Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Chloramphenicol encapsulated in poly-ε-caprolactone–pluronic composite: nanoparticles for treatment of MRSA-infected burn wounds
Authors Kalita S, Devi B, Kandimalla R, Sharma KK, Sharma A, Kalita K, Ch Kataki A, Kotoky J
Received 27 September 2014
Accepted for publication 20 December 2014
Published 15 April 2015 Volume 2015:10(1) Pages 2971—2984
DOI https://doi.org/10.2147/IJN.S75023
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Sanjeeb Kalita,1 Banasmita Devi,1 Raghuram Kandimalla,1 Kaustav Kalyan Sharma,1 Arup Sharma,2 Kasturi Kalita,3 Amal Chandra Kataki,4 Jibon Kotoky1
1Institute of Advanced Study in Science and Technology (IASST), Division of Life Sciences, Paschim Boragaon, Garchuk, Guwahati, Assam, India; 2College of Veterinary Science, Assam Agriculture University, Khanapara, Guwahati, Assam, India; 3Hyat Hospital, Lalganesh, Guwahati, Assam, India; 4Dr B Borooah Cancer Institute, Guwahati, Assam, India
Abstract: The emergence of methicillin-resistant Staphylococcus aureus (MRSA) infection has increased precipitously over the past several decades, with far-reaching health care and societal costs. MRSA infections in the context of burn wounds lead to invasive disease that could potentially cause mortality. Chloramphenicol is a well-known broad-spectrum bacteriostatic antibiotic that has been used since 1949, but due to its hydrophobicity, poor penetration in skin, fast degradation, and toxicity, its application has been hindered. Furthermore, it has been demonstrated that old antibiotics such as chloramphenicol remained active against a large number of currently prevalent resistant bacterial isolates due to their low-level use in the past. Recently, the novel nanoparticulate drug-delivery system has been used and reported to be exceptionally useful for topical therapeutics, due to its distinctive physical characteristics such as a high surface-to-volume ratio and minuscule size. It helps to achieve better hydrophilicity, bioavailability, and controlled delivery with enhanced therapeutic index, which has resulted in decreased toxicity levels compared to the crude drug. Here, we report a novel chloramphenicol loaded with poly(ε-caprolactone) (PCL)-pluronic composite nanoparticles (CAM-PCL-P NPs), physicochemical characterizations, and its bioactivity evaluation in a MRSA-infected burn-wound animal model. CAM-PCL-P NPs could encapsulate 98.3% of the drug in the nanoparticles and release 81% of the encapsulated drug over 36 days with a time to 50% drug release of 72 hours (51%). Nanoparticle suspensions maintained the initial properties with respect to size and encapsulation efficiency, even after 6 months of storage at 4°C and 25°C, respectively (P>0.05). Significant reduction in the level of toxicity was observed for CAM-PCL-P NPs compared with that of free drug as confirmed from hemolytic activity against human blood erythrocytes and cytotoxicity assay against an MCF-7 breast cancer cell line. In vitro antibacterial activities were performed by zone of inhibition, minimum inhibitory concentrations, minimum bacterial concentration, and time-kill assays, which showed that CAM-PCL-P NPs exhibited significantly enhanced anti-MRSA activity against ten clinical isolates of MRSA strains. The augmented activity of CAM-PCL-P NPs was further tested on a MRSA-infected burn-wound animal model and achieved quicker efficacy in MRSA clearance and improved the survival rate compared with free-chloramphenicol treatment. Thus, we propose CAM-PCL-P NPs as a promising novel antimicrobial candidate that may have a good potential for preclinical applications.
Keywords: chloramphenicol, PCL-pluronic, nanoparticle, methicillin-resistant Staphylococcus aureus, anti-MRSA activity, burn-wound animal model
Introduction
Despite major advances in the management of severe burn injury, thermally injured patients still suffer significant mortality and morbidity from sepsis and its related complications.1 Thermal destruction of the skin barrier and concomitant depression of local and systemic host cellular and humoral immune responses are the pivotal factors contributing to infectious complications in patients with severe burns.2–6 Staphylococcus aureus, especially methicillin-resistant S. aureus (MRSA), is a major cause of sepsis in patients who are immunosuppressed by their burns. MRSA was represented in 40% of wound isolates, and the wounds of 14%–17% of thermally injured patients became infected once they were colonized with MRSA.7,8
In spite of the constantly increasing need and the alarming epidemic of multidrug-resistant bacteria, antibiotic drug discovery and development seem to have greatly decelerated in recent years, which has forced clinicians to reintroduce forgotten antibiotics into their practice. Since 2000, only three antimicrobials of a new class or subclass have been approved: the oxazolidinone linezolid, the cyclic lipopeptide daptomycin, and the glycylcycline tigecycline. To date, no topical broad-spectrum antibiotic with US Food and Drug Administration approval is available for the treatment of skin-wound infections.9
Although research on the anti-MRSA properties of various nanoparticulate systems is ongoing, no references were found on nanoparticulate systems for MRSA-infected burn wounds. Due to the low levels of use of many of the old antibiotic compounds, these have remained active against a large number of currently prevalent bacterial isolates. MRSA clinical isolates have shown susceptibility to chloramphenicol with minimum inhibitory concentrations (MICs) of ≤8 μg/mL.10 Thus, clinicians are beginning to reevaluate their use in various patient populations and infections, regardless of the fact that they were previously thought to be less effective and/or more toxic than newer agents. The novel nanoparticulate biodegradable polymer-based drug delivery system can help to attain better penetration, controlled release, better bioavailability, and the pharmacokinetic parameters of medicinal entities with enhanced therapeutic index; this has resulted in decreased levels of toxicity compared to the crude drug.11,12 Moreover, the availability of novel genetic and molecular modification methods provides hope that the toxicity and efficacy drawbacks presented by some of these agents can be surpassed in the future. A number of old antibiotic compounds, such as polymyxins, fosfomycin, fusidic acid, cotrimoxazole, aminoglycosides, and chloramphenicol, are reemerging as valuable alternatives for the treatment of difficult-to-treat infections.
Chloramphenicol is a broad-spectrum bacteriostatic antibiotic drug that stops bacterial growth by inhibiting protein-chain elongation by inhibiting the peptidyl transferase activity of the bacterial ribosome. Along with its broad-spectrum antibacterial nature, chloramphenicol is also effective against Enterococcus faecium, which has led to its being considered for treatment of vancomycin-resistant Enterococcus. Because of its excellent blood–brain barrier penetration, chloramphenicol remains the first-choice treatment for staphylococcal brain abscesses.
The most serious adverse effect associated with chloramphenicol intravenous and oral administration is bone marrow toxicity. Nonetheless, chloramphenicol has still been administered in increasing dosages in recent years due to the increased incidence of antibiotic resistance. The avascularity of the burn eschar further restricts delivery of systemically administered antimicrobial agents. Therefore, topical administration of antibacterials is more advantageous for effective treatment of locally invasive diseases due to its inherent ability to circumvent systemic cytotoxicity and the ease of rapid delivery at the site of infection.13 Currently, the efficacy of the very few options available for the treatment of MRSA-infected burn wounds (eg, nanocrystalline silver dressing, moxifloxacin) is hindered by low-to-moderate eschar penetration capacity and toxicity issues.2 Chloramphenicol ointment has been used to treat bacterial conjunctivitis, but little evidence exists for its effectiveness in the prophylaxis or treatment of burn wound infections. Chloramphenicol is not used for topical treatment of skin infections due to its hydrophobic chemical structure, which hinders adequate cutaneous penetration. Furthermore, human skin being thicker, it becomes an impassable barrier that inhibits transdermal transport of the hydrophobic drug. The problem becomes more serious in the case of an infected burn wound, where the drug should absorb both through the eschar and intact skin surrounding the burn wound to achieve a better therapeutic index.
In the past years, different strategies have been proposed to increase drug skin permeation and to circumvent the inadequate physicochemical characteristics of several substances.14 Nanometric systems, especially nanospheres, have a great surface area, which renders them highly satisfactory for the application of lipophilic substances promoting a homogeneous drug release.15,16 Nanospheres are able to modify the activity of drugs by altering the physicochemical properties of formulations, to delay and control the drug release and increase the drug adhesivity or its longevity in the skin.
Poly(ε-caprolactone) (PCL) is the most-used polymer among subcutaneous drug delivery systems, due to its biocompatibility, biodegradability, nontoxicity, nonimmunogenicity, high physical stability, simple preparation methods, sustained, controlled drug release, and higher probability for surface functionalization.17–20 Pluronic F127 (PF127) is a biocompatible, nonionic, surfactant polyol (molecular weight approximately 12,500 Da) that has been found to facilitate the solubilization of water-insoluble dyes and other materials in physiological media and may also be useful for dispersing other lipophilic probes.
In this study, we for the first time proposed that the encapsulation of chloramphenicol into PCL-pluronic composite nanoparticles (NPs) should intensify its subcutaneous penetration and the sustained release of chloramphenicol. We also report that encapsulation results in more direct and prolonged contact between the antibiotic and MRSA and thereby increased therapeutic index with reduced level of toxicity. Furthermore, we demonstrated its successful application in the treatment of MRSA-infected burn wounds.
Materials and methods
Materials
PCL of molecular weight 14,000 Da, PF127, and chloramphenicol were purchased from Sigma-Aldrich Chemicals Private Ltd (Bangalore, India). Ten MRSA clinical isolates (MRSA1–MRSA10) were collected from Hayat Hospital, Guwahati, Assam, India, courtesy of Dr Paromita C Borua. Identification of MRSA was confirmed according to the recommendations of the National Committee for Clinical Laboratory Standards. All of the cultures were grown on nutrient agar plates and maintained in the nutrient agar slants at 4°C. All other solvents used in the study were of analytical grade and obtained from Merck (Mumbai, India). Milli-Q water was used throughout all the experiments.
Preparation of blank PCL NPs
Nanoprecipitation (solvent displacement) was employed to prepare blank PCL NPs (PCL-P NPs) with minor modifications.21,22 Briefly, 62.5 mg of PCL was dissolved in 10 mL of acetone by mild heating (60°C) and sonication for approximately 15 minutes. The polymer solution was gently added drop by drop into the 20 mL double-distilled water containing 62.5 mg of PF127 as the hydrophilic surfactant under moderate magnetic stirring for approximately 15 minutes. The resulting suspension was centrifuged at 12,600× g for 20 minutes and the supernatant, consisting of acetone and water, was carefully separated from the pellet and discarded. At last, the obtained pellet was washed three times with double-distilled water, centrifuged to remove any residual acetone, and redistributed in a minimal volume of double-distilled water.
Preparation of chloramphenicol-loaded PCL-pluronic NPs
To encapsulate chloramphenicol into chloramphenicol-loaded PCL-pluronic NPs (CAM-PCL-P NPs), three different types of cosolvents, namely dimethyl sulfoxide (DMSO), methanol, and acetone, were used. Different volumes of chloramphenicol stock solution (1–10 mg/mL) were prepared and mixed with the polymer solution (PCL in acetone). PF127 was used here in the expectation that it might help in encapsulation and the dispersion of the drug, which improves the stability of the nanoformulation. NPs were prepared from this drug–polymer–PF127 mixture, as described above.
Characterization of CAM-PCL-P NPs
CAM-PCL-P NPs were studied for shape, size, and aggregation by scanning electron microscopy (SIGMA VP; Carl Zeiss Meditec AG, Jena, Germany). To know the role of different cosolvents (ie, methanol, DMSO, acetone) in the preparation of CAM-PCL-P NPs, were analyzed by field emission scanning electron microscopy. The particle size distribution and zeta potential analysis of CAM-PCL-P NPs dissolved in physiological saline were conducted with dynamic light scattering measurements (Malvern Instruments Ltd., Malvern, UK). The photophysical property of the CAM-PCL-P NPs were characterized by ultraviolet-visible (UV-Vis) absorption spectra analysis. UV-Vis spectra of 100 μL, 200 μL, and 1,000 μL of aqueous solution of CAM-PCL-P NPs prepared with methanol as cosolvent (volumes were made up to 1 mL with Milli-Q water) and PCL-P NPs were recorded (Shimadzu 1800 UV-Vis spectrophotometer; Shimadzu Scientific Instruments, Columbia, MD, USA). Fourier-transform infrared spectroscopy (FT-IR) analysis was done for lyophilized PCL-P NPs and CAM-PCL-P NPs (Nicolet iS 10 FT-IR spectrometer; Thermo Fisher Scientific, Waltham, MA, USA). The same were obtained for PCL and chloramphenicol as standards. Studies on crystallographic structure of PCL-P NPs, CAM-PCL-P NPs, and chloramphenicol isolated from loaded PCL NPs were done using an ADVANCE X-ray powder diffractometer (Bruker AXS Inc., Madison, WI, USA) using CuKα (λ=1.54 Å) source in the region of 2θ from 5° to 30°.
Encapsulation efficiency
Encapsulation efficiency (EE) of PCL-P NPs were determined by spectrophotometry.23 Different required amounts of chloramphenicol solution in acetone was added to the polymeric solution of PCL in acetone to obtain different polymer:drug ratio. The drug encapsulated in PCL NPs was formed and separated from nonencapsulated free chloramphenicol by centrifugation (Sigma 30K refrigerated centrifuge; Sigma-Aldrich Co., St Louis, MO, USA) at 18,000 rpm at 4°C for 20 minutes. Separated NPs were redispersed in distilled water and given a wash by centrifuging at 18,000 rpm at 4°C for 5 minutes. The pellets of each eppendorf containing different polymer:drug NPs were washed with 200 μL of methanol, which was then vortexed vigorously and centrifuged. The yellowish supernatant was collected and quantified spectrophotometrically (Shimadzu 1800 UV-Vis spectrophotometer) at 263 nm. The amount of encapsulated chloramphenicol was calculated (in mg) as the difference between the total amount of drug used to prepare loaded NPs and that recovered by methanol extraction. Chloramphenicol EE was calculated as cited below:
| (1) |

In vitro release studies and release kinetics
Lyophilized CAM-PCL-P NPs were redispersed in 75 mL of 0.01 M phosphate-buffered saline (PBS) solution (pH 7.4) at a final concentration of 1 mg/mL. Total volume was divided into 75 eppendorf tubes giving 25 different sets (each set with 3 eppendorf tubes) for time-dependent release study at time intervals of 0, 2, 4, 6, 8, 12, 18, 24, 36, 48, 60, 72, 96, 120, 144, 168, 192, 216, 240, 264, 288, 312, 336, and 360 hours using a UV-Vis spectrophotometer. At proper time intervals, intake amounts of chloramphenicol in CAM-PCL-P NPs were first extracted in methanol and quantified by UV-Vis spectra. The release was quantified as follows:
| (2) |
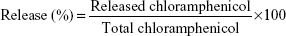
To analyze release kinetics and mechanism, data were fitted to the following four mathematical models.2–29
1. Zero order: Mt/M∞ = k0t | (3) |
2. First order:Mt/M∞ =1 − exp (−k1t) | (4) |
3. Higuchi model:Mt/M∞ = kHt1/2 | (5) |
4. Power law model:Mt/M∞ = ktn | (6) |
We also derived a mathematical model by using SPSS Statistics software for our drug-release data to obtain best fit.
Storage stability
Nanoparticle suspensions were stored under static conditions at 4°C and 25°C during a period of 6 months. Stability was assessed by comparing the initial EE and particle size with those obtained after 6-month storage at 4°C and 25°C. The results of experiments were checked for statistical significance using the statistical analysis (Student’s t-test) where the differences are considered insignificant when P>0.05.
Cytotoxicity study of free chloramphenicol, PCL-P NPs, and CAM-PCL-P NPs
To know whether the encapsulation of chloramphenicol into PCL-P NPs reduce toxicity of chloramphenicol toward mammalian cells or not, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) dye conversion assay30 of free chloramphenicol, PCL-P NPs, and CAM-PCL-P NPs were performed against MCF-7 breast cancer cells. MCF-7 cells at a density of 1×104 per well were cultured in a 100 μL volume of cell culture medium (Dulbecco’s Modified Eagle’s Medium [DMEM]) supplemented with 10% fetal bovine serum in a 96-well cell culture plate. After 24 hours, cultured cells were treated with a series of different concentrations (5 μg/mL, 10 μg/mL, 20 μg/mL, 40 μg/mL, 50 μg/mL, 100 μg/mL, 200 μg/mL, and 250 μg/mL) of filter sterilized, free chloramphenicol, PCL-P NPs and CAM-PCL-P NPs in 100 μL/well DMEM without serum and incubated further for 24 hours. This was followed by removal of the media and treatment with MTT dye at a final concentration of 0.5 mg/mL and further incubated for 4 hours. Finally, 100 μL of DMSO was added to each well to dissolve the blue formazan precipitate, and absorbance was measured at 570 nm using a microplate reader (Bio-Rad Model 680; Bio-Rad Laboratories Inc., Hercules, CA, USA). The cell viability was expressed as a percentage of the control by the following equation:
Viability (%) = Nt/Nc ⨯100 | (7) |
Here: Nt is the absorbance of the cells treated with samples
Nc is the absorbance of the untreated cells (n=5; where n is the number of independent experiments).
Assessing the hemolytic activity of CAM-PCL-P NPs against human red blood
In vitro hemolytic activity was performed by spectrophotometer.31,32 Five milliliters of venous blood were collected from a healthy volunteer. The blood was centrifuged (8 minutes at 1,600× g) and the pellet was washed three times with sterile isotonic PBS solution (pH 7.2±0.2) by centrifugation at 1,500 rpm for 5 minutes. The cells were resuspended in normal saline to 0.5%. A volume of 0.5 mL of the cell suspension was mixed with 0.5 mL of the samples (125 μg/mL, 250 μg/mL, 500 μg/mL, and 1,000 μg/mL concentrations in saline). The mixtures were incubated for 30 minutes at 37°C and centrifuged at 1,500 rpm for 10 minutes. The free hemoglobin in the supernatants was measured in a UV-Vis spectrophotometer at 540 nm. PBS and distilled water were used as minimal and maximal hemolytic controls. Each experiment was performed in triplicate at each concentration. The level of percentage hemolysis by the extracts was calculated according to the following formula:
| (8) |
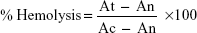
Here: At is the absorbance of test sample
An is absorbance of the control (saline control)
Ac is the absorbance of the control (water control).
The possible protection effect of PCL-pluronic nanoencapsulation was studied by comparison of the hemolysis of the CAM-PCL-P NPs (CN) with the corresponding free chloramphenicol (FC) and expressed as hemolysis reduction (HR).33
| (9) |
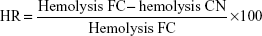
Antibacterial assay
In vitro assay with agar well diffusion
The in vitro antibacterial screening is done using the agar well-diffusion method on nutrient agar plates.34 All the cultures were grown on nutrient agar plates and maintained in the nutrient agar slants at 4°C. In brief, 1 mL (1.0×107 colony-forming units [CFUs]) of 24-hour-old bacterial culture strains suspension were uniformly spread over solidified nutrient agar plates with the help of a sterilized spreader. Wells of 6 mm diameter were made in the center of these agar plates with the help of a sterile cork borer. Using a micropipette, we added 4 μg CAM-PCL-P NPs aqueous preparations and concentration-equilibrated amounts of chloramphenicol to the different wells and allowed them to diffuse at 25°C for 1 hour. Then the plates were incubated at 38°C±2°C for 24–48 hours.
Assessment of increase in fold area
The increase in fold area was assessed by calculating the mean surface area of the inhibition zone of chloramphenicol and CAM-PCL-P NPs. The fold increase area of different test bacteria for free drug and for drug encapsulated NPs was calculated by the equation:
(B2 − A2)/A2 | (10) |
where A and B were zone of inhibition for chloramphenicol and CAM-PCL-P NPs, respectively.35
Determination of minimum inhibitory concentration and minimum bactericidal concentration
MIC and minimum bactericidal concentration (MBC) were determined according to the reported methods with minor modifications.36,37 MIC was determined by using various concentrations of CAM-PCL-P NPs (4–32 μg/mL) with respective equilibrated concentrations of chloramphenicol in nutrient broth. To evaluate the increase in solubility, we used water and not an organic solvent as a dispersion media for both samples; 100 μL of each bacterial inoculum was added to each tube and incubated at room temperature for 24–48 hours. Chloramphenicol was used as the positive control. The MIC was regarded as the lowest concentration of the tested sample that did not permit any visible growth after 24–48 hours of incubation.
The MBC was determined using reported methods. The tube that showed no visible growth after 48 hours of incubation when subcultured on a nutrient agar plate at using an inoculum size of 0.5 mL is considered to be the MBC.
Time-kill assay
The rate at which PCL-P NPs and CAM-PCL-P NPs killed MRSA was determined.38 Two MRSA strains (MRSA1, MRSA2) included in this experiment were grown overnight at 37°C with shaking in nutrient broth. The next day, the bacteria were washed twice in PBS, added to nutrient broth with or without antibiotics to achieve a final concentration of approximately 2×105 CFU/mL log phase of the tested MRSA, and then incubated at 37°C with shaking. CAM-PCL-P NPs at an MIC of 32 μg/mL and free chloramphenicol (equilibrated concentration) was added to bacterial cultures. Aliquots were removed at 0 hour, 4 hours, 8 hours, and 12 hours and analyzed to determine the number of viable bacteria that remained after treatment. The CFUs of MRSA recovered posttreatment were compared for both free chloramphenicol and CAM-PCL-P NPs and plotted as the CFU/mL over time posttreatment. The nutrient broth media without any antibacterial agents was used as the control for MRSA growth at each time point. The relationship between the treatment time and the viable cell count (CFU/mL) was assessed by the time-kill curve. Three independent experiments were run in triplicate.
CAM-PCL-P NPs applied in an in vivo burn-wound model
CAM-PCL-P NP performance as a topically applied antibiotic agent was evaluated in an in vivo burn-wound model. All procedures for animal experimentation were approved by the Institutional Animal Care and Use Committee at the Institute of Advanced Study in Science and Technology (Guwahati, India). All mice were anesthetized with a ketamine–xylazine cocktail, shaved, and cleansed with antiseptics. Uniform, reproducible burn wounds were created with a heated brass knob (1.27 cm diameter; applied for 45 seconds until reaching 180°C–200°C measured by calorimeter); 24 hours after wounding, the infected control, free chloramphenicol, and CAM-PCL-P NP groups were inoculated with 107 MRSA1 cells. The remaining group was not inoculated with MRSA and served as an uninfected control. On odd numbered days, topical treatment was administered by directly applying 5 mg of CAM-PCL-P NPs and free chloramphenicol (equilibrated concentration) onto the burn wound and moistened with 10 μL of PBS. Wounds were measured by two investigators independent of one another using vernier calipers and were photographed every other day to track the progression of wound closure.39
CFUs/wound determination
On days 3, 7, 11, and 15 after wounding, burn wounds were excised, pulverized, and homogenized in sterile PBS. Samples were diluted 100-fold and plated on nutrient agar in order to tally MRSA bacterial CFUs grown after 48 hours of incubation at 37°C. All results were normalized based on excised tissue weight.
Histological examinations
At days 7 and 11 after wounding, excised burn tissues were fixed in 10% formalin for 24 hours, processed, and embedded in paraffin. Four-micron vertical sections were fixed to glass slides and subjected to hematoxylin and eosin staining. Slides were examined by light microscopy with a Leica LEITZ BIOMED microscope (Leica Microsystems, Wetzlar, Germany), and images were obtained by using LAS EZ software.
Statistical analysis
Data are represented as mean ± SD of five independent experiments. For the time-kill assay, comparisons were made using an unpaired Student’s t-test. For the in vivo burn-wound study, analysis of variance for the means of the groups was calculated and P-values ≤0.05 were considered statistically significant.
Results and discussion
Preparation, microscopy, zeta potential, and particle size analysis of CAM-PCL-P NPs
The solvent-displacement method was followed for the preparation of PCL-P NPs containing chloramphenicol, without the use of toxic chlorinated organic solvents to incorporate the drug into the NPs. Here, the first challenge was the selection of an organic phase to solubilize both chloramphenicol and the polyester polymer PCL. Acetone (a water-miscible and low-boiling-point solvent) was selected for PCL-P NPs preparation, and chloramphenicol-loaded PCL-P NPs in acetone yielded an amorphous precipitate of nonassociated drug. Therefore, we used different cosolvents to optimize the solubility of both chloramphenicol and the polymer. It was observed that CAM-PCL-P NPs produced without a cosolvent mostly resulted in an amorphous precipitate, whereas when methanol was used as a cosolvent it gave regular-shaped, spherical solid, dense structured particles, and amorphous entities were highly reduced in comparison to DMSO. The cosolvent was used to assist in solubility of chloramphenicol and thus got incorporated into the hydrophobic core of PCL-P NPs. Due to the great hydrophobicity of PCL, pure hydrophobic PCL NPs tend to form aggregates in water because of its high specific surface area and high surface energy.
PF127, a nonionic coemulsifier, gets adsorbed strongly onto the surface of hydrophobic PCL-P NPs via their hydrophobic polypropylene oxide center block. This mode of adsorption leaves the hydrophilic polyethylene-oxide sidearms in a mobile state because they extend outward from the particle surface. These sidearms provide stability to the particle suspension by a repulsion effect through a steric mechanism of stabilization, involving both enthalpic and entropic contribution. This leads to the formation of fine particles, avoiding aggregation and resulting in smaller particle size with narrow size distribution (Figure 1A and B). Particle sizes of CAM-PCL-P NPs and PCL-P NPs obtained from a particle-size analyzer were 123.5 nm and 121 nm, respectively. The CAM-PCL-P NPs showed a negative surface charge of around −29.6 mV, whereas the NPs prepared without pluronic showed zeta potential value of −22.4 mV. The significant increase in the absolute zeta potential value may refer to a higher dispersion stability that resulted in a more stable suspension. Again, a lower zeta potential value indicates colloid instability, which could lead to aggregation of NPs.40,41 The particle size and surface properties play a major role in bioactivity by influencing the in vitro drug release, interaction with bacterial cells, cellular uptake, cytotoxicity of these NPs, as well as their in vivo pharmacokinetics and biodistribution, and thereby the therapeutic efficacy of the encapsulated drug.42–44
 | Figure 1 Images of CAM-PCL-P NPs viewed under field emission scanning electron microscopy.
|
Spectroscopic analysis
CAM-PCL-P NPs showed distinct UV-Vis absorption bands with a sharp characteristic peak of chloramphenicol at around 263 nm, whereas PCL-P NPs did not show such an absorption pattern (Figure 2). A gradual increase in the chloramphenicol concentration leads to increased intensities of spectral peaks and hence confirmed the successful loading of chloramphenicol into the PCL-P NPs with intake photophysical properties.
 | Figure 2 UV-Vis spectra of PCL-P NPs and CAM-PCL-P NPs. |
FT-IR analysis
FT-IR spectra were analyzed to ensure that no chemical interactions between the drug and the polymer had occurred in the nanoparticle. FT-IR spectra of chloramphenicol, PCL-P NPs, and CAM-PCL-P NPs are shown in Figure 3. The characteristic infrared peaks of chloramphenicol indicates the presence of free hydroxyl group, N-H stretching, aromatic stretching, CH2 asymmetric and symmetric stretching, and C-O stretching. The spectra of the PCL-P NPs showed the characteristic bands corresponding to C=O lactone stretching, C-O lactone stretching, terminal hydroxyl (OH) group stretching, and overlapping symmetric and asymmetric C-H stretching. The absorption peaks of the chloramphenicol were not observed in the CAM-PCL-P NPs, which explains that no chloramphenicol was interacted onto the surface of the NPs but was completely encapsulated in the PCL-P NPs. Almost all the positions of the peaks of PCL-P NPs and CAM-PCL-P NPs were the same, with no observed changes in the intensities. This seems to indicate the absence of chemical interaction between the polymer and the drug in chloramphenicol NP preparation.
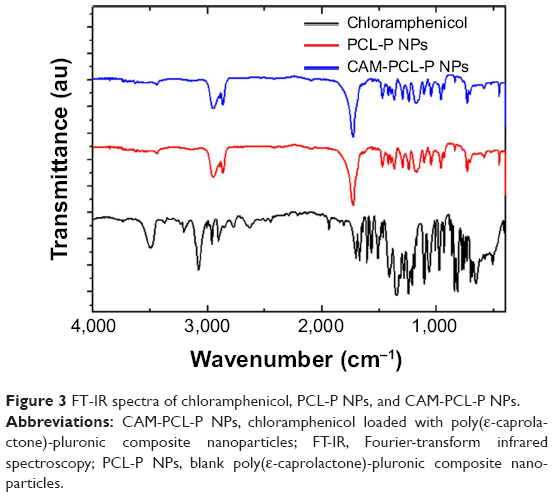 | Figure 3 FT-IR spectra of chloramphenicol, PCL-P NPs, and CAM-PCL-P NPs. |
X-ray diffraction analysis
The intact crystalline nature of chloramphenicol extracted from loaded NPs were confirmed by X-ray diffraction analysis (Figure 4). The diffraction patterns of extracted chloramphenicol showed several peaks similar to that of pure chloramphenicol (2θ at 10.76, 12.82, 18.90, 25.88 and 31.62). The PCL-P NPs and CAM-PCL-P NPs showed a similar type of Bragg peaks with that of the pure PCL (2θ at 23.7 and 21.4), which suggested that the crystalline structure of PCL is maintained and not changed into amorphous phase during the preparation of NPs. No Bragg peaks of chloramphenicol were seen in the CAM-PCL-P NPs, which is in agreement with FT-IR results that most of the chloramphenicol is encapsulated inside the nanoparticle but not interacted onto the surface of the NPs.
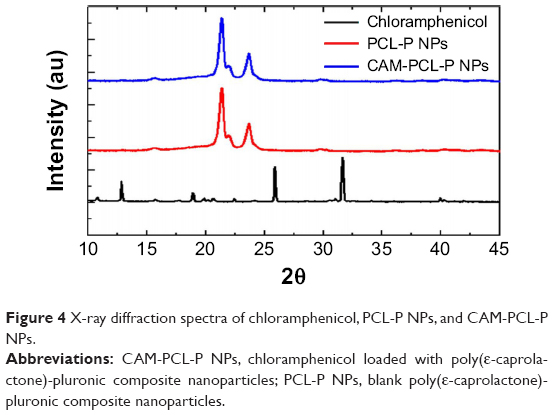 | Figure 4 X-ray diffraction spectra of chloramphenicol, PCL-P NPs, and CAM-PCL-P NPs. |
Encapsulation efficiency
The EE of chloramphenicol in PCL-P NPs was dependent on the drug-to-polymer ratio (Table 1). As mentioned above, PCL concentration (62.5 mg) was maintained as constant in combination with ten different concentrations of drug. The highest EE was found to be 98.3%, when the drug-to-PCL ratio (w/w) was 7.5:62.5, ie, 7.3725 μg of chloramphenicol was encapsulated. The EE increased with the increasing amount of drug until it reached a plateau at 7.5 mg of chloramphenicol, after which it started decreasing. Change in the EE with added drug amount ascertained the successful encapsulation of chloramphenicol into PCL-P NPs. A significant increase in EE was observed in NPs prepared with PF127 compared with NPs prepared without PF127 (data not shown), which may be because of its self-assembling property in an aqueous environment with hydrophobic core. Thus, PF127 might have encapsulated chloramphenicol while its hydrophilic parts interacted with PCL.
 | Table 1 EE (%) of chloramphenicol in PCL-P NPs |
In vitro drug release and release kinetics
The in vitro release profile of chloramphenicol in the first 16 days (384 hours) is shown in Figure 5. A biphasic release pattern of drug was observed from the polymeric NPs. In the first 12 hours, 21% of chloramphenicol was released; this slowly increased up to 26% in 24 hours. Thereafter, a drop in the rate of release was observed, and finally 84% of chloramphenicol was released over the entire 384 hours. For CAM-PCL NPs, the release was a little less, at 19%, 23%, and 82%, respectively, for the same time periods. This may be because of increased hydrophilicity of the CAM-PCL-P NPs due to presence of pluronic. The drug-loaded NPs exhibited sustained-release properties, followed by a slight initial burst release due to drug adsorbed on the NP surface. Prolonged release in the later stage can be attributed to the slow diffusion of the drug from the NPs. The t50 (time taken for 50% drug release) was calculated as 72 hours (51%). The t50 value suggested a highly controlled and sustained release of chloramphenicol from the CAM-PCL-P NPs. The sustained release characteristics of PCL-P NPs could be a beneficial delivery system for topical applications.
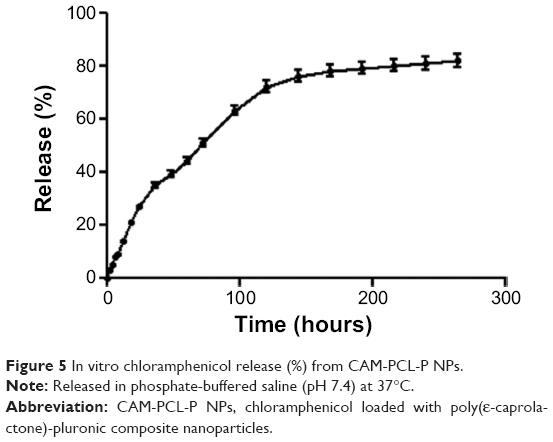 | Figure 5 In vitro chloramphenicol release (%) from CAM-PCL-P NPs. |
The calculated values for release constants and regression coefficients (R2) for the release data are shown in Table 2. We analyzed regression coefficients for different kinetic models to find out the mechanism of chloramphenicol release from CAM-PCL-P NPs. The Higuchi model was best fitted with release kinetic data of chloramphenicol. Based on our release data analysis, the following mathematical model was derived to obtain best curve fit for our drug-release data.
 | Table 2 Mathematical models for drug-release kinetics |
| (11) |
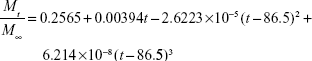
Where Mt/M∞ is the fraction of drug released at time t. The calculated regression coefficient (R2) 0.997 showed that the derived mathematical model is best fitted with release kinetic data of chloramphenicol from CAM-PCL-P NPs.
Storage stability
Nanoparticle suspensions maintained the initial properties with respect to size and EE after 6 months of storage at 4°C and 25°C, respectively (P>0.05). This shows that the developed CAM-PCL-P NPs have a good stability, which is a characteristic of an ideal drug.
Hemolytic activity
Hemolytic activity of CAM-PCL-P NPs in comparison to the crude drug was screened against normal human erythrocytes and exhibited low to mild hemolytic effect. Hemolytic activity of the test samples was expressed in % hemolysis and reported as mean ± standard deviation of three replicates. The result indicated that the CAM-PCL-P NPs (at dose 1,000 μg/mL) possess lesser hemolytic activity (4.12%±0.19%) with an IC50 (half maximal inhibitory concentration) value of 5,025 μg/mL compared to the crude drug (8.87%±0.29%) with an IC50 value of 1,259 μg/mL (equilibrated chloramphenicol concentration). We also observed that encapsulation of chloramphenicol helps in lowering the hemolytic activity, which can be attributed to an ideal drug characteristic. Dose-dependent increase in percentage hemolysis was observed (Table 3). An average of 33.14% hemolysis reduction was observed for the tested concentrations of CAM-PCL-P NPs compared to free chloramphenicol.
 | Table 3 Dose-dependent increase in percentage hemolysis |
Cytotoxicity study results of free chloramphenicol, PCL-P NPs, and CAM-PCL-P NPs
In the cytotoxicity assay, after 24 hours of posttreatment with CAM-PCL-P NPs (250 μg/mL), HeLa cells showed a much higher viability (83%) than did free chloramphenicol (76%) (equilibrated concentration), whereas the PCL-P NPs showed no sign of toxicity toward MCF-7 breast cancer cells, which is in support of the previous reports. Prior to any in vivo application of the CAM-PCL-P NPs, high dose tolerance exhibited by a mammalian cell line is very important.
Antibacterial activity
For therapeutic drugs, retention of biological activity is very important, since instability (chemical modification, denaturation) may occur in the manufacturing process of the NPs as well as during the release. The antibacterial activity of the developed CAM-PCL-P NPs was studied by determining the zone of inhibition, MIC, MBC, killing kinetics, and interaction study. It is evident from the obtained data that CAM-PCL-P NPs show enhanced antibacterial activity by showing a greater zone of inhibition against the ten clinical isolates of MRSA (Table 4). PCL-encapsulated chloramphenicol has the highest activity against MRSA1 and lowest against MRSA5. It was observed that CAM-PCL-P NPs showed an average of 1.007 increase in the fold area. From all the zone of inhibition and increase in fold area data, it is clear that for all the cases, the CAM-PCL-P NPs are more effective than the chloramphenicol alone. This may be because of the enhanced solubility and stability of chloramphenicol in the nanoparticulate system.
 | Table 4 Antibacterial activity of CAM-PCL-P NPs (zone of inhibition against the ten clinical isolates of MRSA) |
Minimum inhibitory concentration and minimum bactericidal concentration
The MIC/MBC values of the CAM-PCL-P NPs ranged from 4 to 12 μg/mL, which is much lower than the MIC/MBC values shown by the concentration-equilibrated chloramphenicol in free form (Table 5). According to Clinical and Laboratory Standards Institute recommendations for MIC, ≤8 μg/mL was taken as susceptible, ≤16 μg/mL as intermediate, and ≥32 μg/mL as resistant.45
 | Table 5 MIC/MBC values of free CAM and CAM-PCL-P NPs |
CAM-PCL-P NPs used at 32 μg/mL rapidly kill MRSA (killing kinetics)
CAM-PCL-P NPs at 32 μg/mL concentration demonstrated significantly enhanced killing of the tested MRSA compared to free chloramphenicol. As shown in Figure 6, the nontreated MRSA showed significant increase of the CFU count throughout all the time points. This excellent killing rate shown by CAM-PCL-P NPs is just as important as the bactericidal nature of the agent, because the faster the drug kills, the more efficient it is at the blockage of biofilm formation.
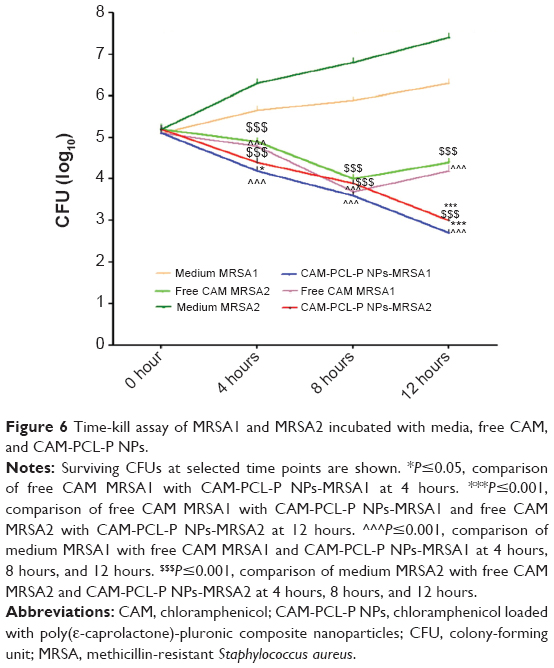 | Figure 6 Time-kill assay of MRSA1 and MRSA2 incubated with media, free CAM, and CAM-PCL-P NPs. |
In vivo MRSA burden reduction on topical application of CAM-PCL-P NPs
Postinfection CFU data of the in vivo experiment indicated a significant reduction in the MRSA burden in the group treated with CAM-PCL-P NPs in comparison to the group treated with free chloramphenicol solution (Figure 7A). Moreover, no significant difference in CFU count was seen between the group treated with PCL-P NPs and the nontreated group. Significantly, two animals from the PCL-P NP group and one animal from the free chloramphenicol group died due to the severity of MRSA infection during the course of treatment. CAM-PCL-P NPs exhibited significantly better results at MRSA killing as well as increased survivability rate and wound closure in comparison to the concentration-equilibrated free chloramphenicol-solution group at days 3, 7, and 14 (Figure 7B). No significant differences in relative wound area (Figure 7B) were seen across all groups due to variability in eschar size and healing times.
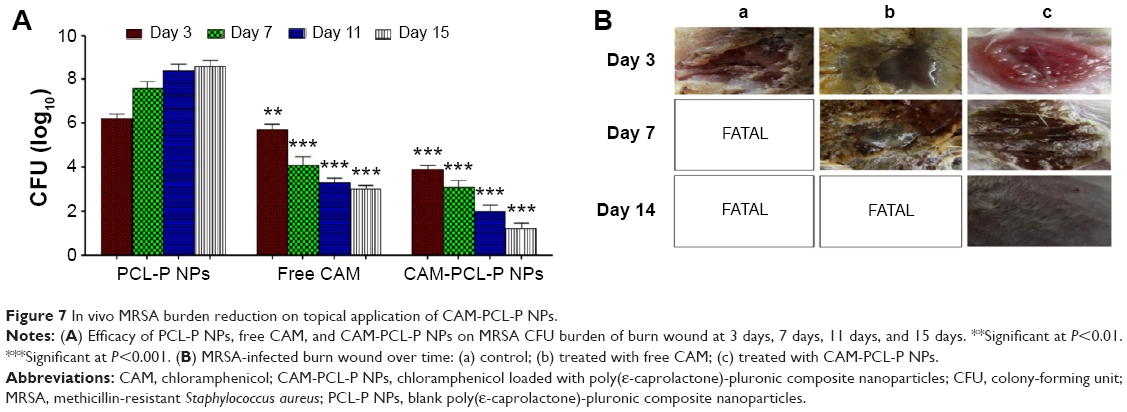 | Figure 7 In vivo MRSA burden reduction on topical application of CAM-PCL-P NPs. |
Histologic and clinical evaluation of wounds
To evaluate wound healing progression, tissue sections were obtained on days 3, 7, and 14 for control, free chloramphenicol solution treated, and CAM-PCL-P NP treated burn wounds, respectively. Control burn on day 3 (Figure 8A) revealed complete disruption, necrosis, and disorganized inflammation of epidermis, subepidermis, and dermis layers, and only some kind of bulbous elements were observed. In cases of burn wound treated with free chloramphenicol solution (as observed on day 7), epidermis was ulcerated with a sign of little epidermal reepithelialization and presence of inflammatory granulation (Figure 8B). It was observed that the group treated with CAM-PCL-P NPs showed more-improved healing than did the group treated with free chloramphenicol, as the various cellular ultrastructures were better organized in the former. On day 14, the CAM-PCL-P NPs group (Figure 8C) showed a pronounced reepithelialization with intact maturing subepidermis and dermis, along with normal skin adnexal structures.
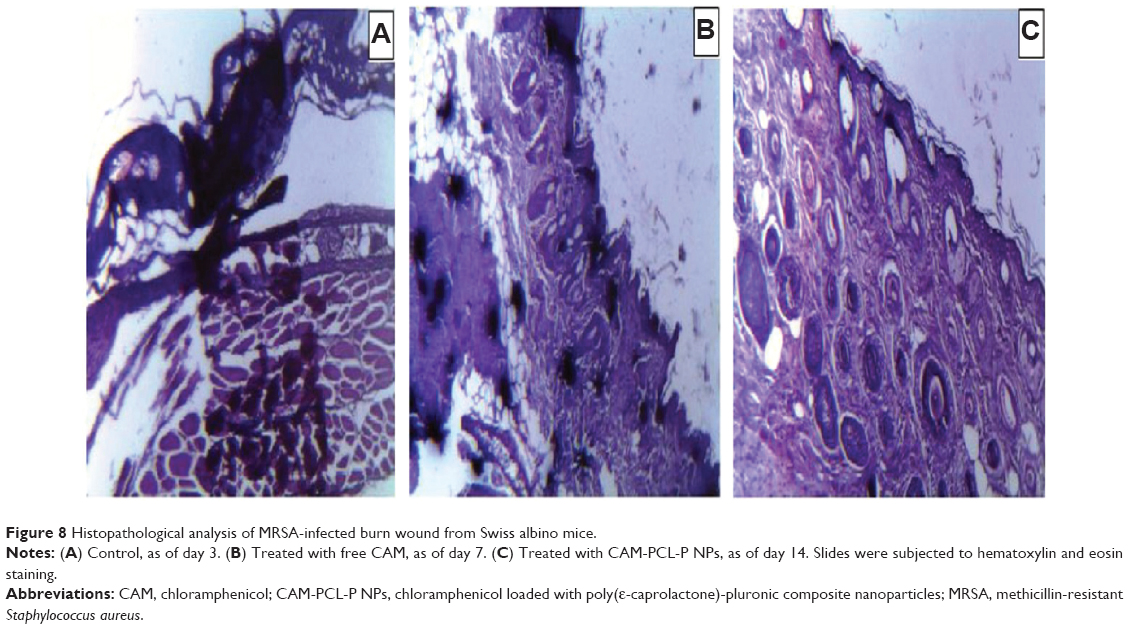 | Figure 8 Histopathological analysis of MRSA-infected burn wound from Swiss albino mice. |
Conclusion
In this study, a novel biodegradable, biocompatible polymeric PCL-P NP was developed to attain hydrophilicity with the better skin penetration and sustained-release properties of broad-spectrum lipophilic antibiotic chloramphenicol. PCL-P NPs containing chloramphenicol were successfully fabricated using a modified solvent-displacement method. Chloramphenicol loaded with PCL nanocarriers was characterized in terms of photophysical properties, mean size, morphology, physical state of encapsulated drug, and in vitro drug release. The prepared NPs possess the following advantages: small particle size (123.5 nm), suitable size distribution, moderate preparation condition, high drug loading, and sustained drug-release properties. The prepared nanoformulation showed significantly enhanced antimicrobial activity against ten clinical isolates of MRSA, compared with the free drug. Further, it was observed that the nanoencapsulation significantly decreased the toxicity of chloramphenicol against human red blood erythrocytes and tested HeLa cells. In vitro anti-MRSA activity evaluation of CAM-PCL-P NPs revealed significant enhancement in activity compared to free chloramphenicol.
We were also able to demonstrate in vivo efficacy of CAM-PCL-P NPs against MRSA infected in burn wounds, as the nanoform was able to diffuse into thermally injured skin and tissue to significantly reduce MRSA burden and accelerate wound closure. Intuitively, localized coverage provided by CAM-PCL-P NPs will facilitate less exposure of chloramphenicol to mammalian cells systemically and allows for targeting of MRSA; colonizing beneath skin minimizes chloramphenicol’s deleterious side effects. Compared to systematically administrated NPs, topically applied NPs are of lesser concern because in the earlier case it is not subjected to variable metabolism pathways for drug excretion and thus avoid end-organ accumulation. Moreover, CAM-PCL-P NPs can be systematically dispersed when applied to a full-thickness burn wound, although this needs to be further evaluated.
In conclusion, CAM-PCL-P NPs can be a promising novel antimicrobial drug candidate. Also, PCL-P NPs holds immense potential in delivery of hydrophobic drugs in general. Further in vivo investigations, including pharmacokinetics, biodistribution, pharmacodynamics, and toxicity, will be carried out to evaluate the potential of these copolymer NPs to be used as drug delivery vehicles.
Acknowledgments
The authors are grateful for the support from the Director of the Institute of Advanced Study in Science and Technology (IASST) and the Department of Science and Technology, Government of India for the funding of the research. We acknowledge Sushmita Gupta for helping in English language copy editing of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Horvath EE, Murray CK, Vaughan GM, et al. Fungal wound infection (not colonization) is independently associated with mortality in burn patients. Ann Surg. 2007;245(6):978–985. | ||
Church D, Elsayed S, Reid O, Winston B, Lindsay R. Burn wound infections. Clin Microbiol Rev. 2006;19(2):403–434. | ||
Alexis A, Carrer DP, Droggiti DI, et al. Immune responses in relation to the type and time of thermal injury: an experimental study. Injury. Epub 2014 Oct 31. | ||
Chamania S, Hemvani N, Joshi S. Burn wound infection: Current problem and unmet needs. Indian J Burns. 2012;20(1):18–22. | ||
Lederer JA, Rodrick ML, Mannick JA. The effects of injury on the adaptive immune response. Shock. 1999;11(3):153–159. | ||
Fong J, Wood F. Nanocrystalline silver dressings in wound management: a review. Int J Nanomedicine. 2006;1(4):441–449. | ||
Zorgani AA, Shahen A, Zaidi M, Franka M. A profile and spectrum of four cases of methicillin-resistant Staphylococcus aureus in a burns intensive care unit. Ann Burns Fire Disasters. 2006;19(1):5–10. | ||
Katakura T, Yoshida T, Kobayashi M, Herndon DN, Suzuki F. Immunological control of methicillin-resistant Staphylococcus aureus (MRSA) infection in an immunodeficient murine model of thermal injuries. Clin Exp Immunol. 2005;142(3):419–425. | ||
Jacobsen F, Fisahn C, Sorkin M, et al. Efficacy of topically delivered moxifloxacin against wound infection by Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55(5):2325–2334. | ||
Fayyaz M, Mirza IA, Ahmed Z, Abbasi SA, Hussain A, Ali S. In vitro susceptibility of chloramphenicol against methicillin-resistant Staphylococcus aureus. J Coll Physicians Surg Pak. 2013;23(9):637–640. | ||
Govender T, Riley T, Ehtezazi T, et al. Defining the drug incorporation properties of PLA-PEG nanoparticles. Int J Pharm. 2000;199(1):95–110. | ||
Atyabi F, Moghaddam FA, Dinarvand R, Zohuriaan-Mehr MJ, Ponchel G. Thiolated chitosan coated poly hydroxyethyl methacrylate nanoparticles: synthesis and characterization. Carbohydr Polym. 2008;74(1):59–67. | ||
Ting WW, Vest CD, Sontheimer RD. Review of traditional and novel modalities that enhance the permeability of local therapeutics across the stratum corneum. Int J Dermatol. 2004;43(7):538–547. | ||
Bonina FP, Puglia C, Barbuzzi T, et al. In vitro and in vivo evaluation of polyoxyethylene esters as dermal prodrugs of ketoprofen, naproxen and diclofenac. Eur J Pharm Sci. 2001;14(2):123–134. | ||
Bouchemal K, Briançon S, Perrier E, Fessi H, Bonnet I, Zydowicz N. Synthesis and characterization of polyurethane and poly(ether urethane) nanocapsules using a new technique of interfacial polycondensation combined to spontaneous emulsification. Int J Pharm. 2004;269(1):89–100. | ||
Shim J, Seok Kang H, Park WS, Han SH, Kim J, Chang IS. Transdermal delivery of mixnoxidil with block copolymer nanoparticles. J Control Release. 2004;97(3):477–484. | ||
Alvarez-Román R, Naik A, Kalia YN, Guy RH, Fessi H. Enhancement of topical delivery from biodegradable nanoparticles. Pharm Res. 2004;21(10):1818–1825. | ||
Alvarez-Román R, Barré G, Guy RH, Fessi H. Biodegradable polymer nanocapsules containing a sunscreen agent: preparation and photoprotection. Eur J Pharm Biopharm. 2001;52(2):191–195. | ||
Jiménez MM, Pelletier J, Bobin MF, Martini MC. Influence of encapsulation on the in vitro percutaneous absorption of octyl methoxycinnamate. Int J Pharm. 2004;272(1–2):45–55. | ||
Ma Y, Zheng Y, Zeng X, et al. Novel docetaxel-loaded nanoparticles based on PCL-Tween 80 copolymer for cancer treatment. Int J Nanomedicine. 2011;6:2679–2688. | ||
Kim DY, Rhee YH. Biodegradation of microbial and synthetic polyesters by fungi. Appl Microbiol Biotechnol. 2003;61(4):300–308. | ||
Suksiriworapong J, Sripha K, Kreuter J, Junyaprasert VB. Functionalized (poly(ε-caprolactone))2-poly(ethylene glycol) nanoparticles with grafting nicotinic acid as drug carriers. Int J Pharm. 2012;423(2):562–570. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. | ||
Das RK, Kasoju N, Bora U. Encapsulation of curcumin in alginate-chitosan-pluronic composite nanoparticles for delivery to cancer cells. Nanomedicine. 2010;6(1):153–160. | ||
Li P, Dai NY, Zhang JP, Wang AQ, Wei Q. Chitosan-alginate nanoparticles as a novel drug delivery system for nifedipine. Int J Biomed Sci. 2008;4(3):221–228. | ||
Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv Drug Deliv Rev. 2001;48(2–3):139–157. | ||
Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60(4):110–111. | ||
Kassem MA, ElMeshad AN, Fares AR. Lyophilized sustained release mucoadhesive chitosan sponges for buccal buspirone hydrochloride delivery: formulation and in vitro evaluation. AAPS Pharm Sci Tech. Epub 2014 Nov 6. | ||
Mathew ST, Devi SG, KV S. Formulation and evaluation of ketorolac tromethamine-loaded albumin microspheres for potential intramuscular administration. AAPS Pharm Sci Tech. 2007;8(1):14. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Yang ZG, Sun HX, Fang WH. Haemolytic activities and adjuvant effect of Astragalus membranaceus saponins (AMS) on the immune responses to ovalbumin in mice. Vaccine. 2005;23(44):5196–5203. | ||
Kalita S, Kumar G, Karthik L, Rao KVB. Phytochemical composition and in vitro hemolytic activity of Lantana camara L. (Verbenaceae) leaves. Pharmacologyonline Newsletter. 2011;1:59–67. | ||
Serrano DR, Hernández L, Fleire L, et al. Hemolytic and pharmacokinetic studies of liposomal and particulate amphotericin B formulations. Int J Pharm. 2013;447(1–2):38–46. | ||
Murugan K, Senthilkumar B, Senbagam D, Al-Sohaibani S. Biosynthesis of silver nanoparticles using Acacia leucophloea extract and their antibacterial activity. Int J Nanomedicine. 2014;9:2431–2438. | ||
Gajbhiye M, Kesharwani J, Ingle A, Gade A, Rai M. Fungus-mediated synthesis of silver nanoparticles and their activity against pathogenic fungi in combination with fluconazole. Nanomedicine. 2009;5(4):382–386. | ||
Irobi ON, Daramola SO. Antifungal activities of crude extracts of Mitracarpus villosus (Rubiaceae). J Ethnopharmacol. 1993;40(2):137–140. | ||
Rotimi VO, Laughon BE, Bartlet JG, Mosadomi HA. Activities of Nigerian chewing stick extracts against Bacteroides gingivalis and Bacteroides melaninogenicus. Antimicrob Agents Chemother. 1998;32(4):598–600. | ||
Sande L, Sanchez M, Montes J, et al. Liposomal encapsulation of vancomycin improves killing of methicillin-resistant Staphylococcus aureus in a murine infection model. J Antimicrob Chemother. 2012;67(9):2191–2194. | ||
Sanchez DA, Schairer D, Tuckman-Vernon C, et al. Amphotericin B releasing nanoparticle topical treatment of Candida spp. in the setting of a burn wound. Nanomedicine. 2014;10(1):269–277. | ||
Scholes PD, Coombes AG, Illum L, et al. Detection and determination of surface levels of poloxamer and PVA surfactant on biodegradable nanospheres using SSIMS and XPS. J Control Release. 1999;59(3):261–278. | ||
Zhang Y, Tang L, Sun L, et al. A novel paclitaxel-loaded poly(epsilon-caprolactone)/Poloxamer 188 blend nanoparticle overcoming multidrug resistance for cancer treatment. Acta Biomater. 2010;6(6):2045–2052. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Florence AT. Nanoparticle uptake by the oral route: fulfilling its potential? Drug Discov Today Technol. 2005;2(1):75–81. | ||
Yan F, Zhang C, Zheng Y, et al. The effect of poloxamer 188 on nanoparticle morphology, size, cancer cell uptake, and cytotoxicity. Nanomedicine. 2010;6(1):170–178. | ||
Clinical and Laboratory Standards Institute (CLSI). Performance standards for antimicrobial susceptibility testing; twenty-second informational supplement (M100-S22). Wayne, PA: CLSI; 2012. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.