel (1%) and left to run at 80 V for 45 min. Another group was incubated with a final concentration of 1% SDS to release the pDNA from the complexes and then was electrophoresed. The mobility of plasmid was visualized by Ethidium bromide staining and UV illumination. Integrity of the plasmid in each formulation was compared with untreated pDNA as a control.
Cell culture, transfection and gene expression analysis
HEK293 cell line was purchased from the cell bank of the Pasture Institute in Iran. HEK293 was grown in the medium supplemented with 10% FBS, penicillin (100 units/mL) and streptomycin (100 mg/mL). Cells were grown in 25 cm2 polystyrene tissue culture flasks in a humidified atmosphere of 5% CO2 and 37°C. Cells were seeded in 24-well sterile culture plates at a density of 3×105 cells/well and were grown overnight to approximately 70% confluence ready to use the next day. The growth medium was removed and cells were washed twice with pre-warmed phosphate-buffered saline (PBS) and replaced with fresh serum-free DMEM medium. Various formulations of CLs with different N/P ratios were added to the wells and cells were incubated for 6 hours. Lipofectamine 2,000 as a control was prepared in serum-free DMEM then added to the cells (pGL3 1.0 mg/3.0 mL Lipofectamine per well). After 6 hours, the serum-free medium was replaced with DMEM supplemented with 10% FBS and further incubated for 48 hours (in a humidified atmosphere of 5% CO2 and 37°C) prior to evaluation of transfection efficiency using luciferase assay. The luciferase assay was performed 48 hours later.
To analyze luciferase activity, the transfected cells were washed twice gently with PBS and lysed by cell lysis buffer (Promega Corporation, Fitchburg, WI, USA). The luciferase activity was measured in the presence of firefly luciferase substrates (2 mM Luciferin, 4 mM ATP and 100 mM MgSO4 [pH 7.8]) with a luminometer (Berthold Detection Systems, GmbH, Pforzheim, Germany). The results were normalized with respect to their protein concentrations. The data are indicated as the relative light unit (RLU/Sec)/mg of total proteins and presented as mean ± SD (n=3).
Cell viability assay
HEK293 (7×103) cells were seeded in 96-well plates and incubated for 24 h at 37°C in a humidified incubator with 5% CO2 atmosphere. Cells were attached to the plate surface and were then treated with lipoplexes at the same concentrations used for transfection experiments. Then, the cells were incubated for another 24 hours. Cell viability was assayed using MTT according to a reported method.21 Briefly, MTT 10 µl (5.0 mg/mL) was added to each well and the cells were incubated at 37°C for 4 hours. Then the MTT-containing medium was removed and the formazan crystals formed by living cells were dissolved in 100 µl DMSO. The absorbance at 570 nm was determined by a microplate reader (ELx800TM, BioTek Instruments, Winooski, VT, USA) at test and reference wavelengths of 570 and 630 nm, respectively. The absorbance of untreated cell was considered as 100% viable.
Serum stability of lipoplexes
The stability of the (PEGylated) lipoplexes was investigated by incubation in 10% FBS. Then 1 µg/µl of pGL3, as a control, was incubated in the same concentration of FBS for 6 hours at 37°C. Then, nucleases in the FBS were inactivated with EDTA solution for 10 min at room temperature. To separate DNA from lipoplexes, 1% SDS was added to the suspensions. FBS-treated samples were evaluated using agarose gel electrophoresis. In addition, to investigate the effect of serum on the stability of lipoplexes, 1.0 µg of pGL3 was transfected to HEK293 in different concentrations of serum (10, 20 and 40%) and luciferase activity was measured 48 hours later.
sgRNA design, EGFP disruption assay and indel analysis
A pre-validated sgRNA 5´-GGCGAGGGCGATGCCACCTA-3´ targeting EGFP oligoes were cloned in MLM3636-derived vector Addgene (#3860) according to the protocol previously described in the literature. The target site starts at position 119 by the protospacer adjacent motif (PAM) CGG. Cas9-expression vector Addgene #41815 was a kind gift of Concordet’s lab. Plasmids were amplified by transformation in the TOP10 cells following Midi prep. Then, 1 µg of Cas9 and 1 µg of sgRNA expression plasmids were transfected in 7×105 HEK293 cells by prepared post-PEGylated four-component lipoplexes and Cas9/sgRNA alone as a control according to the previously described method. Finally, 48 hours after transfection, cells were studied by microscopy and flow cytometry.
Cells were trypsinized and resuspended single-cells were transferred to tubes and washed with phosphate-buffered saline (PBS). They were analyzed by flow cytometer using a FACS BD biosciences Accuri 6 gating EGFP-positive cells using the blue laser (488 nm) and detecting fluorescent light in each cell at 538 nm.
Finally, 48 hours after transfection by PEGylated four-component lipoplex and lipo2000 as a control, cells were harvested to extract genomic DNA using Qiagen Genomic DNA extraction kit. Indel assay was carried out using T7 endonuclease-I according to the standard protocol by the designed primers EGFP-Fw 5´- ATGGTGAGCAAGGGCGAG-3´ and EGFP-Rv 5´-TTACTTGTACAGCTCGTCCATGC-3´ toward the N-terminus of EGFP, which interrupt GFP production via Indel in the targeting sequence by NHEJ repair.
Statistical analysis
The statistically significant differences between the groups were estimated by one-way ANOVA and two-sample t-test. Data were expressed as mean ± standard error and a p-value < 0.05 was considered statistically significant.
Results
Physicochemical properties of (PEGylated) CLs and lipoplexes
Formulated liposomes consist of a constant monovalent cationic lipid (DOTAP), two neutral lipids (DOPE, Cholesterol) and PEGylated moieties (DSPE-PEG2000, Chol-PEG2000). The size, PDI and zeta potential of particles were assessed before and after DNA entrapment. Cholesterol domains formation happens at higher Cholesterol concentrations ≥52% molar ratio. Two-components and three-component CLs depicted particle size around 163±10 with low PDI (eg 0.1±0.035). After mixing with DNA an increase in size occurred in all CL forms (eg two components: ≥185±1.7, three components: ≥190±2.4) that were totally N/P ratio dependent. Also, shrinkage in size is obvious after PEGylation in all formulations. PDI of different compositions was generally around 0.2±0.03 which reflects the homogeneity of the prepared particles dispersion. Table 1 shows the details of lipid compositions, size and PDI for both (PEGylated) CLs and lipoplexes. Increasing the N/P ratios in all formulations raised the size of the lipoplexes of non-PEGylated and PEGylated either by DSPE-PEG or Chol-PEG. Table 1 demonstrates the zeta potential related to all designed CLs and lipoplexes. All of the studied CLs kept their net positive charge after DNA entrapment and PEGylation. In almost all formulations, zeta potential was raised to a higher N/P ratio (5 and 7); while, PEGylation of particles by either DSPE-PEG or Chol-PEG decreased the zeta potential. No statistical differences were observed between surface charge shielding by DSPE-PEG and Chol-PEG. Lipoplexes of CL formulations (PEGylated and Non-PEGylated) were prepared by pGL3 incubation in water at various N/P ratios (3, 5, 7). In this assay, non-entrapped pGL3 moved equal to the control plasmid, while entrapped pGL3 remained in the wells. Gel retardation analysis of formulated lipoplexes demonstrated CLs ability for pGL3 entrapment, either PEGylated or non-PEGylated. (PEGylated) CLs indicated complete DNA entrapment and no plasmid band was detectable on the gel (Figure 2A). Therefore, the concentration of liposomes at each N/P ratio was enough to neutralize the negative charge of pDNA. Additionally, lipoplexes treatment by SDS (1%) resulted in the release of entrapped plasmids which were detectable in agarose gel with the same pattern as the pGL3 control. Thus, based on the gel retardation, the capability of all formulated liposomes was the same for plasmid entrapment and binding force between the carrier and plasmid. In other words, PEGylation type would not act as a differential parameter in delivery rate of the vectors (Figure 2B).
 | Table 1 Particle size and zeta potential of CLs and corresponding lipoplexes at various N/P ratios: DOTAP/DOPE (1:1), DOTAP/Cholesterol (1:1), DOTAP/DOPE/Cholesterol (1:1:2.1), 2 mol% of DSPE-PEG and 0.4 mol% of Chol-PEG. (A) Size and PDI, (B) Zeta potential |
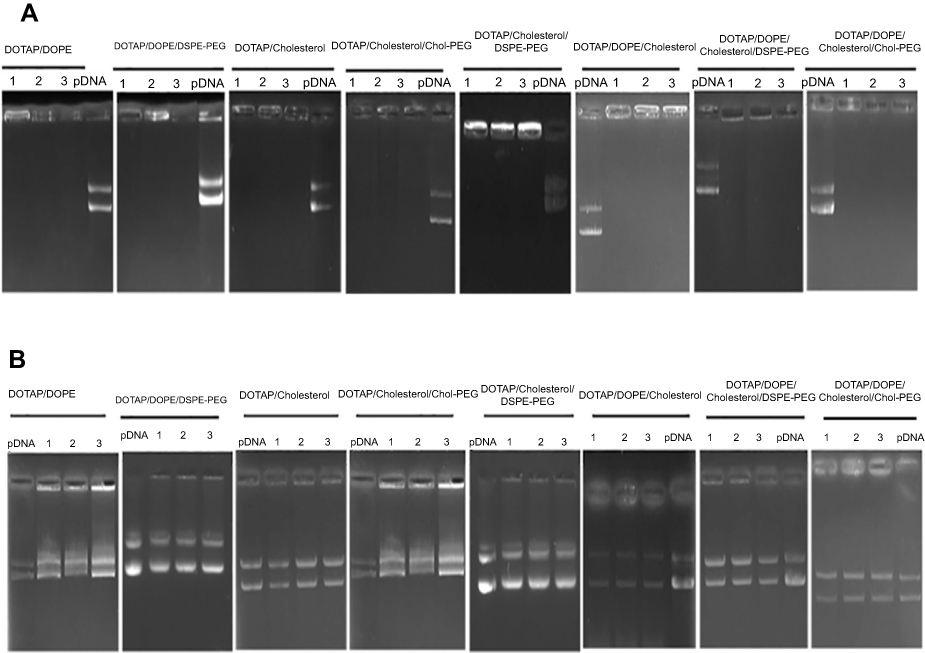 | Figure 2 Gel retardation assay of lipoplexes at different N/P ratios (3, 5, 7).Notes: (A) Agarose gel (1%) retardation assay of all (PEGylated) CLs formulations mixed by 1 µg pGL3 plasmid at various N/P ratios. pDNA control: pGL3, lanes 1–3: N/P ratios 3, 5 and 7, respectively. (B) The mobility pattern of released pGL3 from the complexes by SDS 1% at different N/P ratios. pGL3 control: pGL3, lanes 1–3: N/P ratios 3, 5 and 7, respectively.Abbreviation: CLs, cationic liposomes. |
Transfection efficiency of (PEGylated) lipoplexes and endosomal release
Transfection capacity of (PEGylated) lipoplexes was determined by transferring 1 µg of luciferase expression plasmid (pGL3) to the HEK293 and luminescence signal assessment 48 hours later. All formulations depicted the highest and lowest rate of transfection at N/P 3 and 7, respectively. DSPE-PEG incorporation in the formulation dramatically decreased the rate of transfection compared to Chol-PEG (0.4 mol %) integrated in the Cholesterol domain of lipoplexes (>1.5 folds) at N/P 3 (Figure 3A). Luciferase expression explained why Chol-PEGylated lipoplexes were more (1.3 times) efficient than commercial lipofectamine2000 reagent in HEK293 cells. According to our data Chol-PEG incorporation in Cholesterol domains of lipoplex enhanced the entrance of particles to the cells. But for the four-component lipoplexes (DOTAP/DOPE/Cholesterol/Chol-PEG) in comparison to three-component ones (DOTAP/Cholesterol/Chol-PEG) a higher level of luciferase expression was detected that could be related to the synergistic effect of pH sensitive lipid, DOPE, known to enhance the transfection efficiency of CLs by facilitating endosomal escape and Chol-PEG arrangement in Cholesterol domains. This hypothesis was evaluated by measuring the stability of DOTA/Cholesterol/Chol-PEG and DOTAP/DOPE/Cholesterol/Chol-PEG at different pH values, from 7 to 4.5. The increase in diameter of DOTAP/DOPE/Cholesterol/Chol-PEG particles at lower pH values suggested disruption of lipoplexes under acidic conditions while fewer changes were detected in the formulation without DOPE (Figure 3B and C). It was exciting to see that in similar low pH situations like lysosomes in the cells, the structure of the lipoplexes can break down and the trapped pGL3 could release in the cytoplasm.
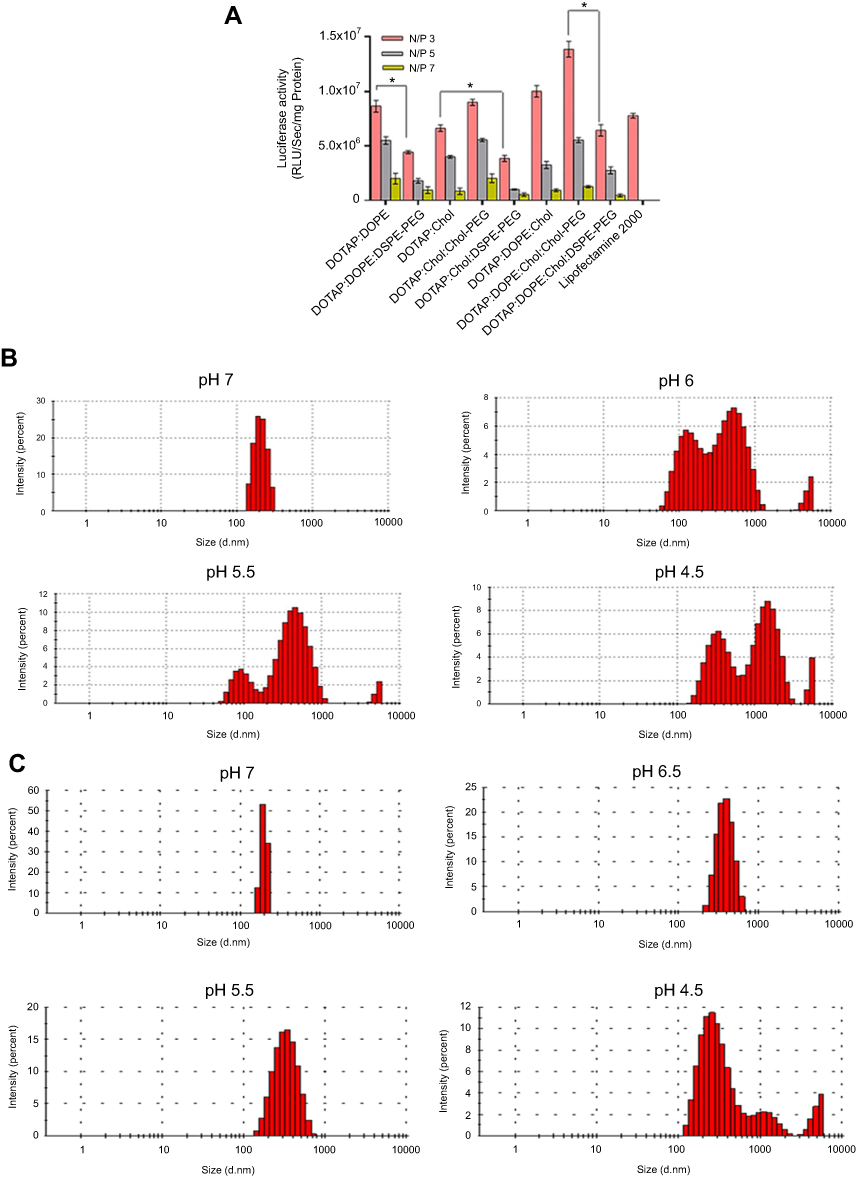 | Figure 3 (A) Transfection efficiency of (PEGylated) lipoplexes. Rate of transfection of 1 µg pGL3 by (PEGylated) Lipoplexes in HEK293 measured by luminescence signal assessment after 48 hours. Total protein (mg) from luciferase assay was used for normalization of relative light Unit/Sec (RLU/Sec) values. Data are mean ± SD from luciferase assay (n=3). *p < 0.05. (B) and (C) DOTAP/DOPE/Cholesterol/Chol-PEG and DOTAP/Cholesterol/Chol-PEG stability at various pH values reducing from 7 to 4.5 based on diameters' determination, respectively. |
The cytotoxicity and serum stability of CLs
Nanoparticle impact on cell viability is another effective factor which should not be underestimated because their cytotoxicity can reduce the rate of transfection efficiency. In this regard, the viability of HEK293 was examined by MTT assay under treatment of lipoplexes at N/P 3 which brought about the highest luciferase expression in all formulations. No significant toxic effect of nanocomplexes was observed on HEK293 cells especially compared to lipofectamin 2000/pGL3 mixture (Figure 4A). The average cell viability of our lipoplex formulations (90%) was significantly higher than commercial lipofectamine (75%) after 48 hours. In other words, the lipoplexes in all formulations at N/P 3 were safe enough for gene delivery. The other essential requirement of a carrier for gene delivery is protection of DNA against serum nucleases. Through various designed (PEGylated) lipoplexes at different N/P ratios DOTAP/DOPE/Cholesterol/Chol-PEG possessed the highest capacity of gene delivery selected to evaluate for stability in the serum by gel retardation assay. As Figure 4B shows, naked pGL3 was completely degraded after 6 hours in the serum. SDS treatment brought about release of pGL3, which preserved its integrity and size in the complex carrier. Non-PEGylated lipoplex could not protect pGL3 and it was degraded in the serum by the nuclease activity, while PEGylated lipoplexes protected pGL3 against nuclease break. According to this result, PEGylated formulations preserved pGL3 from degradation by cytoplasm nucleases and most likely kept it stable, in vivo. Moreover, luciferase activity of transfected cells by DOTAP/DOPE/Cholesterol/Chol-PEG lipoplex at N/P 3 in different concentrations of serum (10, 20 and 40%), depicted no significant impact at 10% or 20% but a statistically significant difference at 40% serum after 48h (Figure 4C). It is noteworthy that further cell death analysis using a positive control like Triton –X-100 and also validation of cell death modality using flow cytometry will also be required in future applications of the current lipoplexes.22,23
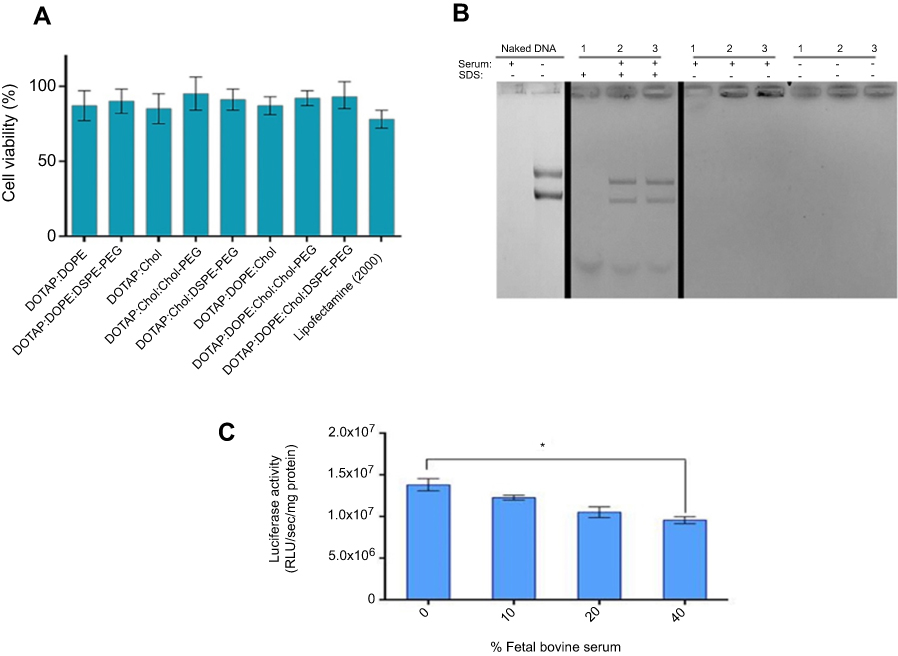 | Figure 4 Cytotoxicity and serum stability of CLs.Notes: (A) Cytotoxicity profile of (PEGylated)lipoplexes at N/P 3. HEK293 cells were treated by formulated lipoplexes for 48 hours. Normalization of the data performed based on considering viability of untreated cell as 100%. Data are mean ± SD (n=3). (B) Serum stability of Lipoplex at N/P 3. (1) DOTAP/DOPE/Cholesterol, (2) DOTAP/DOPE/Cholesterol/DSPE-PEG, and (3) DOTAP/DOPE/Cholesterol/Chol-PEG at N/P 3 were incubated with 10% FBS for 6 hours. Serum nuclease activity was haltered by EDTA and SDS treatment to release protected pGL3. Naked and serum treated pGL3 and untreated lipoplexes acted as controls. (C) luciferase activity measurement by transfecting 1 µg pGL3 by DOTAP/DOPE/Cholesterol/Chol-PEG at N/P 3 to HEK293 in different concentrations of FBS (10, 20 and 40%) after 48 hours. Total protein (mg) from luciferase assay was used for normalization of relative light Unit/Sec (RLU/Sec) values. Data are mean ± SD from luciferase assay (n=3). *p < 0.05.Abbreviation: CLs, cationic liposomes. |
EGFP disruption assay and indel analysis
Stably expressing EGFP HEK293 cells applied to evaluate the potential of DOTAP/DOPE/Cholesterol/Chol-PEG to deliver Cas9 and sgRNA plasmids, were designed to target EGFP gene. Using fluorescence microscopy, it simply turned out that EGFP expression was lower in transfected cells than in un-transfected ones (Figure 5A). Nonotable effect was observed by transfection of Cas9/sgRNA alone, without any carrier, as a control. To quantify this result by flow cytometer, 10,000 cells were screened and 39% reduction in the fluorescence signal of transfected cells by DOTAP/DOPE/Cholesterol/Chol-PEG lipoplex was observed in comparison to un-transfected cells and Cas9/sgRNA alone (Figure 5B and C).
 | Figure 5 EGFP knockout assessment.Notes: (A) Fluorescent and bright field (BF) microscopy of HEK293 cells stably expressing EGFP 48 hours after transfection of Cas9/sgRNA alone and DOTAP/DOPE/Cholesterol/Chol-PEG/Cas9/sgRNA. (B) Flow cytometry histograms of EGFP expression in Cas9/sgRNA transfected cells; (1) no treatment, (2) Cas9/sgRNA alone, and (3) DOTAP/DOPE/Cholesterol/Chol-PEG/Cas9/sgRNA. (C) Cas9/sgRNA activity quantification. Significant effect of Cas9/sgRNA delivered by DOTAP/DOPE/Cholesterol/Chol-PEG is obvious in comparison to Cas9/sgRNA alone. Error bars indicate SE (n=3). |
The genomic DNA was extracted from transfected and control cell lines. The flanking region of the sgRNA targeted site was amplified by the specific primers to assess the rate of NHEJ in the disrupted EGFP gene. Denaturation and renaturation of the PCR product resulted in the formation of mismatched DNA which was detectable by T7 endonuclease I (T7EI) (Figure 6). Analysis of the cut at this region indicated that disruption at the target region had happened which proves the results of FACS and microscopy.
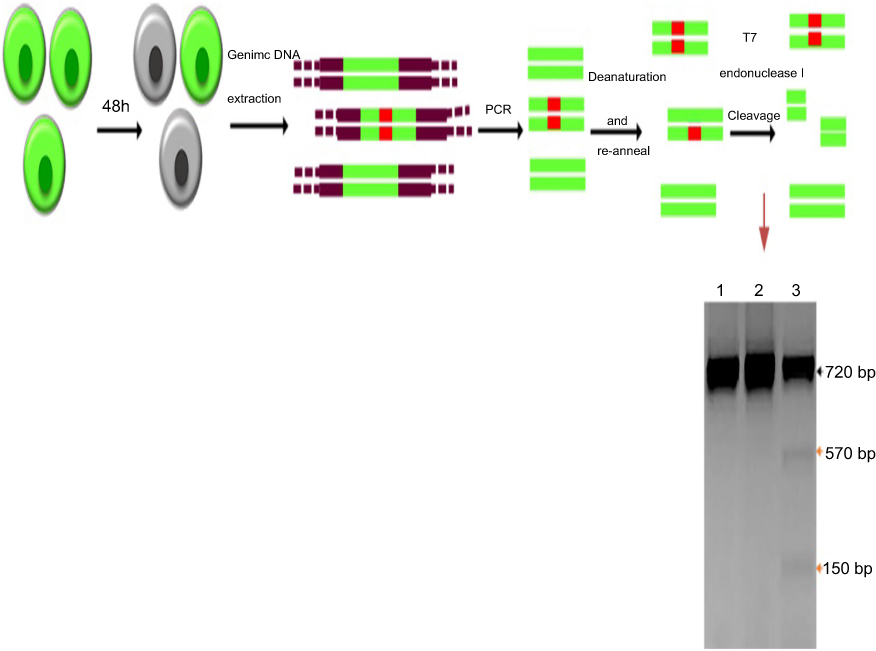 | Figure 6 T7 endonuclease I assay for indels detection in EGFP knockout cells schematic representation.Notes: The order of assay is as follows: transfection by DOTAP/DOPE/Cholesterol/Chol-PEG/Cas9/sgRNA; incubation for 48 hours; genomic DNA extraction; targeted segment amplification; denaturation and re-annealing; T7 endonuclease I treatment and gel electrophoresis of the cleavage products. Lane 1, untreated; lane 2, Cas9/sgRNA; lane 3, DOTAP/DOPE/Cholesterol/Chol-PEG/Cas9/sgRNA; the black and orange arrows imply wild- and mutant bands, respectively. |
Discussion
Gene delivery, introduction of DNA to the targeted cells, is a critical step in gene therapy strategies. Optimization of delivery systems for gene therapy complexes, specifically CRISPR-Cas9 is in progress.24 Various delivery systems have been developed which work well in some aspects. However, development of an efficient and safe carrier is one of the challenges in this area.25 In the present study, PEGylated and Non-PEGylated, two-, three- and four-component lipoplexes were prepared, characterized and the combination effect of PEGylated Cholesterol domains and DOPE on transfection efficiency and serum stability were investigated. Our results due to the size and charge of lipoplexes are consistent with previous studies.26 No significant difference was observed between PEGylated liposomes and Non-PEGylated ones which could be related to the concentration of grafted PEG in the formulations contained DSPE-PEG. At higher N/P ratios (5 and 7), the size of lipoplexes increased.27
It should be emphasized that delivery of designed lipoplexes to a specific tissue is largely dependent on the size of lipoplexes. For example, in liver-directed gene delivery, the average size of liver sinusoidal fenestrae in humans is 105 nm.28,29 Therefore, it can be concluded that the size of current lipoplexes for in vivo gene transfer to liver cells should be modified.
In addition, PEGylation seems to have a shield effect on the charge of the lipoplexes and caused a significant reduction in their positive charge. Safe delivery of the genetic tools in the presence of serum proteins and nucleases is another significant factor affecting transfection rate. Electrostatic interactions between the negative charge of the pDNA and the positive charge of the CLs maintained the lipoplexes constant in the physiological conditions. According to the serum stability result, entrapped pGL3 in the PEGylated liposome structure was completely shielded while the Non-PEGylated one depicted degradation (Figure 4B). Transfection yield of lipoplexes was determined by delivery of plasmid contained in luciferase gene as a quick, simple and effective method to the HEK293. Transfection rate of designed lipoplexes was totally N/P ratio dependent. Generally, in all PEGylated-lipoplexes at various N/P ratios, the ones which were equipped with Chol-PEG in Cholesterol domains expressed more luciferase in comparison to the ones PEGylated by DSPE-PEG2000 which is consistent with previous studies.30 It is well-known that PEGylation enhances serum stability and circulation lifetime of nanoparticles owing to steric stabilization. Nevertheless, it can interfere with intracellular trafficking which prevent effective endosomal escape of nanocarriers.31 Various studies recommended several clues to this challenge like using a pH-sensitive linker between PEG-linked parts.32 Cholesterol domains form at higher concentrations of Cholesterol (>52%).33 In 2011, Long et al indicated the effect of PEGylation on liposomes with and without Cholesterol domain for transfection in different cell lines. According to their results conjugation of PEG to Cholesterol and incorporation of PEG in the Cholesterol domains reduce the adverse effect of PEG in intracellular trafficking which can trigger a different approach regarding the detrimental effect of DSPE-PEG in transfection. At lower concentrations of the Cholesterol, 36% and 45%, Cholesterol domains did not form and Chol-PEGs should distribute equally all over the surface of the particles. Yield of transfection was totally lower than the ones including Cholesterol domains. Moreover, they showed that the microenvironment of the ligand impact on efficiency of gene delivery means that carriers by multiple microenvironments are more advanced than the ones with uniform surface attributions.30 In addition to the serum stability, overcoming the endosomal barrier is significant as it has a higher rate of transfection. Therefore, impact of DOPE as a pH-sensitive lipid in the formulations was evaluated. Kim et al rendered more efficient transfection by involvement of DOPE in the CL formulation. Formation of inverted hexagonal structures encouraged by the conical structure of DOPE, resulted in fusion with endosomal membrane that caused rapid release of DNA in the cytoplasm.34 The post-PEGylation method of adding DSPE-PEG or Chol-PEG after lipoplex preparation resulted in more plasmid entrapment than mixing PEGylated lipids with other components at the same time.35 In the present study fusogenic properties of DOPE in the lipoplex formulation combined with the design of its surface by Chol-PEG in Cholesterol domains significantly enhanced the transfection efficiency. To figure out the potency of the optimized formulation to transfer gene editing tools, transfection by DOTAP/DOPE/Cholesterol/Chol-PEG/Cas9/sgRNA lipoplex was performed and its efficacy was evaluated by knocking out intracellular GFP sequence. According to the qualitative and quantitative estimations of GFP signal by fluorescence microscopy and flow cytometry, 39% decrease in GFP signal was observed. It should be pointed out as outlined in the introduction, that challenges related to stable gene therapy in specific tissues like liver show efficient targeting of hepatocytes, stability of the vector genome, and persistent high level expression. Many of these obstacles can be overcome with adeno-associated viral (AAV) gene transfer vectors in liver hepatocytes. The first AAV gene transfer vector developed for in vivo use was based on the AAV2 serotype.36 However, due to possible pro-oncogenic activity of viral-based gene delivery, non-viral vectors have attracted attention. So, we presume the correct liposome-based gene delivery accompanied with gene editing machinery (CRISPR-Cas9) can be used for stable gene delivery in future.
Thus, development of this highly promising lipid-mediated nanoparticle for knocking out the targeted genes in vivo is recommended due to the serum stability and transfection efficiency.
Conclusion
In conclusion, PEGylated Cholesterol domain lipoplexes including a cationic lipid (DOTAP) and a fusogenic lipid (DOPE) possess characteristics which are beneficiary for the carriers in the gene therapy field. Increased stability in the serum, complete protection of DNA against nucleases and efficient endosomal escape lead to a higher yield of transfection. This implies that DOTAP/DOPE/Cholesterol/Chol-PEG liposome can be a powerful candidate for delivery of CRISPR-Cas9 plasmids to the targeted cells, especially to the tumor cells to knock out the oncogenes which are overexpressed in most of cancers and make the tumor therapy by the CRISPR promising.
Acknowledgment
Financial support of this work was provided by the Research Council of Tarbiat Modares University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nayerossadat N, Maedeh T, Ali PA. Viral and nonviral delivery systems for gene delivery. Adv Biomed Res. 2012;1:27. doi:10.4103/2277-9175.98152
2. Ramamoorth M, Narvekar A. Non viral vectors in gene therapy an overview. J Clin Diagn Res. 2015;9(1):GE01–GE06. doi:10.7860/JCDR/2015/13028.5790
3. Alipour M, Majidi A, Molaabasi F, Sheikhnejad R, Hosseinkhani S. In vivo tumor gene delivery using novel peptideticles: pH-responsive and ligand targeted core–shell nanoassembly. Int J Cancer. 2018;143(8):2017–2028. doi:10.1002/ijc.31577
4. Ghafary SM, Nikkhah M, Hatamie S, Hosseinkhani S. Simultaneous gene delivery and tracking through preparation of photo-luminescent nanoparticles based on graphene quantum dots and chimeric peptides. Sci Rep. 2017;7(1):9552. doi:10.1038/s41598-017-09890-y
5. Majidi A, Nikkhah M, Sadeghian F, Hosseinkhani S. Development of novel recombinant biomimetic chimeric MPG-based peptide as nanocarriers for gene delivery: imitation of a real cargo. Eur J Pharm Biopharm. 2016;107:191–204. doi:10.1016/j.ejpb.2016.06.017
6. Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–355. doi:10.1038/nbt.2842
7. Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16(5):299–311. doi:10.1038/nrg3899
8. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. doi:10.1016/j.cell.2014.05.010
9. Bhaya D, Davison M, Barrangou R. CRISPR-cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45(1):273–297. doi:10.1146/annurev-genet-110410-132430
10. Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:6213. doi:10.1126/science.1255826
11. Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168(1–2):20–36. doi:10.1016/j.cell.2016.10.044
12. Wang H-X, Li M, Lee CM, et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem Rev. 2017;117(15):9874–9906. doi:10.1021/acs.chemrev.6b00799
13. Lino CA, Harper JC, Carney JP, Timlin JA. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 2018;25(1):1234–1257. doi:10.1080/10717544.2018.1474964
14. Majumder P, Bhunia S, Bhattacharyya J, Chaudhuri A. Inhibiting tumor growth by targeting liposomally encapsulated CDC20siRNA to tumor vasculature: therapeutic RNA interference. J Controlled Release. 2014;180:100–108. doi:10.1016/j.jconrel.2014.02.012
15. Yanez Arteta M, Kjellman T, Bartesaghi S, et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc Natl Acad Sci U S A. 2018;115(15):E3351–E3360. doi:10.1073/pnas.1720542115
16. Majumder P, Bhunia S, Chaudhuri A. A lipid-based cell penetrating nano-assembly for RNAi-mediated anti-angiogenic cancer therapy. Chem Commun. 2018;54(12):1489–1492. doi:10.1039/c7cc08517f
17. Samadikhah HR, Majidi A, Nikkhah M, Hosseinkhani S. Preparation, characterization, and efficient transfection of cationic liposomes and nanomagnetic cationic liposomes. Int J Nanomedicine. 2011;6:2275–2283. doi:10.2147/IJN.S23074
18. Ghanbari Safari M, Hosseinkhani S. Lipid composition of cationic nanoliposomes implicate on transfection efficiency. J Liposome Res. 2013;23(3):174–186. doi:10.3109/08982104.2013.779703
19. Zhang Y, Anchordoquy TJ. The role of lipid charge density in the serum stability of cationic lipid/DNA complexes. Biochim Biophys Acta (BBA) – Biomembr. 2004;1663(1):143–157. doi:10.1016/j.bbamem.2004.03.004
20. Zhao XB, Muthusamy N, Byrd JC, Lee RJ. Cholesterol as a bilayer anchor for PEGylation and targeting ligand in folate‐receptor‐targeted liposomes. J Pharm Sci. 2007;96(9):2424–2435. doi:10.1002/jps.20885
21. Zhao Z, Li Y, Jain A, et al. Development of a peptide-modified siRNA nanocomplex for hepatic stellate cells. Nanomedicine. 2018;14(1):51–61. doi:10.1016/j.nano.2017.08.017
22. Lu X, Qian J, Zhou H, et al. In vitro cytotoxicity and induction of apoptosis by silica nanoparticles in human HepG2 hepatoma cells. Int J Nanomedicine. 2011;6:1889–1901. doi:10.2147/IJN.S24005
23. Jain A, Barve A, Zhao Z, Jin W, Cheng K. Comparison of avidin, neutravidin, and streptavidin as nanocarriers for efficient siRNA delivery. Mol Pharm. 2017;14(5):1517–1527. doi:10.1021/acs.molpharmaceut.6b00933
24. Ibraheem D, Elaissari A, Fessi H. Gene therapy and DNA delivery systems. Int J Pharm. 2014;459(1):70–83. doi:10.1016/j.ijpharm.2013.11.041
25. Li L, Hu S, Chen X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: challenges and opportunities. Biomaterials. 2018;171:207–218. doi:10.1016/j.biomaterials.2018.04.031
26. Majzoub RN, Chan CL, Ewert KK, et al. Uptake and transfection efficiency of PEGylated cationic liposome-DNA complexes with and without RGD-tagging. Biomaterials. 2014;35(18):4996–5005. doi:10.1016/j.biomaterials.2014.03.007
27. Garbuzenko O, Barenholz Y, Priev A. Effect of grafted PEG on liposome size and on compressibility and packing of lipid bilayer. Chem Phys Lipids. 2005;135(2):117–129. doi:10.1016/j.chemphyslip.2005.02.003
28. Wisse E, Jacobs F, Topal B, Frederik P, De Geest B. The size of endothelial fenestrae in human liver sinusoids: implications for hepatocyte-directed gene transfer. Gene Ther. 2008;15:1193. doi:10.1038/gt.2008.60
29. Jacobs F, Wisse E, De Geest B. The role of liver sinusoidal cells in hepatocyte-directed gene transfer. Am J Pathol. 2010;176(1):14–21. doi:10.2353/ajpath.2010.090136
30. Xu L, Wempe MF, Anchordoquy TJ. The effect of cholesterol domains on PEGylated liposomal gene delivery in vitro. Ther Deliv. 2011;2(4):451–460. doi:10.4155/tde.11.13
31. Nakamura K, Yamashita K, Itoh Y, Yoshino K, Nozawa S, Kasukawa H. Comparative studies of polyethylene glycol-modified liposomes prepared using different PEG-modification methods. Biochim Biophys Acta (BBA) – Biomembr. 2012;1818(11):2801–2807. doi:10.1016/j.bbamem.2012.06.019
32. Li W, Huang Z, MacKay JA, Grube S, Szoka FC. Low-pH-sensitive poly(ethylene glycol) (PEG)-stabilized plasmid nanolipoparticles: effects of PEG chain length, lipid composition and assembly conditions on gene delivery. J Gene Med. 2005;7(1):67–79. doi:10.1002/jgm.634
33. Xu L, Anchordoquy TJ. Cholesterol domains in cationic lipid/DNA complexes improve transfection. Biochim Biophys Acta (BBA) – Biomembr. 2008;1778(10):2177–2181. doi:10.1016/j.bbamem.2008.04.009
34. S̆Misterová J, Wagenaar A, Stuart MCA, et al. Molecular shape of the cationic lipid controls the structure of cationic lipid/dioleylphosphatidylethanolamine-DNA complexes and the efficiency of gene delivery. J Biol Chem. 2001;276(50):47615–47622. doi:10.1074/jbc.M106199200
35. Peeters L, Sanders NN, Jones A, Demeester J, De Smedt SC. Post-pegylated lipoplexes are promising vehicles for gene delivery in RPE cells. J Controlled Release. 2007;121(3):208–217. doi:10.1016/j.jconrel.2007.05.033
36. Sands MS. AAV-mediated liver-directed gene therapy. Methods Mol Biol 2011;807:141–157. doi:10.1007/978-1-61779-370-7_6
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.