Back to Journals » Vascular Health and Risk Management » Volume 15
Cholesterol-embolization syndrome: current perspectives
Authors Ozkok A
Received 7 February 2019
Accepted for publication 10 May 2019
Published 8 July 2019 Volume 2019:15 Pages 209—220
DOI https://doi.org/10.2147/VHRM.S175150
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Abdullah Ozkok
Department of Internal Medicine and Nephrology, Memorial Şişli Hospital, Istanbul, Turkey
Abstract: Cholesterol-embolization syndrome (CES) is a multisystemic disease with various clinical manifestations. CES is caused by embolization of cholesterol crystals (CCs) from atherosclerotic plaques located in the major arteries, and is induced mostly iatrogenically by interventional and surgical procedures; however, it may also occur spontaneously. Embolized CCs lead to both ischemic and inflammatory damage to the target organ. Therefore, anti-inflammatory agents, such as corticosteroids and cyclophosphamide, have been investigated as treatment for CES in several studies, with conflicting results. Recent research has revealed that CES is actually a kind of autoinflammatory disease in which inflammasome pathways, such as NLRP3 and IL1, are induced by CCs. These recent findings may have clinical implications such that colchicine and IL1 inhibitors, namely canakinumab, may be beneficial in the early stages of CES.
Keywords: cholesterol crystals, atherosclerosis, inflammation, autoinflammation, corticosteroids, interleukin 1, NLRP3, colchicine, canakinumab
Introduction
Cholesterol-embolization syndrome (CES) is a systemic disease caused by showering of atherosclerotic plaque materials, such as cholesterol crystals (CCs), from the aorta and its major branches to distal circulation, leading to ischemic and inflammatory damage to multiple organs.1 This syndrome is also called atheroembolism, atheromatous embolization syndrome, and cholesterol-crystal embolization. Renal involvement of CES is referred to as atheroembolic renal disease (ARD) or cholesterol ARD.2
CES should be differentiated form a more frequent form of arterial embolization syndrome — arterioarterial thromboembolism — in which a sudden release of thrombus from an atheromatous plaque causes acute ischemia and infarction of the distal organ. However, CES is characterized by embolization of smaller CCs, resulting in more gradual end-organ damage caused by both ischemic and inflammatory mechanisms.3 CES is a frequently underdiagnosed disease. However in recent years CES has been diagnosed more frequently, probably due to increased clinical awareness, increased life expectancy of patients with atherosclerosis, and an increase in the number of invasive vascular procedures.2
Epidemiology
Although there has been significant variability among studies, the incidence of clinically evident CES has been reported to be 0.09%–2.9%.4–6 In autopsy series, CES was found at a frequency of 0.31%–2.4%.7,8 However CES frequency was significantly higher (12%–77%) in autopsy studies performed on selected populations ,such as elderly patients who had died after aortic surgery or aortography.9,10
In a study of 519 patients with thoracic aortic atherosclerotic plaques determined on transesophageal echocardiography (TEE), CES was found in 1% of patients during follow-up of >3 years.5 In a prospective observational study of 1,786 patients undergoing cardiac catheterization, CES was found in 1.4% of patients, with 64% of those having renal damage, and definite CES was established in 0.8% of patients.11 Abdominal aortic aneurysms are important sources of cholesterol emboli. In a prospective study of 660 patients with abdominal aortic aneurysms that were followed for a mean of 15 months, CES was diagnosed in 2.9%.6 In a retrospective study, only 15 of 16,223 patients (0.09%) who had undergone vascular procedures were found to have CES.4 In three autopsy studies, incidence of spontaneous CES was found to be 0.79%–3.4% which was most frequently observed in elderly patients.7
However the diagnosis of CES is easily overlooked in most cases, and exact incidence is probably much higher than has been reported. In a prospective study performed on 60 patients presenting with acute myocardial infarction who underwent coronary artery–bypass–graft surgery, two muscle-biopsy and one skin-biopsy specimens were obtained during surgery.12 A total of seven patients (12%) had pathological evidence of CES in the muscle-biopsy specimens; however, clinically evident disease was present in only one.
ARD was found at a frequency of about 1% in series of 755 and 4,580 consecutive kidney biopsies.13,14 However, in a study performed on renal biopsies of patients >65 years of age, 14 cases of ARD were found in 334 biopsies (4.2%). 15 ARD may be an important cause of acute kidney injury (AKI) in elderly patients. In a study performed on 259 patients >60 years of age who underwent kidney biopsy for AKI, 7% were found to have ARD.16 It should be emphasized that retrospective biopsy studies may overestimate the incidence of CES, due to inclusion of many subclinical cases.2
Pathophysiology of CES
Atherosclerotic plaques are usually composed of platelets, fibrin, necrotic cell debris, and CCs.1 Hemodynamic changes, inflammation, and intraplaque hemorrhage, which may occur spontaneously or due to invasive procedures, may induce plaque erosion and rupture that expose the components of the plaque to systemic circulation. Subsequent showering of CCs to distal circulation leads to obstruction of arterioles with diameters of 100–200 μm.17
Initially, embolization of CCs causes ischemic injury; however subsequent inflammatory reaction aggravates and perpetuates the injury. Endothelial injury, complement activation, oxidative stress, activation of the renin–angiotensin–aldosterone system (RAAS), leukocyte aggregation, and release of leukocyte enzymes are all considered responsible for end-organ injury encountered in the course of CES.18,19 Mechanical obstruction of arcuate arteries, interlobular arteries, and glomerular capillaries may reduce regional blood perfusion and in turn activate the RAAS, leading to oxidative stress, apoptosis, inflammation, and fibrosis.20 Therefore, clinically RAAS inhibitors may have beneficial effects on kidney survival in CES. A summary of the pathophysiological mechanisms of CES is presented in Figure 1.
 |
Figure 1 Pathophysiological mechanisms of cholesterol embolization syndrome. |
CES and inflammation
CCs are known to cause inflammatory reactions around the arterioles resembling a foreign-body giant-cell reaction. CCs are accepted as danger-associated molecular patterns that have been shown to activate IL1β pathway via the NLRP3 inflammasome molecule.21 Furthermore, CCs have been shown to induce TNF and MIP2 secretion.22
In Fukumoto et al,11 CES was found to be independently associated with preprocedural CRP levels. Inflammation is well known to be an important factor in the pathogenesis of atherosclerosis.23,24 The vulnerable atherosclerotic plaques contain a large amount of inflammatory cells, which can be the source of CES. Therefore, increased CRP levels may represent increased inflammatory activity in these atherosclerotic plaques. Furthermore, complement activation may also be an important aspect of CES. In an in vitro study, human atheromatous plaque extracts were shown to activate a complement pathway.25
Experimental animal models
Animal models of CES have been developed in which atherosclerotic plaque suspensions are injected into the animal's left carotid artery or aorta.26 On the day of injection, atheromatous particles and focal fibrin deposits can usually be identified in renal vessels and glomeruli. On the third day, panarteritis with perivascular mononuclear and eosinophilic infiltrations develops. Foreign-body giant cells also appear around the CCs. On the sixth day of infusion, intimal proliferation and luminal occlusion of the vascular structures occur.2,27 After the tenth day, intimal fibrosis and encasement of the CCs by foreign-body giant cells occur. After 5 months, CCs can still be found in histiocytes, which may show that CCs are irremovable by phagocytosis.28
Diagnosis
The gold standard of CES diagnosis is tissue biopsy, which may be obtained from skin, muscle, kidney, bone marrow, and gastric and colonic mucosa. With kidney biopsy, CES has been reported to be diagnosed in >75% of cases.29 CES involves the kidney in a patchy pattern, and thus a kidney biopsy may not always demonstrate the characteristic histological findings of ARD.30 Different stages of CES may be observed in a single biopsy sample, because embolization of CCs may occur in different time intervals. Findings of ischemic injury, tissue infarction, focal segmental necrotizing glomerulonephritis, and crescentic glomerulonephritis may also be observed in kidney biopsies of patients with ARD.31
Skin biopsy is relatively noninvasive, especially when obtained from the feet and legs, which has high sensitivity — around 92%.32 The histologically pathognomonic feature of CES is the biconvex and needle-shaped “ghosts” of CCs or “cholesterol clefts" within arterioles, which are caused by dissolution of CCs during fixation of the biopsy sample.33 CCs can be seen only if biopsy samples are prepared with liquid nitrogen, by which technique CCs demonstrate double refraction under polarized light.34
However, in clinical practice a diagnosis of CES can usually be established when a combination of an inciting event and characteristic manifestations of the disease are present. For example, after a percutaneous angiography, if a patient has a delayed-onset of AKI together with cutaneous manifestations, such as livedo reticularis or blue-toe syndrome, a clinical diagnosis of CES can be established. Tissue biopsy may not be necessary if CCs are detected in the retinal vessels (Hollenhorst plaques), which can be observed in 10%–25% of CES cases.28,35
Differential diagnosis
Differential diagnosis of CES is presented in Table 1. Since clinical manifestations of CES are varied, with non-specific features, the list of differential diagnoses is long, and thus CES may be considered one of the “great imitators” (Table 1). First of all, CES should be differentiated from arterial thromboembolism, which usually causes acute ischemia and infarction of the distal organ. Although these two entities have a common risk factor, which is advanced atherosclerosis, differential diagnosis is important, because the prognosis and treatments of these diseases are different.36 Thromboembolism is usually abrupt in onset and usually causes acute organ dysfunction due to ischemia and infarction. However in CES clinical manifestations are usually subacute and chronic, and end-organ dysfunction is slow in nature. Treatment of thromboembolism should be started promptly with appropriate anticoagulation, thrombolytics, or interventional procedures. If optimal treatment is started early, successful recovery may be expected. However, treatment of CES is more difficult, prognosis is usually worse, and anticoagulation, thrombolytics, and invasive procedures may be harmful, rather than beneficial. One of the most prominent clinical features of CES is livedo reticularis, differential diagnosis of which is varied, including Raynaud's phenomenon, vasculitis, such as polyarteritis nodosa, systemic lupus erythematosus, dermatomyositis, leukocytoclastic angiitis, rheumatoid vasculitis, thromboangiitis obliterans, infections, cryoglobulinemia, and antiphospholipid syndrome.36
 |
Table 1 Differential diagnosis of cholesterol-embolization syndrome |
Blue-toe syndrome is a characteristic but not pathognomonic finding of CES that may also be observed in vasculitis and endocarditis. Peripheral pulses are usually palpable in the affected regions in CES. As such, palpable pulses may be beneficial in differentiating CES from other diseases. ARD should also be differentiated from contrast-induced AKI in patients who have undergone interventional angiographic procedures. In contrast-induced AKI, kidney dysfunction usually occurs within 48–72 hours after the procedure, and renal function usually improve within 4–7 days. By contrast, renal dysfunction due to ARD usually follows a subacute course with gradually increasing creatinine levels within 2 months.38
Ischemic acute tubular necrosis (ATN) is also included in the differential diagnosis of ARD, and may occur due to hypotension, hypovolemia, and blood loss in patients who have undergone angiography and cardiovascular surgery. Ischemic ATN is usually associated with acute and progressive renal dysfunction after the procedure, and kidney functions usually improve within 2–3 weeks if underlying etiology is properly managed.39 Multisystemic manifestations of CES and ARD, such as extrarenal emboli and cutaneous findings, may be beneficial in differentiation of these entities from contrast-induced AKI and ischemic ATN.
Risk factors for CES
Risk factors for CES are presented in Table 2. The most important risk factor for CES is atherosclerosis and most of the listed risk factors, such as diabetes mellitus, hypertension, hyperlipidemia, and smoking are actually also risk factors for atherosclerosis. Severity of atherosclerosis is the determinant of the risk of CES. It has been shown that if atherosclerotic plaques have ulceration, mobile thrombi, and thickness ≥4 mm in TEE, the risk of CES is very high.40,41 In addition to TEE, computed tomography and magnetic resonance imaging are becoming increasingly popular to determine the characteristics of the atherosclerotic plaques, which may allow us to stratify the risk of CES in high-risk populations.3
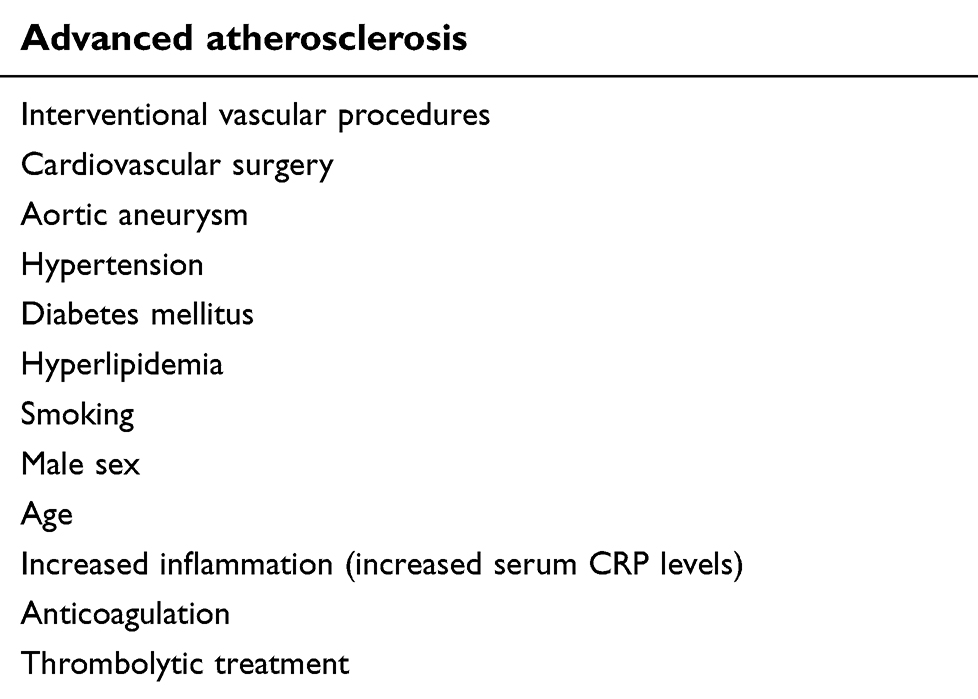 |
Table 2 Risk factors of cholesterol-embolization syndrome |
Although CES most commonly occurs iatrogenically after interventional procedures or cardiovascular surgery in 70% of cases, it may also occur spontaneously.35,42 In an autopsy study by Ramirez et al,10 incidence of spontaneous CES was only 4%, in contrast to 25%–30% in patients who died within 6 months after coronary angiography and aortography. Angiography seems to be the most frequent procedure causing CES (80% of iatrogenic cases).35,42 Among angiographic procedures, coronary angiography is the most common intervention causing CES, with incidence of 0.06%–1.8%.4,43
In Ascione et al,44 retinal and cerebral microembolization were compared between off-pump and traditional cardiopulmonary bypass techniques. In off-pump group, fewer microembolization events were found compared to the traditional surgery group. As we have previously mentioned, increased inflammation is an important risk factor for CES. In Fukumoto et al,11 plasma CRP levels were significantly higher in patients with CES than those without CES (0.7 vs 2.4 mg/dL). Furthermore, on multivariate analysis increased CRP was found to be an independent predictor of CES (OR 4.6). Presence of atherosclerotic renal artery stenosis was found to be an important risk factor for postprocedural ARD after renal arteriography, with incidence of 2%.45
Anticoagulation and fibrinolytics as a risk factor of CES
Anticoagulants and fibrinolytics have been accused of causing CES through rupture of plaques by causing internal hemorrhage and disruption of fibrous caps, which causes exposure of CCs to systemic circulation.38,46 However, these treatments rarely lead to CES in the absence of vascular intervention or surgery, and most of these patients who have anticoagulation-induced CES have an additional inciting event, such as angiography.
In Blankenship et al,12 60 patients with acute myocardial infarction who underwent coronary artery bypass–graft surgery were enrolled: 29 patients received thrombolytic therapy for myocardial infarction and 31 were treated conservatively. CES frequency was not different between the groups (14% vs 10%, respectively) which might denote that thrombolytics were not associated with induction of CES.
In several studies, warfarin has been reported to be related to CES, with incidence of 0.7%–1.0%.46 However, there have been other studies that have investigated whether anticoagulant therapy leads to CES in patients with aortic plaque determined by TEE, and in these studies no increased risk of CES was found with anticoagulation.11,47 In conclusion, a causal association between anticoagulants/fibrinolytics and CES has not been established.
Route of angiography as a risk factor of CES
Abdominal aorta may be the most severely involved location with atherosclerotic plaques, and thus mechanical injury and disruption of plaques by catheters has been considered to lead to CES.48 Therefore, it is hypothesized that the femoral approach may be associated with a higher risk of CES than the brachial approach.49 Brachial and femoral approaches of angiography have been compared in terms of risk of CES in several studies. In Fukumoto et al,11 there was no significant difference in prevalence of the femoral approach with and without CES. It was concluded that ascending aorta may also be a main embolic source leading to CES. Similarly, Johnson et al50 did not find a difference in peripheral vascular complications, such as CES, between the brachial and femoral approaches. However, in this study only one patient developed CES at follow-up, which may be regarded an important limitation of the study.
However, several other studies have shown less frequent AKI and possibly less ARD after a brachial/transradial approach compared to the femoral approach. In Kooiman et al, the risk of AKI was found to be significantly lower with the brachial route than the femoral approach.51 Similarly, in a large randomized multicenter trial (AKI-MATRIX), AKI occurred in 15% of patients with the radial approach and 17% with the femoral approach (OR 0.87, 95% CI 0.77–0.98; P=0.01).52 It was concluded that this lower risk of AKI might have been due to lower incidence of contrast-induced AKI and/or ARD. In another study performed on 69,214 patients after coronary angiography, development of chronic kidney disease within 6 months of intervention was significantly lower with the transradial approach than the transfemoral approach.53
Clinical manifestations
Clinical manifestations of CES are summarized in Table 3. Systemic constitutional symptoms, such as fever, fatigue, anorexia, weight loss, and myalgia, may be frequently observed during the course of CES. CES is characterized by a relatively long prodromal period between the inciting event and appearance of symptoms. For example, in a review of CES cases, skin findings were found to develop after more than a month of the inciting event.54 Most frequent manifestations of CES seem to be cutaneous and renal.
 |
Table 3 Clinical and laboratory manifestations of cholesterol-embolization syndrome |
Cutaneous manifestations
In a review of CES cases, skin findings were commonly observed, and reported to be present in 34% of cases.1 Cutaneous findings included livedo reticularis, cyanosis, gangrene, skin ulcers, purpura, erythematous nodules, and blue-toe syndrome. Since CES involves the small arteries and arterioles, arterial pulses are usually palpable in involved areas, which may be a discriminating feature of CES.55,56
Atheroembolic renal disease
ARD may develop in an acute, subacute, or chronic fashion. Massive embolization of CCs may cause acute ARD within 1 week of the inciting event. However, most commonly ARD follows a subacute clinical course, with progressive kidney dysfunction within several weeks. In a study, mean duration between vascular intervention and diagnosis of ARD was found to be 5.3 weeks.32 Another clinical form is chronic ARD, in which slow and progressive kidney dysfunction occurs. Chronic ARD is difficult to diagnose and frequently underdiagnosed, because it is clinically silent and extrarenal manifestations usually absent.57 Renal outcomes of ARD can be variable. Dialysis has been reported to be needed in 28%–61% of patients, with 20%–30% partially recovering renal function after several dialysis sessions.28,35,38,58,59
In patients with ARD, preexisting chronic kidney disease and long-standing hypertension have been found to be associated with increased risk of progression to end-stage renal disease (ESRD).35,60 Since CCs mostly involve the small arcuate and interlobar arteries and arterioles, symptoms and signs of ARD are usually bland when compared to thromboembolically induced renal infarction, which presents with acute severe flank pain.36
Mild–moderate proteinuria is commonly seen in ARD. However nephrotic-range proteinuria has also been reported in cases of CES-induced focal segmental glomerulosclerosis. In such cases, focal segmental glomerulosclerosis is usually a cellular variant with podocyte hypertrophy and capillary-loop collapse.61 CES-induced malignant hypertension may also cause significant proteinuria.62
ARD may also be seen after renal transplantation, with incidence of 0.39%–0.47%.62,63 It may occur very early or late after transplantation.64 The source of CCs may be donor or recipient vessels.65 CES of donor origin usually occurs early after transplantation, leading to primary allograft failure; however, CES of recipient origin usually occurs years after transplantation, causing chronic allograft dysfunction.66,67 CES of donor origin has been reported to have poorer prognosis.62
Hypertension
CES is known to be associated with resistant and malignant hypertension.68,69 In a review by Lye et al70 that included 129 patients, 48% of these were found to have severe hypertension. The pathophysiology of this hypertension seems to be caused by obstruction of renal arterioles by CCs, which in turn leads to activation of the RAAS. In a case series by Scolari et al,28 a history of hypertension exacerbated by CES was found in 46 patients (88%). In six patients (12%), de novo hypertension developed after CES.
Gastrointestinal manifestations
Gastrointestinal symptoms related to CES include abdominal pain, diarrhea, and bleeding.71 In addition to gastric and colonic ischemia and infarction, necrotizing pancreatitis, focal hepatic cell necrosis, and acalculous necrotizing cholecystitis have also been observed in the course of CES.72,73
Central nervous system and ocular manifestations
Central nervous system manifestations of CES include transient ischemic attack, stroke, confusion, headache, dizziness, paraparesis, mononeuropathy, spinal cord infarction, amaurosis fugax, eye pain, and blurred vision. CES usually leads to diffuse brain injury clinically represented by confusion and memory loss, rather than focal neurological signs and symptoms. Minor ischemic lesions and border-zone infarcts are characteristically observed in imaging studies in CES.74 In contrast, thromboembolism characteristically leads to acute focal neurologic symptoms.
Hollenhorst plaques are pathognomonic features of CES that can be seen as bright and refractive lesions in the retina. The most common source of these plaques is the carotid artery.75,76 However it should be noted that the presence of Hollenhorst plaques does not necessarily confirm that acute clinical picture is due to CES, because these plaques may represent a prior CES attack.77 Hollenhorst plaques have been found to persist for more than a year.76
Other manifestations
Myocardial infarction,78 adrenal insufficiency,55,79 penile necrosis,80 myositis,81 and splenic infarcts55,70,79 have been reported in the course of CES. Pulmonary involvement in the course of CES is rare, but may occur when CCs pass through systemic circulation into the venous system and pulmonary capillaries. Alveolar hemorrhage may also be seen as a manifestation of CES, which may be considered as new cause of pulmonary–renal syndromes.82
Laboratory testing
Leukocytosis, anemia, thrombocytopenia, eosinophilia, hypocomplementemia, increased erythrocyte-sedimentation rate, CRP, and fibrinogen may be seen in the course of CES.11,36 Incidence of eosinophilia has been reported to be 14%–71%. It is usually transient and induced by secreted IL5 due to activated T cells.2 CES with kidney dysfunction has been found to show a greater increase in eosinophil counts compared to patients without kidney dysfunction.11 Urinalysis in patients with CES is typically bland, with few cells or casts.42 Although mild proteinuria is usually seen, nephrotic-range proteinuria has rarely been reported.29,61 Proteinuria and active urinary sediment are suggested to be associated with glomerular embolization, rather than typical CES cases.2 Eosinophiluria may also be found when urine sediment is stained with Hansel's stain.83 ANA may become positive, due to antigenic stimulation by CCs in the course of CES84
Treatment and management
General measures and prevention of CES
Treatment strategies are presented in Table 4. CES is a manifestation of advanced atherosclerosis, and thus secondary prevention of cardiovascular disease is of utmost importance in these patients.85 These measures include aspirin, statins, cessation of smoking, and control of weight, blood pressure, and glycemia. Invasive interventional studies should be avoided as much as possible in patients with CES. Although not proven, the radial artery approach may be preferred to the femoral approach when arterial intervention is absolutely necessary. Although no casual relationship between anticoagulants/thrombolytic agents and CES has been proven, they have been reported to induce CES and thus they should not be used, unless these drugs have any other indication, such as atrial fibrillation or prosthetic valve. Although antiplatelet agents have not been proven as treatment of CES, they should be used for secondary prevention of cardiovascular diseases.1
 |
Table 4 Treatment and management of cholesterol-embolization syndrome |
Statins may have three basic beneficial effects as a treatment of CES: they lower low-density lipoprotein (LDL) levels, they may stabilize atherosclerotic plaques, and they may have pleiotropic anti-inflammatory effects.86,87 Statin treatment has been reported to be beneficial in CES, with improved cutaneous manifestations and renal outcomes.88,89 In a study performed on 354 patients with ARD, 116 needed dialysis and 102 died in a follow-up of 2 years.35 In this study, baseline statin treatment was found to be associated with a lower risk of ESRD and improved 1-year cumulative survival. LDL apheresis was reported to decrease the need for dialysis in 49 patients with CES after 6 months.90 Also, in other studies it was reported that LDL apheresis improved clinical manifestations in patients with CES.91,92
Anti-inflammatory treatments
Inflammation is one of the cornerstones in the pathophysiology of CES, and thus anti-inflammatory agents may be hypothesized to be effective in CES. Although several case reports and series have shown beneficial effects with such anti-inflammatory drugs as corticosteroids and cyclophosphamide, there has been no randomized controlled trial evaluating these drugs in the treatment of CES.59,93–96 Dahlberg et al18 reported that high-dose corticosteroids were effective in reducing symptoms in two patients with ARD. In another study, with oral prednisolone 1 mg/kg/day, kidney outcomes were improved in patients with CES.94 In contrast, several studies have shown that corticosteroids are not effective, especially in the long term.97
Colchicine is known to inhibit chemotaxis and phagocytosis of polymorphonuclear lymphocytes.98 Furthermore, colchicine has also been reported to block autoinflammatory pathways, including NLRP3 and IL1.99,100 Recently, colchicine has been reported to reduce the risk of cardiovascular events.101 A case of leg ulceration caused by CES was reported to improve with colchicine and corticosteroids.102
Interventional and surgical treatments
Endovascular interventions and surgical treatments, such as endarterectomy and bypass procedures, may be beneficial if the embolic source can be localized exactly.6,102 However, frequently the source of CES is not certain and embolization risk of the existing plaques is not predictable. In a study by Keen et al103 performed on 100 patients with CES, correction of the embolization source was achieved with bypass surgeries and endarterectomies. The survival rate was found to be 89% at 1 year. All seven early deaths occurred in patients with suprarenal aortic disease, 6 of whom were under hemodialysis treatment. In this study, it was concluded that surgical elimination of the embolization source can be performed with low mortality only when the source is located in the infrarenal aorta. Since the risk of morbidity and mortality is high, surgery should be considered only as rescue therapy in life-threatening situations in CES. Angioplasty and stent implantation have been reported in a small number of patients with aortoiliac and femoral arterial sources.6,104 There is always a high risk of further induction of CES in these vascular interventions, and thus embolic protection devices may decrease the risk of further embolization.
Prognosis
Prognosis of patients with CES is usually poor, probably due to advanced atherosclerosis and related comorbid cardiovascular diseases. In a recent study, 648 cases of iatrogenic CES were reviewed and composite incidence of mortality was reported to be 63%.105 In a review of 221 cases of histologically proven CES, mortality rates were as high as 80% when cases that had been diagnosed postmortem were included.55 In Fukumoto et al,11 in which patients who had undergone left-heart catheterization were enrolled, the in-hospital mortality rate of patients with CES was found to be 16% (four patients), which was significantly higher than the group without CES (0.5%; P<0.01). In this study, all cases of death had renal dysfunction after catheterization, suggesting that renal involvement in CES may have important prognostic implications. In another study performed on 354 patients diagnosed with ARD, at 2-year follow-up, >30% of patients had progressed to ESRD and 28% had died.42 Furthermore, most importantly, 1-year and 2-year survival was markedly reduced in these patients (83% and 75%, respectively).
Conclusion
CES is a multisystemic autoinflammatory disease that is frequently underdiagnosed. It may have a silent clinical course, and diagnosis is difficult in patients without prominent characteristic signs and symptoms. Therefore, high clinical suspicion is required for diagnosis. Its autoinflammatory nature and implications of NLRP3/IL1 pathways in CES pathogenesis have been very recently discovered. These autoinflammatory pathways are also important in atherosclerosis, and IL1 antagonists (canakinumab) have been investigated in atherosclerotic patients, with very promising results.106 These specific targeted treatments may also be beneficial in selected patients with CES in early phases of the disease. Future randomized trials are needed in this context. Other conventional anti-inflammatory agents, such as corticosteroids and cyclophosphamide, may be used as treatment of CES; however, serious side effects associated with these agents and lack of randomized studies limit their use.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation. 2010;122:631–641. doi:10.1161/CIRCULATIONAHA.109.886465
2. Modi KS, Rao VK. Atheroembolic renal disease. J Am Soc Nephrol. 2001;12:1781–1787.
3. Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15(4):315. doi:10.1007/s11883-013-0315-y
4. Scolari F, Bracchi M, Valzorio B, et al. Cholesterol atheromatous embolism: an increasingly recognized cause of acute renal failure. Nephrol Dial Transplant. 1996;11:1607–1612.
5. Tunick PA, Nayar AC, Goodkin GM, et al. Effect of treatment on the incidence of stroke and other emboli in 519 patients with severe thoracic aortic plaque. Am J Cardiol. 2002;90:1320. doi:10.1016/S0002-9149(02)02870-9
6. Carroccio A, Olin JW, Ellozy SH, et al. The role of aortic stent grafting in the treatment of atheromatous embolization syndrome: results after a mean of 15 months follow-up. J Vasc Surg. 2004;40:424. doi:10.1016/j.jvs.2004.06.036
7. Cross S. How common is cholesterol embolism? J Clin Pathol. 1991;44:859–861. doi:10.1136/jcp.44.12.1018
8. Drost H, Buis B, Haan D, Hillers JA. Cholesterol embolism as a complication of left heart catheterisation. Report of seven cases. Br Heart J. 1984;52:339–342. doi:10.1136/hrt.52.3.339
9. Moolenaar W, Lamers CB. Cholesterol crystal embolization in the Netherlands. Arch Intern Med. 1996;156:653–657. doi:10.1001/archinte.1996.00440060081009
10. Ramirez G, O’Neill WM, Lambert R, Bloomer HA. Cholesterol embolization: a complication of angiography. Arch Intern Med. 1978;138:1430–1432. doi:10.1001/archinte.1978.03630340096035
11. Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211. doi:10.1016/S0735-1097(03)00579-5
12. Blankenship JC, Butler M, Garbes A. Prospective assessment of cholesterol embolization in patients with acute myocardial infarction treated with thrombolytic vs conservative therapy. Chest. 1995;107:662. doi:10.1378/chest.107.3.662
13. Jones DB, Iannacone PM. Atheromatous emboli in renal biopsies. An ultrastructural study. Am J Pathol. 1975;78:261–276.
14. Lie JT. Cholesterol atheromatous embolism. The great masquerader revisited. Pathol Annu. 1992;27:17–50.
15. Preston RA, Stemmer CL, Materson BJ, Perez-Stable E, Pardo V. Renal biopsy in patients 65 years of age or older. An analysis of the results of 334 biopsies. J Am Geriatr Soc. 1990;38:669–674.
16. Haas M, Spargo BH, Wit EJ, Meehan SM. Etiologies and outcome of acute renal insufficiency in older adults: a renal biopsy study of 259 cases. Am J Kidney Dis. 2000;35:433–447. doi:10.1016/S0272-6386(00)70196-X
17. Dizman N, Aydın Bahat K, Özkanlı Ş, Özkök A. Cholesterol embolization syndrome: a report of two cases. Turk Kardiyol Dern Ars. 2016;44(3):251–255. doi:10.5543/tkda.2015.94587
18. Dahlberg PJ, Frecentese DF, Cogbill TH. Cholesterol embolism: experience with 22 histologically proven cases. Surgery. 1989;105(6):737–746.
19. Hitti WA, Wali RK, Weinman EJ, Drachenberg C, Briglia A. Cholesterol embolization syndrome induced by thrombolytic therapy. Am J Cardiovasc Drugs. 2008;8(1):27–34. doi:10.2165/00129784-200808010-00004
20. Li X, Bayliss G, Zhuang S. Cholesterol crystal embolism and chronic kidney disease. Int J Mol Sci. 2017;18(6):1120.
21. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi:10.1038/nature08940
22. Kiyotake R, Oh-Hora M, Ishikawa E, Miyamoto T, Ishibashi T, Yamasaki S. Human mincle binds to cholesterol crystals and triggers innate immune responses. J Biol Chem. 2015;290:25322–25332. doi:10.1074/jbc.M115.645234
23. Fukumoto Y, Shimokawa H, Kozai T, et al. Vasculoprotective role of inducible nitric oxide synthase at inflammatory coronary lesions induced by chronic treatment with interleukin-1 beta in pigs in vivo. Circulation. 1997;96:3104–3111. doi:10.1161/01.CIR.96.9.3104
24. Ridker PM. Intrinsic fibrinolytic capacity and systemic inflammation: novel risk factors for arterial thrombotic disease. Haemostasis. 1997;27:2–11.
25. Hammerschmidt DE, Greenberg CS, Yamada O, Craddock PR, Jacob HS. Cholesterol and atheroma lipids activate complement and stimulate granulocytes. A possible mechanism for amplification of ischemic injury in atherosclerotic states. J Lab Clin Med. 1981;98:68–77.
26. Snyder HE, Shapiro JL. A correlative study of atheroembolism in human beings and experimental animals. Surgery. 1961;49:195–204.
27. Cosio FG, Zager RA, Sharma HM. Atheroembolic renal disease causes hypocomplementaemia. Lancet. 1985;2:118–121. doi:10.1016/S0140-6736(85)90225-9
28. Scolari F, Tardanico R, Zani R, et al. Cholesterol crystal embolism: a recognizable cause of renal disease. Am J Kidney Dis. 2000;36(6):1089–1109.
29. Haqqie SS, Urizar RE, Singh J. Nephrotic-range proteinuria in renal atheroembolic disease: report of four cases. Am J Kidney Dis. 1996;28(4):493. doi:10.1016/S0272-6386(96)90458-8
30. Carvajal JA, Anderson WR, Weiss L, Grismer J, Berman R. Atheroembolism: an etiologic factor in renal insufficiency, gastrointestinal hemorrhages, and peripheral vascular diseases. Arch Intern Med. 1967;119:593–599. doi:10.1001/archinte.1967.00290240115009
31. Goldman M, Thoua Y, Dhaene M, Toussaint C. Necrotising glomerulonephritis associated with cholesterol microemboli. Br Med J. 1985;290:205–206. doi:10.1136/bmj.290.6463.205
32. Frock J, Bierman M, Hammeke M, Reyes A. Atheroembolic renal disease: experience with 22 patients. Nebr Med J. 1994;79:317–321.
33. Meyrier A, Buchet P, Simon P, Fernet M, Rainfray M, Callard P. Atheromatous renal disease. Am J Med. 1988;85(2):139. doi:10.1016/S0002-9343(88)80332-2
34. Eliot RS, Kanjuh VI, Edwards JE. Atheromatous embolism. Circulation. 1964;30:611–618.
35. Scolari F, Ravani P, Pola A, et al. Predictors of renal and patient outcomes in atheroembolic renal disease: a prospective study. J Am Soc Nephrol. 2003;14:1584–1590.
36. Saric MKronzon I. Embolism from atherosclerotic plaque: atheroembolism (cholesterol crystal embolism). 2019. Avaialble from: https://www.uptodate.com/contents/embolism-from-atherosclerotic-plaque-atheroembolism-cholesterol-crystal-embolism. Accessed May 25, 2019.
37. O’Keeffe ST, Woods BO, Breslin DJ, Tsapatsaris NP. Blue toe syndrome: causes and management. Arch Intern Med. 1992;152:2197–2202. doi:10.1001/archinte.1992.00400230023004
38. Thadhani RI, Camargo CA
39. Scolari FGCPalevsky PM. Clinical presentation, evaluation, and treatment of renal atheroemboli. 2018. Available from: https://www.uptodate.com/contents/clinical-presentation-evaluation-and-treatment-of-renal-atheroemboli. Accessed May 25, 2019.
40. Tunick PA, Rosenzweig BP, Katz ES, Freedberg RS, Perez JL, Kronzon I. High risk for vascular events in patients with protruding aortic atheromas: a prospective study. J Am Coll Cardiol. 1994;23(5):1085.
41. Tunick PA, Kronzon I. Protruding atherosclerotic plaque in the aortic arch of patients with systemic embolization: a new finding seen by transesophageal echocardiography. Am Heart J. 1990;120(3):658. doi:10.1016/0002-8703(90)90024-R
42. Scolari F, Ravani P, Gaggi R, et al. The challenge of diagnosing atheroembolic renal disease: clinical features and prognostic factors. Circulation. 2007;116(3):298. doi:10.1161/CIRCULATIONAHA.106.680991
43. Saklayen MG, Gupta S, Suryaprasad A, Azmeh W. Incidence of atheroembolic renal failure after coronary angiography. A prospective study. Angiology. 1997;48(7):609. doi:10.1177/000331979704800707
44. Ascione R, Ghosh A, Reeves BC, et al. Retinal and cerebral microembolization during coronary artery bypass surgery: a randomized, controlled trial. Circulation. 2005;112:3833–3838. doi:10.1161/CIRCULATIONAHA.105.557462
45. Rudnick MR, Berns JS, Cohen RM, Goldfarb S. Nephrotoxic risks of renal angiography: contrast media-associated nephrotoxicity and atheroembolism–a critical review. Am J Kidney Dis. 1994;24(4):713. doi:10.1016/S0272-6386(12)80235-6
46. Scolari F, Ravani P. Atheroembolic renal disease. Lancet. 2010;375(9726):1650. doi:10.1016/S0140-6736(09)62073-0
47. Blackshear JL, Zabalgoitia M, Pennock G, et al. Warfarin safety and efficacy in patients with thoracic aortic plaque and atrial fibrillation. SPAF TEE Investigators. Stroke prevention and atrial fibrillation. Transesophageal echocardiography. Am J Cardiol. 1999;83(3):453. doi:10.1016/S0002-9149(98)00886-8
48. Rosansky SJ, Deschamps EG. Multiple cholesterol emboli syndrome after angiography. Am J Med Sci. 1984;288:45–48. doi:10.1097/00000441-198407000-00012
49. Oda H, Miida T, Sato H, Higuma N. Treatment of unstable angina with cholesterol embolization as a complication of left heart catheterization. Jpn Circ J. 1990;54:487–492. doi:10.1253/jcj.54.487
50. Johnson LW, Esente P, Giambortolomei A, et al. Peripheral vascular complications of coronary angioplasty by the femoral and brachial techniques. Cathet Cardiovasc Diagn. 1994;31:165–172. doi:10.1002/ccd.1810310302
51. Kooiman J, Seth M, Dixon S, et al. Risk of acute kidney injury after percutaneous Coronary interventions using radial versus femoral access. insights from the blue cross blue shield of Michigan cardiovascular consortium. Circ Cardiovasc Interv. 2014;7:190–198. doi:10.1161/CIRCINTERVENTIONS.113.000778
52. Ando G, Cortese B, Russo F, et al. Acute kidney injury after radial or femoral access for invasive acute Coronary syndrome management, AKI-MATRIX. J Am Coll Cardiol. 2017;69:2592–2603. doi:10.1016/j.jacc.2016.11.026
53. Vuurmans T1, Byrne J, Fretz E, et al. Chronic kidney injury in patients after cardiac catheterisation or percutaneous coronary intervention: a comparison of radial and femoral approaches (from the British Columbia Cardiac and Renal Registries). Heart. 2010;96(19):1538–1542. doi:10.1136/hrt.2009.192294
54. Falanga V, Fine MJ, Kapoor WN. The cutaneous manifestations of cholesterol crystal embolization. Arch Dermatol. 1986;122(10):1194. doi:10.1001/archderm.1986.01660220112024
55. Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38(10):769. doi:10.1177/000331978703801007
56. Donohue KG, Saap L, Falanga V. Cholesterol crystal embolization: an atherosclerotic disease with frequent and varied cutaneous manifestations. J Eur Acad Dermatol Venereol. 2003;17(5):504. doi:10.1046/j.1468-3083.2003.00710.x
57. Zucchelli P, Zuccalà A. The diagnostic dilemma of hypertensive nephrosclerosis: the nephrologist’s view. Am J Kidney Dis. 1993;21:87–91. doi:10.1016/0272-6386(93)70100-D
58. Mayo RR, Swartz RD. Redefining the incidence of clinically detectable atheroembolism. Am J Med. 1996;100:524–529. doi:10.1016/S0002-9343(95)00059-3
59. Belenfant X, Meyrier A, Jacquot C. Supportive treatment improves survival in multivisceral cholesterol crystal embolism. Am J Kidney Dis. 1999;33:840–850.
60. Theriault J, Agharazzi M, Dumont M, Pichette V, Ouimet D, Leblanc M. Atheroembolic renal failure requiring dialysis: potential for renal recovery? A review of 43 cases. Nephron Clin Pract. 2003;94:c11–c18. doi:10.1159/000070819
61. Greenberg A, Bastacky SI, Iqbal A, Borochovitz D, Johnson JP. Focal segmental glomerulosclerosis associated with nephrotic syndrome in cholesterol atheroembolism: clinicopathological correlations. Am J Kidney Dis. 1997;29:334–344. doi:10.1016/S0272-6386(97)90193-1
62. Meyrier A. Cholesterol crystal embolism: diagnosis and treatment. Kidney Int. 2006;69(8):1308–1312. doi:10.1038/sj.ki.5000263
63. Lai CK, Randhawa PS. Cholesterol embolization in renal allografts: a clinicopathologic study of 12 cases. Am J Surg Pathol. 2007;31:536–545. doi:10.1097/PAS.0b013e31802b30e3
64. Ripple M, Charney D, Nadasdy T. Cholesterol embolization in renal allografts. Transplantation. 2000;69:2221–2225. doi:10.1097/00007890-200005270-00050
65. Darsee JR. Cholesterol embolism: the great masquerader. South Med J. 1979;72:174–180. doi:10.1097/00007611-197902000-00017
66. Mulay SR, Evan A, Anders HJ. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant. 2014;29:507–514. doi:10.1093/ndt/gft248
67. Gonzalez AP, Juega J, Vazquez C, et al. Late onset of cholesterol embolism leading to graft failure after renal transplantation: report of two cases. Transplant Proc. 2015;47:2361–2363. doi:10.1016/j.transproceed.2015.09.005
68. Saleem S, Lakkis FG, Martinez-Maldonado M. Atheroembolic renal disease. Semin Nephrol. 1996;16:309–318.
69. Dalakos TG, Streeten DH, Jones D, Obeid A. ‘Malignant’ hypertension resulting from atheromatous embolization predominantly of one kidney. Am J Med. 1974;7:135–138. doi:10.1016/0002-9343(74)90778-5
70. Lye WC, Cheah JS, Sinniah R. Renal cholesterol embolic disease. Case report and review of the literature. Am J Nephrol. 1993;13:489–493. doi:10.1159/000168669
71. Moolenaar W, Lamers CB. Gastrointestinal blood loss due to cholesterol crystal embolization. J Clin Gastroenterol. 1995;21(3):220. doi:10.1097/00004836-199510000-00011
72. Ben-Horin S, Bardan E, Barshack I, Zaks N, Livneh A. Cholesterol crystal embolization to the digestive system: characterization of a common, yet overlooked presentation of atheroembolism. Am J Gastroenterol. 2003;98(7):1471–1479. doi:10.1111/j.1572-0241.2003.07532.x
73. Moolenaar W, Lamers CB. Cholesterol crystal embolization to liver, gallbladder, and pancreas. Dig Dis Sci. 1996;41(9):1819. doi:10.1007/BF02088752
74. Ezzeddine MA, Primavera JM, Rosand J, Hedley-Whyte ET, Rordorf G. Clinical characteristics of pathologically proved cholesterol emboli to the brain. Neurology. 2000;54:1681–1683. doi:10.1212/WNL.54.8.1681
75. Hollenhorst RW. Significance of bright plaques in the retinal arterioles. JAMA. 1961;178:23. doi:10.1001/jama.1961.03040400025005
76. Bunt TJ. The clinical significance of the asymptomatic Hollenhorst plaque. J Vasc Surg. 1986;4(6):559. doi:10.1016/0741-5214(86)90169-2
77. Babikian V, Wijman CA, Koleini B, Malik SN, Goyal N, Matjucha IC. Retinal ischemia and embolism. Etiologies and outcomes based on a prospective study. Cerebrovasc Dis. 2001;12(2):108. doi:10.1159/000047689
78. Trono R, Sutton C, Hollman J, Suit P, Ratliff NB. Multiple myocardial infarctions associated with atheromatous emboli after PTCA of saphenous vein grafts. A clinicopathologic correlation. Cleve Clin J Med. 1989;56:581–584. doi:10.3949/ccjm.56.6.581
79. Moolenaar W, Lamers CBHW. Cholesterol crystal embolization and the digestive system. Scand J Gastroenterol. 1991;188:69–72. doi:10.3109/00365529109111232
80. Mondragon P, Descombes E, Bollmann J, et al. Penile necrosis in a haemodialysis patient: a rare manifestation of cholesterol crystal embolism. Nephrol Dial Transplant. 1998;13:3233–3235.
81. Robinson R, Pemberton M, Goddard M. Myositis due to cholesterol emboli. Postgrad Med J. 1993;69:947–949. doi:10.1136/pgmj.69.818.947
82. Sabatine MS, Oelberg DA, Mark EJ, Kanarek DH. Pulmonary cholesterol crystal embolization. Chest. 1997;112:1687–1692.
83. Wilson DM, Salazer TL, Farkouh ME. Eosinophiluria in atheroembolic renal disease. Am J Med. 1991;91(2):186. doi:10.1016/0002-9343(91)90013-N
84. Kumar A, Turney JH. Vasculitis look-alikes: variants of renal atheroembolic disease. Nephrol Dial Transplant. 1999;14:2053. doi:10.1093/ndt/14.8.2053
85. Smith SC
86. Tousoulis D, Psarros C, Demosthenous M, Patel R, Antoniades C, Stefanadis C. Innate and adaptive inflammation as a therapeutic target in vascular disease: the emerging role of statins. J Am Coll Cardiol. 2004;63:2491–2502. doi:10.1016/j.jacc.2014.01.054
87. Akdim F, Van Leuven SI, Kastelein JJ, G. Stroes E. Pleiotropic effects of statins: stabilization of the vulnerable atherosclerotic plaque? Curr Pharm Des. 2007;13:1003–1012. doi:10.2174/138161207780487548
88. Yonemura K, Ikegaya N, Fujigaki Y, Suzuki H, Togawa A, Hishida A. Potential therapeutic effect of simvastatin on progressive renal failure and nephrotic-range proteinuria caused by renal cholesterol embolism. Am J Med Sci. 2001;322:50–52. doi:10.1097/00000441-200107000-00010
89. Woolfson RG, Lachmann H. Improvement in renal cholesterol emboli syndrome after simvastatin. Lancet. 1998;351:1331–1332. doi:10.1016/S0140-6736(05)79058-9
90. Ishiyama K, Sato T, Taguma Y. Low-density lipoprotein apheresis ameliorates renal prognosis of cholesterol crystal embolism. Ther Apheresis Dial. 2015;19:355–360. doi:10.1111/1744-9987.12345
91. Tsunoda S, Daimon S, Miyazaki R, et al. LDL apheresis as intensive lipid-lowering therapy for cholesterol embolism. Nephrol Dial Transplant. 1999;14:1041–1042. doi:10.1093/ndt/14.4.1041b
92. Tamura K, Umemura M, Yano H, et al. Acute renal failure due to cholesterol crystal embolism treated with LDL apheresis followed by corticosteroid and candesartan. Clin Exp Nephrol. 2003;7:67–71. doi:10.1007/s10157-003-0243-1
93. Mann SJ, Sos TA. Treatment of atheroembolization with corticosteroids. Am J Hypertens. 2001;14(8 Pt 1):831.
94. Desai M, Ram R, Prayaga A, Dakshinamurty KV. Cholesterol crystal embolization (CCE): improvement of renal function with high-dose corticosteroid treatment. Saudi J Kidney Dis Transpl. 2011;22:327–330.
95. Stabellini N, Cerretani D, Russo G, Rizzioli E, Gilli P. Renal atheroembolic disease: evaluation of the efficacy of corticosteroid therapy. G Ital Nefrol. 2002;19:18–21.
96. Yucel A, Kart-Koseoglu H, Demirhan B, Ozdemir F. Cholesterol crystal embolization mimicking vasculitis: success with corticosteroid and cyclophosphamide therapy in two cases. Rheumatol Int. 2006;26:454–460. doi:10.1007/s00296-005-0012-4
97. Nakayama M, Izumaru K, Nagata M, et al. The effect of low-dose corticosteroids on short- and long-term renal outcome in patients with cholesterol crystal embolism. Ren Fail. 2011;33:298–306. doi:10.3109/0886022X.2011.618968
98. Sullivan TP, King LE, Boyd AS. Colchicine in dermatology. J Am Acad Dermatol. 1998;39:993–999. doi:10.1016/S0190-9622(98)70275-0
99. Martínez GJ, Celermajer DS, Patel S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis. 2018;269:262–271. doi:10.1016/j.atherosclerosis.2017.12.027
100. Robertson S, Martínez GJ, Payet CA, et al. Colchicine therapy in acute coronary syndrome patients acts on caspase-1 to suppress NLRP3 inflammasome monocyte activation. Clin Sci (Lond). 2016;130(14):1237–1246. doi:10.1042/CS20160090
101. Nidorf SM, Eikelboom JW, Thompson PL. Targeting cholesterol crystal-induced inflammation for the secondary prevention of cardiovascular disease. J Cardiovasc Pharmacol Ther. 2014;19(1):45–52. doi:10.1177/1074248413499972
102. Verneuil L, Ze Bekolo R, Dompmartin A, Comoz F, Marcelli C, Leroy D. Efficiency of colchicine and corticosteroids in a leg ulceration with cholesterol embolism in a woman with rheumatoid arthritis. Rheumatology (Oxford). 2003;42(8):1014–1016. doi:10.1093/rheumatology/keg252
103. Keen RR, McCarthy WJ, Shireman PK, et al. Surgical management of atheroembolization. J Vasc Surg. 1995;21(5):773.
104. Matchett W, McFarland D, Eidt J, Moursi M. Blue toe syndrome: treatment with intra-arterial stents and review of therapies. J Vasc Interv Radiol. 2000;11:585–592. doi:10.1016/S1051-0443(07)61610-8
105. Agrawal A, Ziccardi MR, Witzke C, Palacios I, Rangaswami J. Cholesterol embolization syndrome: an under-recognized entity in cardiovascular interventions. J Interv Cardiol. 2018;31(3):407–415. doi:10.1111/joic.12483
106. Ridker PM, Everett BM, Thuren T, et al.; CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.