Back to Journals » International Journal of Nanomedicine » Volume 14
Cholate-modified polymer-lipid hybrid nanoparticles for oral delivery of quercetin to potentiate the antileukemic effect
Authors Yin J, Hou Y, Song X, Wang P, Li Y
Received 26 March 2019
Accepted for publication 8 May 2019
Published 31 May 2019 Volume 2019:14 Pages 4045—4057
DOI https://doi.org/10.2147/IJN.S210057
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Juntao Yin,1 Yantao Hou,2 Xiaoyong Song,3 Peiqing Wang,1 Yang Li1
1Department of Pharmaceutics, Huaihe Hospital Affiliated to Henan University, Kaifeng, People’s Republic of China; 2Henan Vocational College of Applied Technology, Kaifeng, People’s Republic of China; 3School of Pharmacy, Henan University, Kaifeng, People’s Republic of China
Background: Quercetin (QUE) shows a potential antileukemic activity, but possesses poor solubility and low bioavailability.
Purpose: This article explored the bile salt transport pathway for oral deliver of QUE using cholate-modified polymer-lipid hybrid nanoparticles (cPLNs) aiming to enhance its antileukemic effect.
Methods: QUE-loaded cPLNs (QUE-cPLNs) were developed through a nanoprecipitation technique and characterized by particle size, entrapment efficiency (EE), microscopic morphology and in vitro drug release. In vitro cellular uptake and cytotoxicity of QUE-cPLNs were examined on Caco-2 and P388 cells; in vivo pharmacokinetics and antileukemic effect were evaluated using Sprague Dawley rats and leukemic model mice, respectively.
Results: The prepared QUE-cPLNs possessed a particle size of 110 nm around with an EE of 96.22%. QUE-cPLNs resulted in significantly enhanced bioavailability of QUE, up to 375.12% relative to the formulation of suspensions. In addition, QUE-cPLNs exhibited excellent cellular uptake and internalization capability compared to cholate-free QUE-PLNs. The in vitro cytotoxic and in vivo antileukemic effects of QUE-cPLNs were also signally superior to free QUE and QUE-PLNs.
Conclusion: These findings indicate that cPLNs are a promising nanocarrier able to improve the oral bioavailability and therapeutic index of QUE.
Keywords: quercetin, polymer-lipid hybrid nanoparticles, bile salt, bioavailability, leukemia
Introduction
Quercetin (QUE) is a flavonoid compound widely found in flower, leaf and fruit of a diversity of plants. QUE has been proven to possess various bioactivities and pharmacological actions, such as anticancer, antidiabetes, antioxidation, anti-allergy, anti-anemia, and anti-inflammation.1 Among these, its antileukemic effect has attracted increasing attention in recent years. In a pilot study, QUE demonstrated the potential of stabilizing the rising lymphocyte counts of patients with PIM1 kinase-positive chronic lymphocytic leukemia.2 The antileukemic action of QUE was also verified in a xenograft model of human leukemia HL60 cells.3 In addition, QUE has been shown able to sensitize human myeloid leukemia KG-1 cells against TRAIL-induced apoptosis.4 However, QUE is almost insoluble in water and simultaneously has a poor lipophilicity,5 therefore the oral bioavailability of QUE is fairly inadequate. Clinical development and application of QUE are severely impeded by its limited oral absorption and formulation challenge. To improve the pharmaceutical properties of QUE, a variety of formulation strategies have been developed and tried out, including polysaccharide nanoparticles,6 polymeric micelles,7 phospholipid complexes,8 and nanocrystals.9 Nevertheless, these approaches still take possession of some shortfalls as oral delivery carriers. Polysaccharide-based nanoparticles have inadequate encapsulation rate towards hydrophobic drugs. Polymeric micelles also possess low drug loading capacity. Phospholipid complexes, also known as phytosomes,10 are easy to be oxidized and labile to digestive conditions. It is imperative to develop suitable drug delivery systems to ulteriorly enhance the oral delivery of QUE as well as its therapeutic potency.
Polymer–lipid hybrid nanoparticles (PLNs), structurally composed of a phospholipid shell and a polymer core, are provided with excellent properties as drug delivery carriers, combining the advantages of liposomes with those of polymer nanoparticles.11 The lipid monolayer offers needful stability by forming a low tension of oil–water interface whereby to reduce aggregation of particles. Its polymer core is not only biodegradable, but also can improve the drug loading rate (especially for amphiphobic ones) and the gastrointestinal (GI) stability of carrier.12 PLNs have been shown to be a promising drug nanocarrier for multipurpose delivery of various therapeutic agents.13–15 Although PLNs possess fine biocompatibility and stability, there are still certain limitations for them as oral carriers if only utilizing their nanoscale effect and nonspecific absorption. In this case, the surface engineering of nanocarriers become particularly significant to further promote the oral absorption of the payload. As known, the GI transport of bile salts or cholates greatly depends on the apical sodium-dependent bile acid transporter (ASBT),16 which is an active and high-efficiency transport mechanism. In recent years, the importance of utilizing bile salt transport pathway to deliver poorly bioavailable drugs has been highlighted.17–21 However, cholate-modified PLNs (cPLNs) that take advantage of the bile salt transport pathway and lipid-facilitated absorption for oral delivery of therapeutic agents has not been explored.
In this study, a biomimetic nanocarrier based on PLNs was developed by incorporating sodium taurocholate in PLNs for oral delivery of QUE, aiming to potentiate its antileukemic effect. We prepared QUE-loaded cPLNs (QUE-cPLNs) via a nanoprecipitation technique and characterized them with particle size, entrapment efficiency (EE) and morphology. The in vitro release, in vivo oral pharmacokinetics, cellular uptake and internalization, cytotoxicity on leukemia cells, and in vivo antileukemic effect of QUE-cPLNs were investigated and compared with unentrapped QUE as well as cholate-free QUE-loaded PLNs (QUE-PLNs).
Materials and methods
Materials
Quercetin and sodium taurocholate were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). PLGA (DL-lactide: glycolide =50: 50, Mw ~7,500) was obtained from RESENBio Technology Co., Ltd (Xi’an, China). Lecithin S100 was provided by Lipoid GmbH (Ludwigshafen, Germany). Hoechst 33258 and 3, 3ʹ-dioctadecyloxacarbocyanine perchlorate (DiO) were from Aladdin Reagents (Shanghai, China). Deionized water was prepared by a Milli-Q water purifier (EMD Millipore, Billerica, MA, USA). HPLC-grade methanol was provided by Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All other chemicals were of analytical grade and used as received.
Preparation of QUE-cPLNs
QUE-cPLNs were prepared by the nanoprecipitation technique followed by evaporation to remove the organic solvent according to the reported procedure.22 Typically, 10 mg of QUE, 20 mg of PLGA, and 50 mg of lecithin were dissolved in 2 mL of acetonitrile to form the organic phase. Then, the organic phase was rapidly injected into 5 mL of deionized water dissolving 20 mg of sodium taurocholate under constant stirring. Upon the organic solvent diffusing into water, the carrier materials and drug spontaneously formed hybrid nanoparticles. After that, the resulting mixture was evaporated at 40°C under the normal atmospheric pressure for 24 hours to remove the residual acetonitrile. After removal of the organic solvent, the final suspensions were forced to extrude through a 0.45 μm filter to homogenize the nanoparticles. The formulation factors affecting the formulation performance were screened based on testing at a single level by changing one factor while keeping the other two constant, including the QUE usage, the ratio of PLGA to lecithin and the sodium taurocholate level in the aqueous phase. Cholate-free QUE-PLNs were prepared as a control formulation following the same procedure.
Characterization of QUE-cPLNs
The particle size of QUE-cPLNs was measured by Zetasizer Nano ZS (Malvern, Worcestershire, UK) at 25°C. In operation, QUE-cPLNs were diluted to appropriately 25 times with deionized water and then placed in a disposable cuvette. After equilibrium for 120 seconds, the samples were subjected to laser diffraction for particle size test based on the dynamic light scattering principle. The same sample was proceeded for Doppler velocimetry test to determine the ζ potential.
The morphology of QUE-cPLNs was observed using a transmission electron microscopy (TEM) (Tecnai 10, Philips, Eindhoven, Netherlands). QUE-cPLNs diluted with deionized water to an appropriate level were dropped onto a carbon-coated copper grid and fixed by evaporation under a heating lamp. Afterwards, the fixed nanoparticles were transferred to the TEM probe for inspection. Morphologic micrographs were taken at the acceleration voltage of 100 kV.
Determination of entrapment efficiency
The entrapment efficiency (EE) of QUE-cPLNs was determined by centrifugal ultrafiltration with HPLC quantification.23 Briefly, freshly prepared QUE-cPLNs were firstly centrifuged at 5,000 rpm for 5 minutes to precipitate the unentrapped bulk drug. Then, the upper part of nanosuspensions were carefully separated and centrifuged against a centrifugal filter device (Amicon® Ultra-0.5, MWCO 100 kDa, EMD, Millipore) to collect the filtrate. Free QUE in the filtrate was quantified by HPLC established below. EE was calculated according to the equation: EE (%) = (1–Mfre/Mtot) ×100%, where Mfre and Mtot represent the amount of free QUE in the nanosuspensions and total QUE in the system, respectively.
In vitro release study
The in vitro release of QUE from QUE-cPLNs was studied by a reverse bulk equilibrium dialysis technique.24 Briefly, aliquots of QUE-cPLNs equilibrant to 100 mg of QUE were put into 900 mL of water, 0.1 M HCl solution or pH 6.8 PBScarried out in the dissolution cup, in which 0.1% (w/v) Tween® 80 (Shanghai Aladdin Biochemical Technology Co., Ltd, Shanghai, China) was added as a solubilizing agent. Nine ready-to-use dialysis tubes (Spectra/Por® Float-A-Lyzer® G2, MWCO 100 kDa, Spectrum China, Shanghai, China) filled with blank release medium were placed in the dissolution cup, respectively, three in one cup for each medium. At the predetermined intervals (0.25, 0.5, 1, 2, 4, 6, 8, 10 and 12 hours), 100 μL of medium was withdrawn from the dialysis tube. QUE concentration in the release medium was determined by HPLC described below, and the accumulative release percentage of QUE from QUE-cPLNs was calculated according to the release ratio.
HPLC assay for QUE
QUE quantification for both in vitro and in vivo samples was performed on an Agilent 1100 Series HPLC system equipped with a quaternary pump, a degasser, an autosampler, a column heater, and a tunable ultraviolet detector. Separation was carried out against a Diamonsil C18 column (4 μm, 4.6×250 mm, Agilent, USA) at 40°C with 20 μL of injection volume. A mobile phase composed of 55% methanol and 45% water containing 0.1% trifluoroacetic acid (v/v) pumped at a flow rate of 1.0 mL/minute was used to elute the samples. The eluents were monitored at 365 nm. Quantification was based on a standard curve (y=20.234x+20.853, R2 = 0.9998), fitted by the peak area to QUE sample of different concentrations (0.125, 0.5, 2.5, 5, and 10 μg/mL).
Oral pharmacokinetic study
Sprague Dawley rats weighing 220±20 g were used to study the oral pharmacokinetics of QUE and its preparations. The rats were fasted overnight before administration but allowed free access to water. They were randomly divided into QUE suspensions, QUE-PLNs and QUE-cPLNs groups (N=6). The reference formulation, QUE suspensions, was manufactured by grinding the drug with sodium carboxymethyl cellulose (0.5%) in an appropriate amount of water. The rats were given with three kinds of preparations orally by gavage at a dose of 25 mg/kg. Following administration, approximately 250 μL of blood was sampled from the caudal vein of rats into the heparinized tubes at 0.5, 1, 2, 4, 6, 8, 10 and 12 hours. The blood samples were immediately centrifuged at 5,000 rpm for 5 minutes to collect the plasma. A deproteinization procedure was carried out to withdraw QUE by adding four aliquots of methanol into an aliquot of plasma.24 After condensation against a vacuum concentrator, the residues were reconstituted in 50 μL of mobile phase for HPLC analysis with vanillin as an internal standard. All animal experiments were conducted according to the Guidelines on the Care and Use of Animals for Scientific Purposes (2004) and approved by the Experimental Animal Ethical Committee of Huaihe Hospital Affiliated to Henan University.
Cellular uptake and internalization
Caco-2 cells from American Type Culture Collection (ATCC, Manassas, VA, USA) were used to investigate the cellular uptake and internalization of QUE-cPLNs, and the cell culture refers to the reported protocol.25 When the cells grew to ~80–90% confluence, they were ready for the cellular uptake test. QUE solution (dissolved in 75% ethanol followed by dilution with water), QUE-PLNs and QUE-cPLNs, diluted to 25 μg/mL with the culture medium, were added into the wells loading Caco-2 cells for coincubation. In another uptake test of QUE-cPLNs, sodium taurocholate (100 μg/mL) was adopted as uptake competitor to interpret the absorption mechanism. At the time of 0.5, 1 and 2 hours, the upper media were carefully removed and the cells were washed with cold PBS for two times. After that, the cells were lysed with RIPA lysis buffer. The supernatants were collected after centrifugation, in which the protein content was quantified by BCA protein assay kit. QUE concentration in the supernatant was determined by HPLC and corrected by the cell protein level of each well.
To discriminate the cellular internalization of QUE-PLN and QUE-cPLNs, DiO-labeled QUE-PLNs and QUE-cPLNs were specially prepared by introducing the fluorescer of DiO into corresponding formulations upon preparation. The well-cultured Caco-2 cells incubated with DiO-labeled QUE-PLNs and QUE-cPLNs for 0.5 hour at 37ºC. The media were removed and the cells were washed with PBS three times. The cells were then immobilized by 4% paraformaldehyde and imaged using a LSM510 confocal laser scanning microscopy (CLSM, Zeiss, Oberkochen, Germany) after nuclear staining with Hoechst 33258.
In vitro cytotoxic evaluation on P388 cells
The cytotoxicity of free and formulated QUE was evaluated on the lymphoblastic leukemia P388 cells (Chinese Academy of Sciences Cell Resource Center). P388 cells were cultured in RPMI 1640 medium (Gibco, Waltham, USA) supplemented with 10% FBSin an atmosphere of 5% CO2 at 37ºC. The cells were seeded in 96-well plates with a density of 5×105/mL for 48 hours. After washing with PBS, QUE solution, QUE-PLNs and QUE-cPLNs diluted with culture medium in advance were added into the cells at different concentrations for coincubating for 24 hours at 37ºC. Afterwards, MTT solution (20 μL, 5 mg/mL) was introduced into each well and continued to incubate for 4 hours. To dissolve the resultant formazan, 200 μL of dimethylsulfoxide (DMSO) was subsequently added. The UV absorbance of each well was measured at 570 nm. The cell viability was calculated by the equation: Cell viability (%) = (Atri/Acon)×100%, where Atri and Acon denote the absorbance of living cell treated with different QUE preparations and blank culture medium, respectively.
Antileukemic effect of QUE-cPLNs in vivo
P388 cells in the exponential phase were rinsed with PBS twice and appropriately diluted with RPMI 1640 medium. Then, 0.25 mL of cell suspensions (~1×106 cells) were injected into the abdominal cavity of DBA/2 mice for induction of ascitic leukemia. Ascites check was performed to confirm the successful development of ascitic leukemia. The mice with obvious ventosity and increased ascitic cell density were deemed to be successfully inoculated with P388 cells. All ascitic leukemia model mice were randomly divided into four groups (N=10): saline (control), QUE suspensions, QUE-PLNs and QUE-cPLNs. Mice were treated with QUE preparations or saline every day by gavage for 21 days at a dose of 25 mg/kg. The survival of mice was investigated from the first day of administration to the end of the experiment. The cell density in the ascites of trial and control mice during treatment was measured with a UniCel DxH800 automatic blood-counter system (Beckman Coulter Inc., Bream CA, USA). In addition, to check the histomorphological changes of vulnerable organs, the treated mice were humanly sacrificed and the liver and spleen were taken out, prepared into paraffin sections and inspected after staining with H&E.
Results and discussion
Preparation and characterization of QUE-cPLNs
A variety of techniques are available for the preparation of PLNs, such as solvent diffusion,26 high pressure homogenization,27 and emulsification-evaporation.28 Solvent diffusion method is also referred as nanoprecipitation technique, in which the miscible solvent rapidly diffuses into the aqueous phase, resulting in coprecipitation of polymer and lipid materials due to decline of solubility and formation of nanoparticles. For this preparative process, the formulation variables are critical for formation of eligible nanoparticles. In the previous study, we found that the usage of QUE in the formulation, the ratio of PLGA to lecithin and the sodium taurocholate level in the aqueous phase had significant effects on the particle size and EE of QUE-cPLNs. Figure 1 shows the effects of formulation variables on these two indices. QUE content in the formulation had greater impact on the particle size of QUE-cPLNs, rather than EE. High drug content in the formulation resulted in generation of larger nanoparticles due to limited drug loading capacity of carrier materials. A drug usage of ~10% was appropriate for the preparation of QUE-cPLNs that could produce nanoparticles smaller than 100 nm. Differently, the ratio of PLGA to lecithin exerted certain effects both on particle size and EE of QUE-cPLNs. High ratio of PLGA relative to lecithin was not favorable for formation of smaller nanoparticles with high EE. This may be associated with the high phase transition temperature of PLGA and inferior miscibility with QUE. But PLGA can enhance the structural stability and digestive resistibility of nanoparticles. This requires that the ratio of PLGA to lecithin falls within a rational range. Otherwise, a high cholate concentration in the aqueous phase upon preparation could reduce the particle size of QUE-cPLNs. This is due to high cholate concentration that can facilitate molecular self-assembly by reducing the interfacial tension between hydrophobic materials and water.
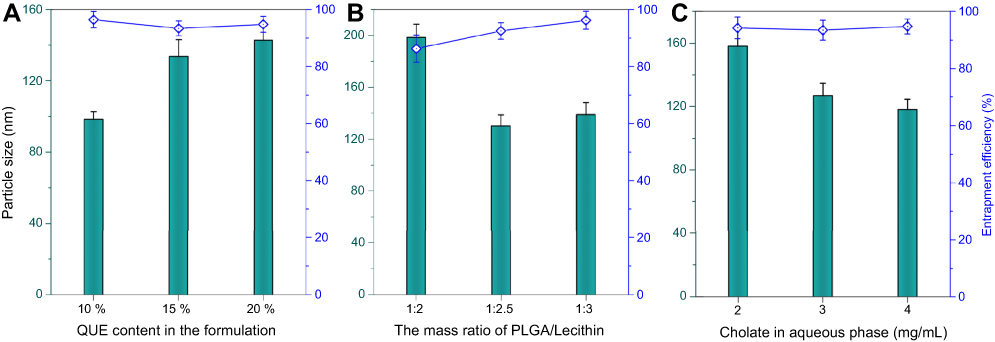 | Figure 1 Formulation variables influencing the formulation performance of QUE-cPLNs, including QUE content in the formulation (A), the ratio of PLGA to lecithin (B), and the cholate concentration in the aqueous phase upon preparation (C). |
The particle size of nanocarriers affects the intestinal absorption of payload,29 and a high drug loading rate can effectively diminish the dosing volume and avoid drug waste. Considering the advantages of small particle size and high EE in oral drug delivery, the formulation was finally determined as 10 mg of QUE, 20 mg of PLGA, and 50 mg of lecithin were formulated into 5 mL of sodium taurocholate solution (4 mg/mL). The resulting QUE-cPLNs possessed a particle size of 110.6 nm with a PDI of 0.237 (Figure 2A). The particle size was slightly smaller than that of QUE-PLNs. But, QUE-cPLNs exhibited a similar morphology to QUE-PLNs, showing a near-spherical microstructure (Figure 2B). The mean ζ potential of QUE-cPLNs was measured to be −31.9 mv, which was lower than that of QUE-PLNs (−12.5 mv), indicating them to be stable as colloidal dispersions. Meanwhile, the difference in ζ potential between QUE-PLNs and QUE-cPLNs demonstrated that there were cholate molecules successfully incorporating into nanoparticles. QUE-cPLNs prepared from the optimum formulation exhibited a high EE, up to 96.22%. TEM revealed that QUE-cPLNs were near-spherical in morphology, which seemed to be more uniform than cholate-free QUE-PLNs due to the incorporation of sodium taurocholate.
 | Figure 2 Characterization of QUE-PLNs and QUE-cPLNs.Note: Particle size distribution (A) and TEM micromorphology (B). Abbreviations: QUE, quercetin; PLNs, polymer-lipid hybrid nanoparticles; cPLNs, cholate-modified polymer–lipid hybrid nanoparticles. |
In vitro drug release
The in vitro release profiles of QUE from QUE-cPLNs in water, 0.1 M HCl and pH 6.8 PBS are shown in Figure 3. QUE-cPLNs exhibited slow QUE release in three media during an approximate GI transport time. The release linearly increased with the time in all media, illustrating that the drug release follows the principle of diffusion-controlled mode. Of note, the accumulative release percentages were rather low in three media, less than 35% within 12 hours, perhaps due to poor solubility and preferential partition of QUE in the lipid phase of nanoparticles. In terms of different release media, QUE release from QUE-cPLNs in pH 6.8 PBS was somewhat faster than in 0.1 M HCl. This may be attributable to the surface potential of QUE-cPLNs that is negatively charged. QUE-cPLNs tend to be ionized in the neutral PBS, whereas QUE-cPLNs will become compact because of counter ion effect in the medium of 0.1 M HCl, resulting in a step-down drug release. Overall, QUE release from QUE-cPLNs was limited. It could be predicted that the great mass of QUE would be retained in the nanoparticles during the GI transport. It may not be a bad thing, because nanoparticle-based oral drug delivery systems generally do not require a drug release for subsequent absorption. The released drug may be subjected to degradation due to unfavorable pH, digestive factors and metabolic enzymes. The sustained release featured by QUE-cPLNs enables them to be transported via integral nanoparticles that could sufficiently exploit the bile acid pathway to overcome the absorption barrier.20 Low drug release rate from PLNs can also be found in other investigators’ reports.30,31
 | Figure 3 In vitro release profiles of QUE from QUE-cPLNs in water, 0.1 M HCl solution and pH 6.8 PBS. |
Improved oral absorption with QUE-cPLNs
The pharmacokinetic profiles of QUE in rats after oral administration of various QUE formulations are shown in Figure 4, and the main pharmacokinetic parameters are summarized in Table 1. The formulation of suspensions produced limited drug absorption both in the rate and the extent. The maximum plasma drug concentration (Cmax) and the area under the plasma drug concentration-time curve from zero to infinity (AUC0–∞) were merely 1.763 μg/mL and 8.882 μg∙hour/mL. In comparison with QUE suspensions, QUE-PLNs promoted the oral absorption of QUE, resulting in higher drug levels in the blood. The fact suggests that plain PLNs can also improve the oral absorption of QUE owing to a supersaturated drug exposure in the GI lumen with physiologically stable nanoparticles.32 However, in the case of QUE-cPLNs, a greater extent of enhancement in the oral absorption of QUE was accomplished. QUE-cPLNs gave rise to higher plasma QUE concentrations in the whole time horizon compared with QUE-PLNs. The Cmax and AUC0–∞ were separately up to 8.807 μg/mL and 33.319 μg∙hour/mL. The relative oral bioavailability of QUE-cPLNs, calculated based on a noncompartment model, was up to 375.12% compared to QUE suspensions, whereas it was just 252.15% for QUE-PLNs. The difference in oral bioavailability between QUE-PLNs and QUE-cPLNs shows that cholate modification is contributive for enhancement of drug absorption. In terms of the time to maximum plasma concentration (Tmax), two kinds of nanocarrier exhibited similar absorption peak time (approximately 1 hour), but significantly different from QUE suspensions. This can be accounted for the slow absorption of coarse drugs in the form of suspensions that causes a long in vivo disposition. In addition, the half-life (T1/2) of QUE among different preparations has changed due to their variant pharmacokinetic behaviors. These results reveal that there are differences in the absorption mode of QUE as formulated into different dosage forms. By contrast, cholate-modified nanoparticles can promote the oral absorption of QUE to a greater extent.
 | Table 1 Comparative pharmacokinetic parameters of QUE in rats after oral administration of QUE suspensions, QUE-PLNs and QUE-cPLNs |
 | Figure 4 Pharmacokinetic profiles of QUE in rats after oral administration of Que suspensions, QUE-PLNs and QUE-cPLNs at a dose of 25 mg/kg (n=6). Abbreviations: QUE-PLNs, quercetin-loaded polymer–lipid hybrid nanoparticles; QUE, quercetin. |
Lipid nanoparticles such as solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) have been proven effective to promote the oral drug absorption, which stand for one of more promising drug delivery systems.33 However, harsh GI digestion and potential drug recrystallization increase variability of drug absorption. PLNs containing a functional polymer are manifested to be more stable and flexibly tunable in drug release, thus avoiding uncertain transport variation in the GI tract. Moreover, PLNs can protect the payload from the intestinal first-pass effect due to steady encapsulation and low drug leakage.34 This study shows that PLNs substantially improve the oral absorption of QUE. But, the oral delivery efficacy of plain PLNs is not sufficient if simply through the nanoscale effect of nanocarriers,35 which involves an increase in the absorption area and nonspecific transmembrane transport (cytosis). Endowing nanoparticles with an active transport represents an important approach to augment the absorption degree of encapsulated drugs. Cholates are various sodium and potassium salts of bile acids and act as critical biological components that support the solubilization of various lipidic nutrients as well as liposoluble compounds in the GI lumen and facilitate their absorption. They are water-soluble compounds and have less chance to pass through the cell membrane via a passive transport mode, thus it requires a corresponding transporter to participate in the intestinal transport of them. Cholate-modified drug delivery systems exactly utilize the function of ASBT to achieve an active drug transport. Our developed cPLNs have already shown a good capability of ameliorating the oral absorption of QUE, which would be favorable for exerting its pharmacological action.
Changes in cellular uptake and internalization
Figure 5A presents the cellular uptake of free QUE, QUE-PLNs and QUE-cPLNs (in the absence or presence of sodium taurocholate). There were significant differences in the cellular uptake between free QUE and nanoparticle-encapsulated QUE. Both QUE-PLNs and QUE-cPLNs exhibited ameliorative cellular uptake of QUE relative to its free form, showing a time-dependent uptake accumulation. Nevertheless, QUE-cPLNs had higher cellular uptake rate than QUE-PLNs. We speculate that it is related to the incorporation of cholate in the nanoparticles, which accelerates the cellular uptake by a transporter-mediated influx. For this point, it can be inferred from the cellular uptake of QUE-cPLNs in the presence of sodium taurocholate. The addition of sodium taurocholate significantly inhibited the cellular uptake of QUE-cPLNs. The intracellular QUE contents were even lower than that of free QUE at parallel time points, indicating that cholate inhibited the cellular uptake of QUE-cPLNs as a result of transport site competition. Moreover, living cells have the preference to take in nontoxic nutrient substance rather than macromolecular aggregates and cytotoxic species. This is the underlying reason that QUE-cPLNs turn up the lowest uptake in the presence of sodium taurocholate. The reduced uptake of QUE-cPLNs in the presence of sodium taurocholate provided evidence of ASBT-mediated uptake. In theory, free QUE should exhibit the highest cellular uptake due to small molecule character and high lipophilicity. However, that is not the case. QUE possesses a certain cytotoxic effect and will experience apical excretion after influx,36 which results in reduced cellular uptake. As loaded into nanoparticles, the detrimental properties of QUE are effectively shielded and thus the cellular uptake increases. The uptake rates of QUE-PLNs were approximately 1.5-fold as high as that of free QUE at three test time points, but the values were close to twofoldin the case of QUE-cPLNs. Obviously, QUE-cPLNs could result in greater QUE uptake, showing them to be of potential in facilitating QUE absorption.
 | Figure 5 Cellular uptake and internalization of free and formulated QUE in Caco-2 cells. Notes: (A) Cellular uptake of free QUE, QUE-PLNs and QUE-cPLNs (in the absense of presence of sodium taurocholate) at different incubation time quantified by the intracellular QUE concentration, data expressed as mean ± SD (n=3), paired t-test, **P<0.01, significantly different comapred with each other. (B) Cellular internalization of QUE-PLNs and QUE-cPLNs characterized by intracellular fluorescence intensity. Abbreviations: QUE, quercetin; QUE-PLNs, quercetin-loaded polymer–lipid hybrid nanoparticles. |
The facilitative effect of cPLNs on QUE transmembrane transport can also be appreciated from the cellular internalization of QUE-cPLNs (Figure 5B). Intense cellular internalization of QUE-cPLNs took place following incubation with Caco-2 cells for a relatively short time. There was considerable nanoparticle-associated fluorescence interspersing within the cytoplasm, even distributing into the nucleus as revealed by the colocalization. The cellular penetrability of QUE-cPLNs was apparently stronger than QUE-PLNs where there was only a small quantity of fluorescence distributing in the cytoplasm. Excellent affinity and permeability to enterocytes allow QUE-cPLNs to be transported in a highly efficient way.
Cytotoxicity of QUE-cPLNs on leukemic cells
The cell viability of P388 cells treated with different QUE formulations is displayed in Figure 6. Although the relative cell viability values of P388 cells for free QUE group were more than 70% at the concentrations from 5 µm to 20 µm, it showed a concentration-dependent downtrend. This indicates that QUE has a definite cytotoxic effect on the leukemic cells, but the concentration that induces P388 cell death is relatively high. The decline in cell viability in the group of QUE-PLNs increased compared with the group of free QUE. However, the cell vitality became worse as treated with QUE-cPLNs. When the concentration of QUE-cPLNs arrived to 20 µm, the cell viability value declined to 44.9%. Low to moderate concentrations of free QUE and QUE-PLNs as well as low concentration of QUE-cPLNs exhibited weak cytotoxicity on P388 cells, though moderate to high concentrations of QUE-cPLNs (10–20 µm) produced strong cytotoxic effect on P388 cells. It can be seen that nanoparticle-encapsulated QUE has a higher cell killing capability than the free one. The underlying reason is supposed to be associated with distinct cellular availability of QUE as far as different formulations concerned. All these results demonstrate that QUE-cPLNs have higher cellular approachability, which also accords well with the cellular uptake events performed in Caco-2 cells.
 | Figure 6 Cytotoxic effects of free QUE, QUE-PLNs and QUE-cPLNs on P388 cells evaluated by the relative cell viability using MTT test. Notes: Data shown as mean ± SD (n=3), paired t-test, **P<0.01, significantly different compared with each other. Abbreviations: QUE, quercetin; QUE-PLNs, quercetin-loaded polymer–lipid hybrid nanoparticles; |
Antileukemic effect of QUE-cPLNs in P388 cell-induced leukemia
In this study, a simple ascetic leukemia model was utilized to examine the in vivo antileukemic effect of QUE-cPLNs.37 Through a peritoneal check and cell density analysis, it can be confirmed that all mice have been successfully transfected with leukemia. The survival of leukemic mice, the cell density in the ascites of surviving mice and the histological morphology were investigated after treatment. QUE-cPLNs significantly prolonged the survival of P388 cell-transfected mice compared with QUE suspensions and QUE-PLNs (Figure 7A). The animals treated saline all died within 15 days after inoculation of P388 cells. Although oral administration of QUE suspensions increased the survival time of mice, the prolongation of life span was relatively inferior. It was different from the formulation of QUE suspensions that QUE-PLNs markedly improved the survival rate of leukemic mice. The mean survival time reached to 26.8 days in the case of QUE-PLNs. A more significant life prolongation took place in the group of mice that were treated with QUE-cPLNs. After 45 days, there were still three mice alive. QUE-cPLNs greatly enhanced the curative activity of QUE. As for this point, it could also be reflected by the cell density in the ascites extracted from the surviving mice after treatment for two weeks (Figure 7B). The control group treated with saline showedconsiderable ascetic cells in the enterocoelia, implying a remarkable proliferation and growth of leukemic cells. For the mice receiving QUE suspensions, QUE-PLNs and QUE-cPLNs, the cell density in the ascites of mice decreased to varying degrees. It indicates that medicinal intervention plays a positive role in suppressing the development of leukemia. Evidently, the ascetic cell level in the group of QUE-cPLNs was the lowest. QUE-cPLNs potentiated the therapeutic effect of QUE.
 | Figure 7 In vivo antileukemic effects of QUE suspensions, QUE-PLNs and QUE-cPLNs in P388 cells-induced murine asceitic leukemia. Notes: (A) The survival curves of leukemic mice treated with saline (control), QUE suspensions, QUE-PLNs and QUE-cPLNs (n=10), ANOVA, **P<0.01. (B) The cell density in the ascetes of surviving mice after treatment for two weeks, paired t-test, **P<0.01, significantly different compared with each other. (C) The histomorphological changes in the liver and spleen after treatment with saline and QUE-cPLNs. |
As the ascetic leukemia progresses, the leukemic cells would infiltrate the corresponding tissues, resulting in swelling of the liver, spleen and lymphatic tissue. We checked the histological morphology of the liver and spleen of the mice after treatment with QUE-cPLNs (Figure 7C). Compared with the control group, the hepatic swelling, albeit not significant, seemed to be alleviated, and the spleen infiltration became less prominent. Because the leukemia model belongs to a mild ascetic leukemia, the tissue infiltration is not apparent. The treatment group showed alleviative damage to the vital organs, proving an intensified antileukemic effect of QUE-cPLNs. QUE can stop or retard the progression of leukemia through a variety of mechanisms, such as inhibiting CK2 and PI3K kinase activities,38 suppressing HSP27 expression,39 and targeting epigenetic regulators of proapoptotic genes.40 This work is not intended to elucidate the antileukemic mechanisms of QUE. It is merely intendedto prove that the pharmacological activity of QUE can be substantially improved through a biomimetic nanodrug delivery system. After all, the pharmacodynamics of a drug depends greatly on its bioavailability after administration. Last but not least, we confirm that cPLNs can enhance the oral bioavailability of QUE, thus the curative effect against leukemia.
Conclusions
In this study, a novel biomimetic nanocarrier, cholate-modified polymer-lipid hybrid nanoparticles (cPLNs), was developed for oral delivery of QUE to potentiate its antileukemic effect. The suitability of cPLNs as a functional delivery vehicle of QUE was evaluated. QUE-cPLNs with smaller particle size (~110 nm) were prepared by incorporating sodium taurocholate in the formulation. High QUE entrapment and slow QUE release were achieved through cPLNs. The in vivo pharmacokinetic results showed that cPLNs significantly enhanced the oral bioavailability of QUE. Increased oral absorption of QUE through cPLNs could be attributed to improvement in the transcellular trafficking due to mediation of bile acid transporter. In addition, QUE-cPLNs demonstrated enhancive in vitro and in vivo antileukemic activities. The cytotoxic effect on P388 cells and the inhibitory effect on leukemia model of QUE-cPLNs were significantly superior to unencapsulated QUE and QUE-PLNs. This work provides fundamental insight into the utilization of the bile acid pathway to deliver poorly bioavailable drugs and potentiate their bioactivities.
Acknowledgments
This work is supported by the Key Research Project of Henan Provincial Science and Technology Department (No. 112102310306), and the authors thank the Department of Pharmaceutics of Huaihe Hospital Affiliated to Henan University for financial assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Anand David AV, Arulmoli R, Parasuraman S. Overviews of biological importance of quercetin: a bioactive flavonoid. Pharmacogn Rev. 2016;10(20):84–89. doi:10.4103/0973-7847.194044
2. Baron BW, Thirman MJ, Giurcanu MC, Baron JM. Quercetin therapy for selected patients with PIM1 kinase-positive chronic lymphocytic leukemia/small lymphocytic lymphoma: a pilot study. Acta Haematol. 2018;139(2):132–139. doi:10.1159/000486361
3. Calgarotto AK, Maso V, Junior GCF, et al. Antitumor activities of Quercetin and Green Tea in xenografts of human leukemia HL60 cells. Sci Rep. 2018;8(1):3459. doi:10.1038/s41598-018-21516-5
4. Naimi A, Entezari A, Hagh MF, Hassanzadeh A, Saraei R, Solali S. Quercetin sensitizes human myeloid leukemia KG-1 cells against TRAIL-induced apoptosis. J Cell Physiol. 2019;234(8):13233–13241. doi: 10.1002/jcp.27995
5. Rich GT, Buchweitz M, Winterbone MS, Kroon PA, Wilde PJ. Towards an understanding of the low bioavailability of quercetin: a study of its interaction with intestinal lipids. Nutrients. 2017;9(2):111. doi:10.3390/nu9020111
6. Barbosa AI, Costa Lima SA, Reis S. Application of pH-responsive fucoidan/chitosan nanoparticles to improve oral quercetin delivery. Molecules. 2019;24(2):346. doi:10.3390/molecules24020346
7. Dian L, Yu E, Chen X, et al. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res Lett. 2014;9(1):2406. doi:10.1186/1556-276X-9-684
8. Zhang K, Zhang M, Liu Z, et al. Development of quercetin-phospholipid complex to improve the bioavailability and protection effects against carbon tetrachloride-induced hepatotoxicity in SD rats. Fitoterapia. 2016;113:102–109. doi:10.1016/j.fitote.2016.07.008.
9. Ghaffari F, Hajizadeh Moghaddam A, Zare M. Neuroprotective effect of quercetin nanocrystal in a 6-hydroxydopamine model of parkinson disease: biochemical and behavioral evidence. Basic Clin Neurosci. 2018;9(5):317–324. doi:10.32598/bcn.9.5.317
10. Zhang X, Xing H, Zhao Y, Ma Z. Pharmaceutical dispersion techniques for dissolution and bioavailability enhancement of poorly water-soluble drugs. Pharmaceutics. 2018;10(3):74. doi:10.3390/pharmaceutics10030074
11. Li Q, Cai T, Huang Y, Xia X, Cole S, Cai Y. A review of the structure, preparation, and application of NLCs, PNPs, and PLNs. Nanomaterials. 2017;7(6):122. doi:10.3390/nano7120458
12. Pandita D, Kumar S, Lather V. Hybrid poly (lactic-co-glycolic acid) nanoparticles: design and delivery prospectives. Drug Discov Today. 2015;20(1):95–104. doi:10.1016/j.drudis.2014.09.018
13. Grigoras AG. Polymer-lipid hybrid systems used as carriers for insulin delivery. Nanomedicine. 2017;13(8):2425–2437. doi:10.1016/j.nano.2017.08.005
14. Bose RJC, Ravikumar R, Karuppagounder V, Bennet D, Rangasamy S, Thandavarayan RA. Lipid-polymer hybrid nanoparticle-mediated therapeutics delivery: advances and challenges. Drug Discov Today. 2017;22(8):1258–1265. doi:10.1016/j.drudis.2017.05.015
15. Jose C, Amra K, Bhavsar C, Momin M, Omri A. Polymeric lipid hybrid nanoparticles: properties and therapeutic applications. Crit Rev Ther Drug Carrier Syst. 2018;35(6):555–588. doi:10.1615/CritRevTherDrugCarrierSyst.2018024751
16. Xiao L, Pan G. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: the apical sodium-dependent bile acid transporter (SLC10A2/ASBT). Clin Res Hepatol Gastroenterol. 2017;41(5):509–515. doi:10.1016/j.clinre.2017.02.001
17. Khatun Z, Nurunnabi M, Cho KJ, Byun Y, Bae YH, Lee YK. Oral absorption mechanism and anti-angiogenesis effect of taurocholic acid-linked heparin-docetaxel conjugates. J Control Release. 2014;177:64–73. doi:10.1016/j.jconrel.2013.12.034.
18. Arzani G, Haeri A, Daeihamed M, Bakhtiari-Kaboutaraki H, Dadashzadeh S. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int J Nanomedicine. 2015;104:797–813. doi:10.2147/IJN.S84703
19. Li Y, Zhu C. Mechanism of hepatic targeting via oral administration of DSPE-PEG-cholic acid-modified nanoliposomes. Int J Nanomedicine. 2017;12:1673–1684. doi:10.2147/IJN.S125047.
20. Fan W, Xia D, Zhu Q, et al. Functional nanoparticles exploit the bile acid pathway to overcome multiple barriers of the intestinal epithelium for oral insulin delivery. Biomaterials. 2018;151:13–23. doi:10.1016/j.biomaterials.2017.10.022.
21. Kim KS, Suzuki K, Cho H, Youn YS, Bae YH. Oral nanoparticles exhibit specific high-efficiency intestinal uptake and lymphatic transport. ACS Nano. 2018;12(9):8893–8900. doi:10.1021/acsnano.8b04315
22. Gou J, Liang Y, Miao L, et al. The promoting effect of enteric materials on the oral absorption of larotaxel-loaded polymer-lipid hybrid nanoparticles. Eur J Pharm Sci. 2018;124:288–294. doi:10.1016/j.ejps.2018.09.002.
23. Deng W, Wang H, Wu B, Zhang X. Selenium-layered nanoparticles serving for oral delivery of phytomedicines with hypoglycemic activity to synergistically potentiate the antidiabetic effect. Acta Pharm Sin B. 2019;9(1):74–86. doi:10.1016/j.apsb.2018.09.009
24. Zhang X, Zhang T, Ye Y, et al. Phospholipid-stabilized mesoporous carbon nanospheres as versatile carriers for systemic delivery of amphiphobic SNX-2112 (a Hsp90 inhibitor) with enhanced antitumor effect. Eur J Pharm Biopharm. 2015;94:30–41. doi:10.1016/j.ejpb.2015.04.023.
25. Natoli M, Leoni BD, D’Agnano I, Zucco F, Felsani A. Good Caco-2 cell culture practices. Toxicol In Vitro. 2012;26(8):1243–1246. doi:10.1016/j.tiv.2012.03.009
26. Zhang X, Chen G, Zhang T, Ma Z, Wu B. Effects of PEGylated lipid nanoparticles on the oral absorption of one BCS II drug: a mechanistic investigation. Int J Nanomedicine. 2014;9:5503–5514. doi: 10.2147/IJN.S73340.
27. Ana R, Mendes M, Sousa J, et al. Rethinking carbamazepine oral delivery using polymer-lipid hybrid nanoparticles. Int J Pharm. 2019;554:352–365. doi:10.1016/j.ijpharm.2018.11.028.
28. Hallan SS, Kaur V, Jain V, Mishra N. Development and characterization of polymer lipid hybrid nanoparticles for oral delivery of LMWH. Artif Cells Nanomed Biotechnol. 2017;45(8):1631–1639. doi:10.1080/21691401.2016.1276920
29. Ran F, Lei W, Cui Y, et al. Size effect on oral absorption in polymer-functionalized mesoporous carbon nanoparticles. J Colloid Interface Sci. 2018;511:57–66. doi:10.1016/j.jcis.2017.09.088.
30. Zhang LJ, Wu B, Zhou W, et al. Two-component reduction-sensitive lipid-polymer hybrid nanoparticles for triggered drug release and enhanced in vitro and in vivo anti-tumor efficacy. Biomater Sci. 2016;5(1):98–110. doi:10.1039/c6bm00662k
31. Zhang L, Zhu D, Dong X, et al. Folate-modified lipid-polymer hybrid nanoparticles for targeted paclitaxel delivery. Int J Nanomedicine. 2015;10:2101–2114. doi:10.2147/IJN.S77667.
32. Zhu C, Gong S, Ding J, et al. Supersaturated polymeric micelles for oral silybin delivery: the role of the Soluplus-PVPVA complex. Acta Pharm Sin B. 2019;9(1):107–117. doi:10.1016/j.apsb.2018.09.004
33. Rajabi M, Mousa SA. Lipid nanoparticles and their application in nanomedicine. Curr Pharm Biotechnol. 2016;17(8):662–672.
34. Joyce P, Prestidge CA. Synergistic effect of PLGA nanoparticles and submicron triglyceride droplets in enhancing the intestinal solubilisation of a lipophilic weak base. Eur J Pharm Sci. 2018;118:40–48. doi:10.1016/j.ejps.2018.03.018.
35. Wang K, Qi J, Weng T, et al. Enhancement of oral bioavailability of cyclosporine A: comparison of various nanoscale drug-delivery systems. Int J Nanomedicine. 2014;9:4991–4999. doi:10.2147/IJN.S72560.
36. Chalet C, Rubbens J, Tack J, Duchateau GS, Augustijns P. Intestinal disposition of quercetin and its phase-II metabolites after oral administration in healthy volunteers. J Pharm Pharmacol. 2018;70(8):1002–1008. doi:10.1111/jphp.12929
37. Alakhova DY, Zhao Y, Li S, Kabanov AV. Effect of doxorubicin/pluronic SP1049C on tumorigenicity, aggressiveness, DNA methylation and stem cell markers in murine leukemia. PLoS One. 2013;8(8):e72238–e72238. doi:10.1371/journal.pone.0072238
38. Russo M, Milito A, Spagnuolo C, et al. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget. 2017;8(26):42571–42587. doi:10.18632/oncotarget.17246
39. Chen X, Dong XS, Gao HY, et al. Suppression of HSP27 increases the antitumor effects of quercetin in human leukemia U937 cells. Mol Med Rep. 2016;13(1):689–696. doi:10.3892/mmr.2015.4600
40. Alvarez MC, Maso V, Torello CO, Ferro KP, Saad STO. The polyphenol quercetin induces cell death in leukemia by targeting epigenetic regulators of pro-apoptotic genes. Clin Epigenetics. 2018;10(1):139. doi:10.1186/s13148-018-0563-3
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.