Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Chitosan-decorated selenium nanoparticles as protein carriers to improve the in vivo half-life of the peptide therapeutic BAY 55-9837 for type 2 diabetes mellitus
Authors Rao L, Ma Y ![]() , Zhuang M, Luo T, Wang Y, Hong A
, Zhuang M, Luo T, Wang Y, Hong A
Received 16 May 2014
Accepted for publication 24 June 2014
Published 17 October 2014 Volume 2014:9(1) Pages 4819—4828
DOI https://doi.org/10.2147/IJN.S67871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Lei Rao,* Yi Ma,* Manjiao Zhuang, Tianjie Luo, Yayu Wang, An Hong
Department of Cell Biology, Guangdong Province Key Lab of Bioengineering Medicine, National Engineering Research Center of Gene Engineering Medicine, Institute of Biological Medicine, Jinan University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Purpose: As a potential protein therapeutic for type 2 diabetes mellitus (T2DM), BAY 55-9837 is limited by poor stability and a very short half-life in vivo. The purpose of this study was to construct a novel nanostructured biomaterial by conjugating BAY 55-9837 to chitosan-decorated selenium nanoparticles (CS-SeNPs) to prolong the in vivo half-life of BAY 55-9837 by reducing its renal clearance rate.
Materials and methods: BAY 55-9837-loaded CS-SeNPs (BAY-CS-SeNPs) were prepared, and their surface morphology, particle size, zeta potential, and structure were characterized. The stability, protein-loading rate, and in vitro release of BAY 55-9837 from CS-SeNPs were also quantified. Additionally, a sensitive high-performance liquid chromatography (HPLC) assay was developed for the quantification of BAY 55-9837 in mouse plasma. Thereafter, mice were injected via the tail vein with either BAY 55-9837 or BAY-CS-SeNPs, and the plasma concentration of BAY 55-9837 was determined via our validated HPLC method at different time intervals postinjection. Relevant in vivo pharmacokinetic parameters (half-life, area under the curve from time 0 to last sampling point, observed clearance) were then calculated and analyzed.
Results: BAY-CS-SeNPs were successfully synthesized, with diameters of approximately 200 nm. BAY-CS-SeNPs displayed good stability with a high protein-loading rate, and the release process of BAY 55-9837 from the CS-SeNPs lasted for over 70 hours, with the cumulative release reaching 78.9%. Moreover, the conjugation of CS-SeNPs to BAY 55-9837 significantly reduced its renal clearance to a rate of 1.56 mL/h and extended its half-life to 20.81 hours.
Conclusion: In summary, our work provides a simple method for reducing the renal clearance rate and extending the half-life of BAY 55-9837 in vivo by utilizing CS-SeNPs as nanocarriers.
Keywords: BAY 55-9837, selenium nanoparticles, clearance, half-life, type 2 diabetes
Introduction
Type 2 diabetes mellitus (T2DM) is a growing health problem across the world, and the number of diagnosed T2DM cases has increased dramatically over the past few decades.1 Traditional therapies for T2DM include treatment with insulin, sulfonylureas, metformin, peroxisome proliferator-activated receptor-γ agonists, and α-glucosidase inhibitors, the majority of which are small molecules. Unfortunately, these therapies display limited efficacy and have significant mechanism-based side effects.2
The use of proteins and peptides as human therapeutics has increased rapidly in recent years, and protein-based products are predicted to represent four of the five top-selling drugs in the global market over the next few years.3,4
BAY 55-9837, a potential protein therapeutic for the treatment of T2DM, was originally engineered by performing iterative rounds of site-directed mutagenesis on vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating peptide (PACAP).5 PACAP is a member of the VIP-glucagon-growth hormone-releasing factor secretin superfamily.6 PACAP exists in two molecular forms: PACAP-27 and PACAP-38.7 PACAP-27 activates both VPAC1 and VPAC2 receptors; VPAC2 activation promotes insulin secretion, whereas VPAC1 activation enhances glucagon secretion.8,9 Both VPAC1 and VPAC2 receptors are expressed within islet cells.10 As such, BAY 55-9837 was engineered to be a potent and highly selective VPAC2 agonist that can stimulate insulin secretion without stimulating glucagon secretion.11 Unfortunately, the potential therapeutic benefit of using BAY 55-9837 is limited by its very short half-life in vivo, which is likely due to the rapid renal clearance of the peptide.12 Therefore, it requires continuous intravenous or subcutaneous infusion to establish efficacy. Recent attempts have been made to overcome these limitations, eg, amino acid substitutions and polyethylene glycolation were used to prolong the half-life of BAY 55-9837 in vivo.13
The in vivo half-life of protein-based products significantly affects both dosing requirements and therapeutic efficacy; therefore, strategies to prolong the half-life of such drugs are highly significant. One such strategy, which utilizes the polyethylene glycolation of a given therapeutic protein, works to prolong the protein half-life by increasing the apparent size of the polypeptide.14 Generally, the conjugation of a given therapeutic protein to polymers or the incorporation of it into a drug carrier for protection and slow release is a commonly utilized method of prolonging their in vivo half-life.15,16
The emergence of nanostructured biomaterials has enabled new prospects for drug therapies in T2DM. Hybrid organic–inorganic nanoparticles (NPs) possess promising and highly controllable properties that have broad application in drug therapies, including targetable drug delivery.17 For example, Liu et al18 demonstrated that the conjugation of selenium NPs (SeNPs) and 5-fluorouracil can achieve anticancer synergism, while Makhluf et al19 showed that modified polyvinyl alcohol–Fe3O4 NPs can successfully serve as protein carriers that are targetable to sperm cells. Furthermore, it has been found that the half-life of some therapeutics can be improved by conjugating them to nanocarriers. For instance, Singh et al20 reported that nanocarriers may facilitate the prolonged ductal retention of diagnostic or therapeutic agents. Moreover, Lee et al21 demonstrated that chitosan (CS)-coated poly(lactic-co-glycolic acid) NPs containing palmitic acid-modified exendin-4 have considerable potential as a long-acting inhalation delivery system for the treatment of T2DM. As nanotechnology is widely used in drug therapies, we aimed to design a nanostructure-based medicine system to improve the life span of the low-molecular-weight protein therapeutic BAY 55-9837 by using SeNPs as carriers.
Selenate, an essential mineral, is an important component of optimal human health.22 Recently, SeNPs have been widely studied because of their excellent photoelectric performance and advanced biological activity.23,24 Se possesses many beneficial properties, including antioxidant, antiapoptotic, anticancer, and immune-system enhancements.25–27 However, its beneficial properties occur within a limited range. In comparison with Se, SeNPs possess lower toxicity as chemotherapeutic agents.28,29 Moreover, the mean serum Se concentrations in patients with DM are significantly lower than normal.30,31 Owing to their high bioavailability, low toxicity, and novel therapeutic properties, SeNPs have been recognized as a promising tool for drug therapies in T2DM.
CS is an effective and naturally occurring material used for synthesizing NPs with therapeutically desirable properties, such as biocompatibility. Furthermore, CS can be used to modify and stabilize the synthesis of SeNPs.32 CS NPs have been widely investigated as carriers for therapeutic proteins and genes in recent years. Moreover, positively charged SeNPs can be constructed with a CS-surface decoration, forming a self-assembled layer, which has a significant impact on their performance in biological applications.33
For these reasons discussed, we utilized CS-SeNPs as nanocarriers to increase the apparent molecular size of BAY 55-9837. As the increased apparent molecular size of BAY 55-9837 can reduce the rate of its renal clearance, BAY-CS-SeNPs may yield longer half-lives in vivo relative to unconjugated BAY 55-9837.
Materials and methods
Materials
CS, sodium selenite (Na2SeO3), and bicinchoninic acid kits were purchased from Sigma-Aldrich (St Louis, MO, USA). Trifluoroacetic acid (TFA) and acetonitrile were purchased from Anpel (Shanghai, People’s Republic of China). Roswell Park Memorial Institute (RPMI) 1640 medium and fetal bovine serum were purchased from Gibco (Gaithersburg, MD, USA). BAY 55-9837 was obtained from R&D Systems (Minneapolis, MN, USA). The internal standard, thymopentin (TP-5) Arg-Lys-Asp-Val-Tyr, was purchased from the Third Affiliated Hospital of Sun Yat-Sen University. All of the solvents used were of high-performance liquid chromatography (HPLC) grade, and Milli-Q water was used in all of the experiments.
Preparation of BAY-CS-SeNPs
SeNPs modified by CS were synthesized according to the method of Yu et al33 by using CS at a concentration of 0.8 mg/mL SeNPs; the concentrations were measured by inductively coupled plasma atomic emission spectrometry. A stock solution of 4 mg/mL BAY 55-9837 was prepared by dissolving 2 mg of BAY 55-9837 powder in 0.5 mL of Milli-Q water. An 8 μL aliquot of a BAY 55-9837 stock solution was mixed with a 24 μL aliquot of CS-SeNPs (0.04368 mg/mL); following this, a 32 μL aliquot of Milli-Q water was added overnight.
Morphology and characterization of BAY-CS-SeNPs
BAY-CS-SeNPs and CS-SeNPs were characterized by transmission electron microscopy and scanning electron microscopy (SEM). Energy dispersive X-ray (EDX) spectrometry was employed to analyze the elemental composition of BAY-CS-SeNPs. Samples were freeze-dried prior to characterization by SEM and EDX spectrometry. To measure zeta potential and size distribution, a Zetasizer Nano (Malvern Instruments, Malvern, UK) was used. The structure of BAY-CS-SeNPs was analyzed and characterized by Fourier-transform infrared spectroscopy (FT-IR). FT-IR spectra of the samples were recorded on an Equinox 55 IR spectrometer in the range 4,000–400 cm−1 using the KBr-disk method. The stability of BAY-CS-SeNPs over 42 days was monitored using the Zetasizer Nano. Changes in the size distribution were used to evaluate stability.
Evaluation of the loading rate and release process
The quantity of free BAY 55-9837 was determined using a combined ultrafiltration–centrifugation technique. Briefly, 2 mL of BAY-CS-SeNPs was added to Ultrafree®-MC (50 kD; Millipore, Bedford, MA, USA) and centrifuged for 15 minutes at 5,000 rpm. The filtrates were collected and immediately measured for protein content using a bicinchoninic acid assay. The loading rate of SeNPs was calculated using the following equation: loading rate = (total amount of BAY 55-9837 − the amount of BAY 55-9837 in the filtrate)/total amount of SeNPs.
To study protein-release behavior in vitro, BAY-CS-SeNPs (50 μg/mL) were incubated in RPMI 1640 medium (pH 7.4) with 10% fetal bovine serum at 37°C under constant shaking. At appropriate intervals, samples (50 μL) were removed from the vial using a pipette. The amount of BAY 55-9837 was evaluated using an Agilent HPLC system, and the detection wavelength was set at 280 nm.
Pharmacokinetic studies of BAY 55-9837 and BAY-CS-SeNPs in mice
HPLC instrumentation and conditions
HPLC analysis was carried out on a Waters Alliance 2695-2487 HPLC system fitted with an Agilent C18 column (Waters, Milford, MA, USA). The components were eluted in gradient mode at a flow rate of 1 mL/min. Chromatography was performed at 40°C with mobile phase A H2O (0.1% TFA) and mobile phase-B CH3CN (0.1% TFA). The gradient of eluent B was as follows: 0–8 minutes, 5%; 8–10 minutes, 5%–25%; 10–15 minutes, 25%–75%; 15–20 minutes, 75%–95%; 20–30 minutes, 95%–5%. The effluent was detected at 280 nm, and the raw data were analyzed by Empower software.
Method validation
Samples for the calibration curve were prepared by combining 20 μL aliquots of mouse plasma with various amounts of BAY 55-9837 (25, 12.5, 3.125, 1.5625, 0.78125, 0.390625, and 0.195313 μg/mL) and a constant amount of internal standard. The peak area ratios of the BAY 55-9837 and internal standard peaks versus the concentration were calculated by least squares linear regression using a weighting factor of 1/x2 (the reciprocal of the squared concentration). The correlation coefficient was used to evaluate the linearity. The calibration curve was obtained in the concentration range of 0.195313–25 μg/mL in mouse plasma, covering the entire range of the expected concentrations.
Accuracy and precision were determined by interday and intraday assays. The accuracy of the method was evaluated by the comparison of the three nominal concentrations with the corresponding mean calculated concentrations. The interday-precision values were determined in triplicate for BAY 55-9837 at concentrations of 0.195313, 0.78125, and 12.5 μg/mL in mouse plasma. The intraday precision was determined across the same three concentrations on different days. Relative standard deviation (RSD) was calculated to evaluate the precision. The recovery of BAY 55-9837 from the extraction procedure was evaluated at concentrations of 0.195313, 0.78125, and 12.5 μg/mL by comparing the peak area ratios of samples subjected to a solid-phase extraction (SPE) column (300 mg; Sepax Technologies, Newark, DE, USA) with those directly spiked into the mobile phase.
Pharmacokinetic studies
Kunming mice (10 weeks old, 18–22 g) were obtained from the Sun Yat-Sen University Animal Studies Committee. All of the animal experiments were conducted in accordance with the guidelines established by the Jinan University Animal Studies Committee. The mice were randomly divided into two groups of six mice each: group I was administered BAY 55-9837 at a dose of 5 mg/kg; group II was administered BAY-CS-SeNPs in accord with the concentration of BAY 55-9837 in group I. Compounds were delivered via a tail-vein injection. At different time intervals postinjection, 20 μL blood samples were collected from the tail vein in heparinized micropipettes and rapidly mixed with 2 mL of phosphate-buffered saline. The collected samples were centrifuged at 10,000 rpm for 10 minutes, and the supernatants were carefully transferred to the SPE column to remove impurities, such as saline ions. The SPE column was used in accordance with manufacturer instructions. Following pretreatment on the SPE column, the eluent was then concentrated using a Termovap sample concentrator (Organomation, South Berlin, MA, USA). Finally, the residues were dissolved in acetonitrile (20 μL, 50%) containing 0.1% (v/v) TFA, and 10 μL of the sample was applied to the HPLC system for protein-content analysis using the validated method. Thereafter, the data analysis of relevant pharmacokinetic parameters (half-life, area under the curve from time 0 to last sampling point, observed clearance) of BAY 55-9837 was processed by the PKSolver software using noncompartmental analysis.34 The data are presented as mean values, with the error depicted as the standard deviation. Data were compared using analyses of variance and Student’s t-test, and P<0.05 was considered significant.
Results and discussion
Morphology and characterization
Transmission electron microscopy and SEM images of CS-SeNPs in the absence and presence of BAY 55-9837 are depicted in Figure 1 and Figure 2, respectively. Figure 1A shows that SeNPs modified by CS were well dispersed, with an average diameter of approximately 70 nm, which can be attributed to the hydroxyl groups in CS, as the hydroxyl groups in CS can react with SeO32− groups to form stable intermediates.33 BAY-CS-SeNPs presented as homogeneous and spherical, with diameters of approximately 200 nm (Figure 1, B and C). BAY-CS-SeNPs were further characterized by SEM and EDX spectroscopy. SEM images of the freeze-dried samples indicated that the morphological characteristics of CS-SeNPs were significantly altered in the presence of BAY 55-9837, demonstrating that BAY 55-9837 was well conjugated to the surface of CS-SeNPs (Figure 2, A and B). Additionally, the elemental analysis of the EDX spectrum identified the presence of a weak peak from an O atom (7.35%) together with a C-atom peak (17.11%) and a sharp peak of SeNPs (75.54%) from BAY-CS-SeNPs (Figure 2C), further confirming the existence of BAY 55-9837.
 | Figure 1 Transmission electron microscopy images of CS-SeNPs (A) and BAY-CS-SeNPs (B, C). |
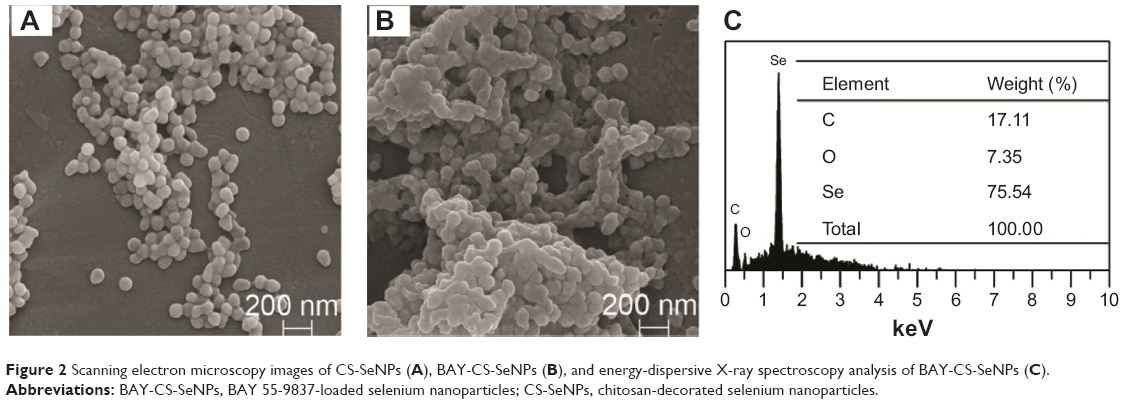 | Figure 2 Scanning electron microscopy images of CS-SeNPs (A), BAY-CS-SeNPs (B), and energy-dispersive X-ray spectroscopy analysis of BAY-CS-SeNPs (C). |
Evaluation of BAY-CS-SeNP surface chemistry
The zeta potential of SeNPs was measured at −3.6 mV and increased to 24.8 mV following modification with CS. BAY 55-9837, a 27-amino acid-long polypeptide, is negatively charged under neutral conditions. Following the attachment of BAY 55-9837 onto the CS-SeNP surface, the zeta potential decreased to −13.8 mV, which confirmed the existence of BAY 55-9837 (Figure 3A). Figure 3B shows the FT-IR spectra analysis of CS-SeNPs, BAY 55-9837, and BAY-CS-SeNPs. The characteristic peaks of CS-SeNPs were in perfect accordance with the study of Yu et al.33 The peak of BAY 55-9837 at 3,305 cm−1 was attributed to O−H groups, and the peaks at 2,922 cm−1 and 1,660 cm−1 corresponded to C−H and CO−NH, respectively. Characteristic peaks of BAY-CS-SeNPs were consistent with that of BAY 55-9837, except for O−H groups. The characteristic O−H group peak in BAY-CS-SeNPs at 3,431 cm−1 was relatively higher than that of BAY 55-9837. The slight shift of the O−H group was likely caused by the conjugation of BAY 55-9837 to the CS-SeNP surface. In conclusion, the electrostatic force was clearly found to be the main driving force behind protein attachment, while chemical bond changes also occurred during the process of conjugation.
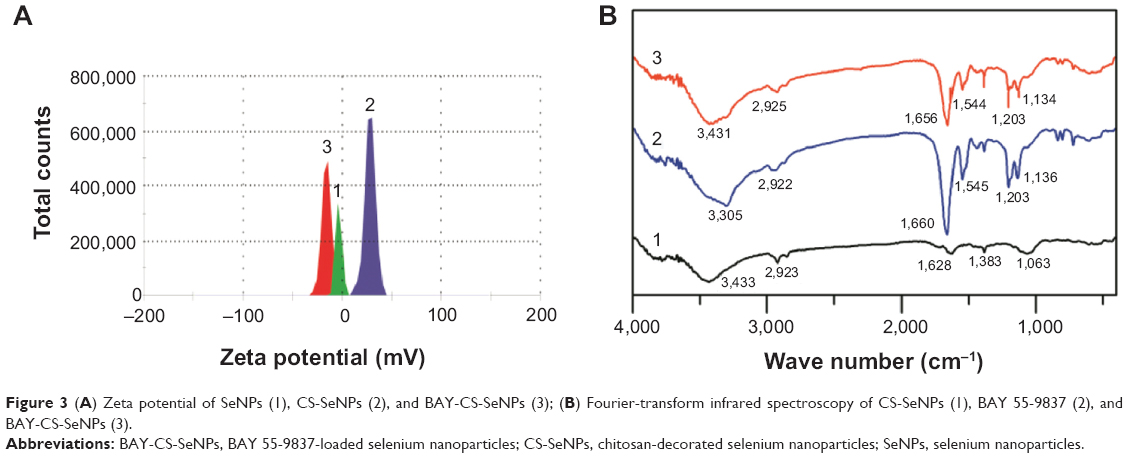 | Figure 3 (A) Zeta potential of SeNPs (1), CS-SeNPs (2), and BAY-CS-SeNPs (3); (B) Fourier-transform infrared spectroscopy of CS-SeNPs (1), BAY 55-9837 (2), and BAY-CS-SeNPs (3). |
Evaluation of BAY-CS-SeNP stability
As depicted in Figure 4A, the average diameter of BAY-CS-SeNPs was approximately 200 nm. The results show that the size of BAY-CS-SeNPs remained stable during the first few days after formation and then demonstrated a gradual decline, while precipitation was barely observed. Nevertheless, BAY-CS-SeNP size increased dramatically on the 37th day after formation (Figure 4B). The gradually declining diameter of the BAY-CS-SeNPs was likely caused by the degradation or release of their surface-layer proteins. At 37 days, the aggregation of SeNPs most likely began to occur, resulting in a radical increase of BAY-CS-SeNP size. The high stability of BAY-CS-SeNPs provides support for translating these findings into future medical applications.35
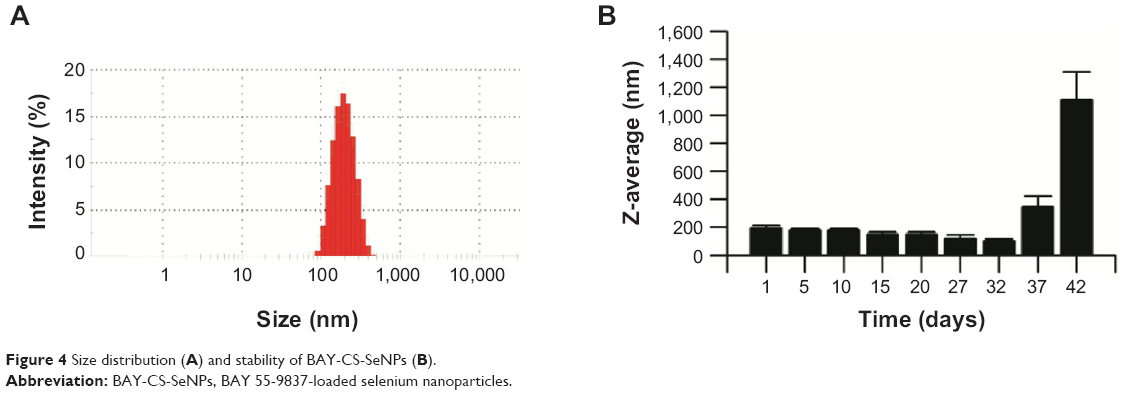 | Figure 4 Size distribution (A) and stability of BAY-CS-SeNPs (B). |
Loading rate and release profile
The protein-loading rate of CS-SeNPs was detected up to 10.79 mg/mg, revealing that CS-SeNPs possessed a high loading capacity. Se displayed a narrow margin between beneficial and toxic effects. The high loading rate of CS-SeNPs can decrease the dose of CS-SeNPs, which minimizes potential side effects. Furthermore, a low dose of SeNPs may be beneficial in the treatment of T2DM. More importantly, SeNPs displayed excellent antioxidant activity, which may prevent pancreatic β-cells from undergoing apoptosis. Nevertheless, the mechanism by which SeNPs function requires further investigation.
Release rates have an impact on pharmacokinetics, biodistribution, therapeutic activity, toxicity, and drug leakage, and sustained release generally confers better therapeutic activity.36 Figure 5 shows the release profiles of BAY 55-9837 from the NPs into the medium. The dissociation of BAY 55-9837 from the nanocomposite was clearly observed within the first 12 hours, and reached 49.2%. Following this, BAY 55-9837 released gradually, with cumulative release reaching 78.9% after 72 hours’ incubation. Protein release was barely observed thereafter. It took over 70 hours for complete release to occur, without a marked burst release. The rapid release observed during the first 12 hours was most likely due to the weak adsorption of the outer-layer protein, whereas the slower release at later time points may be attributed to the stable interaction induced by chemical bonding and the strong electrostatic force. Generally, the human body requires higher insulin levels to overcome postmeal rises in glucose so that its level is kept constant in the blood. BAY 55-9837 induces glucose-dependent insulin secretion, so its rapid release during the first 10 hours may provide enough BAY 55-9837 to promote postmeal insulin secretion during the day. As overnight fasting lowers blood glucose levels, the sustained release measured for BAY 55-9837 may also satisfy insulin needs in the evening.
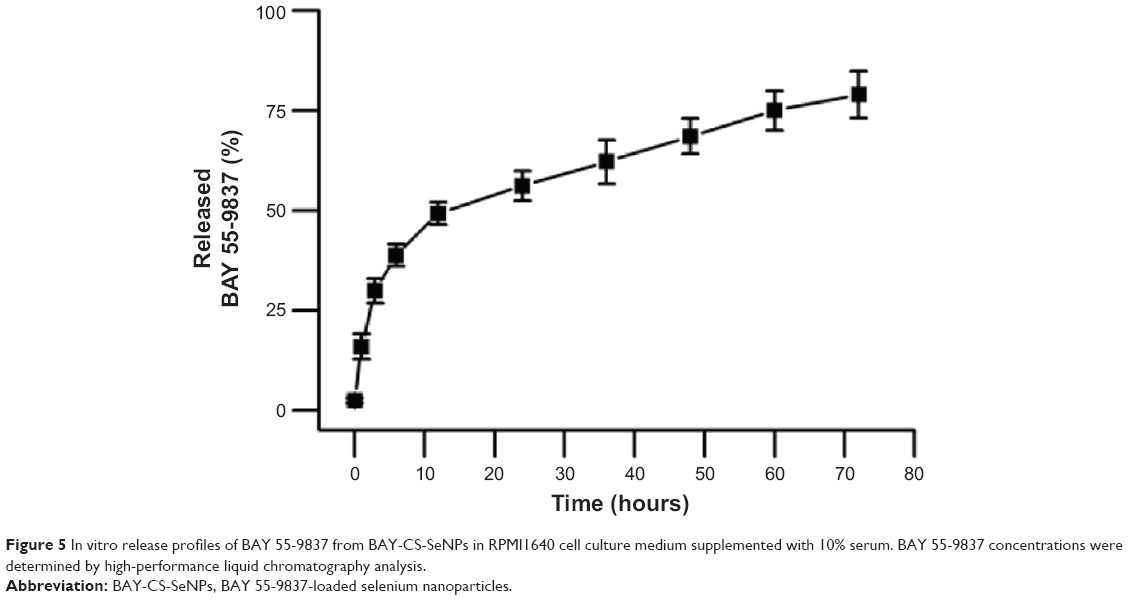 | Figure 5 In vitro release profiles of BAY 55-9837 from BAY-CS-SeNPs in RPMI1640 cell culture medium supplemented with 10% serum. BAY 55-9837 concentrations were determined by high-performance liquid chromatography analysis. |
Based on the excellent protein-loading capacity of CS-SeNPs and the sustained-release profile, BAY-CS-SeNPs can be considered a promising therapeutic with desirable biological properties.
Pharmacokinetic studies of BAY 55-9837 and BAY-CS-SeNPs in mice
HPLC assay validation
BAY 55-9837 and quantitation of substance TP-5 were baseline-separated with retention times of 18.5 and 9.5 minutes without interference from the plasma (Figure 6). Therefore, the specificity of the assay was established. The calibration curve demonstrated excellent linearity over the concentration range of 0.195313–12.5 μg/mL. The representative linear equation was y =15.019x −0.056, with a coefficient of correlation invariably greater than 0.999 (Figure 7). The accuracy and precision values for the assay are summarized in Table 1. The assay was found to be precise, with RSD values within 0.61%–4.05% for interday and within 0.59%–2.37% for intraday measurements. The accuracy was excellent, with interday accuracy ranging from 83.97% to 100.48% and intraday accuracy from 86.53% to 99.59%. Additionally, the recovery rate following application to the SPE column ranged from 81.76% to 106%, which demonstrated agreement between the true and measured values.
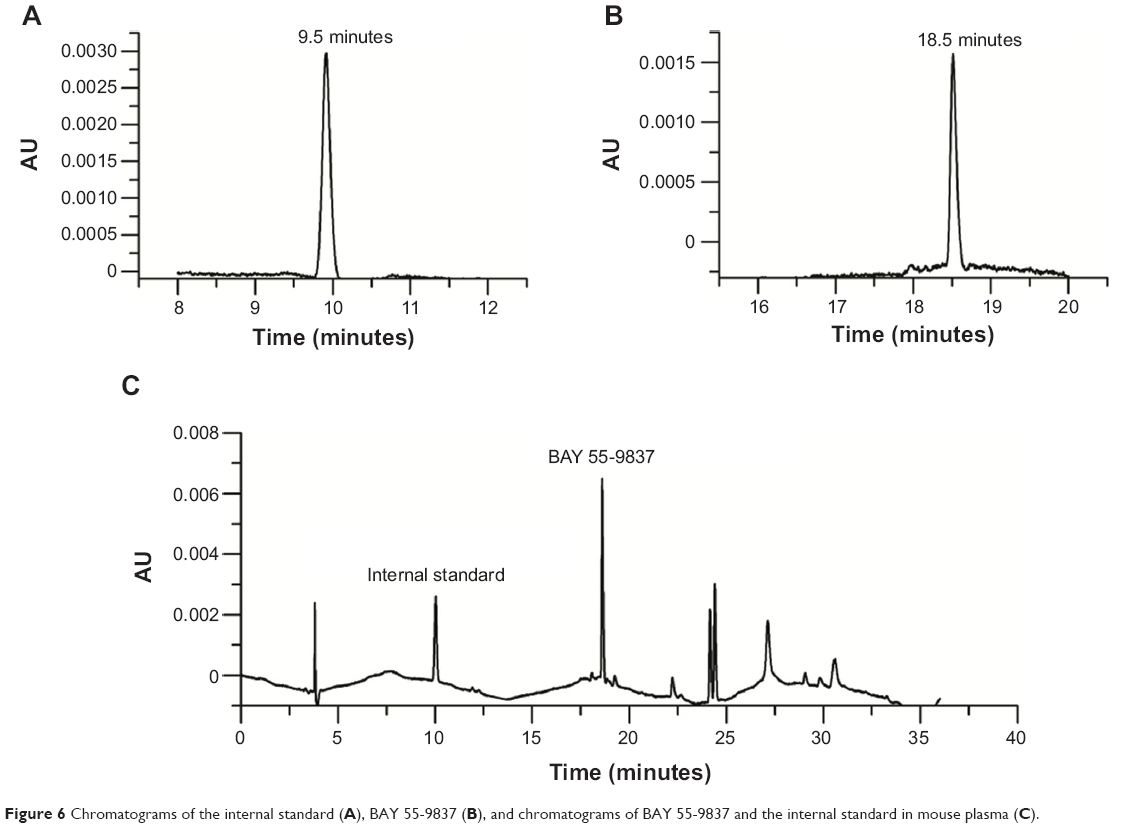 | Figure 6 Chromatograms of the internal standard (A), BAY 55-9837 (B), and chromatograms of BAY 55-9837 and the internal standard in mouse plasma (C). |
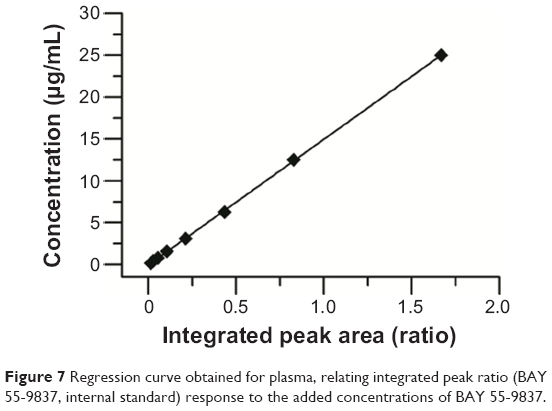 | Figure 7 Regression curve obtained for plasma, relating integrated peak ratio (BAY 55-9837, internal standard) response to the added concentrations of BAY 55-9837. |
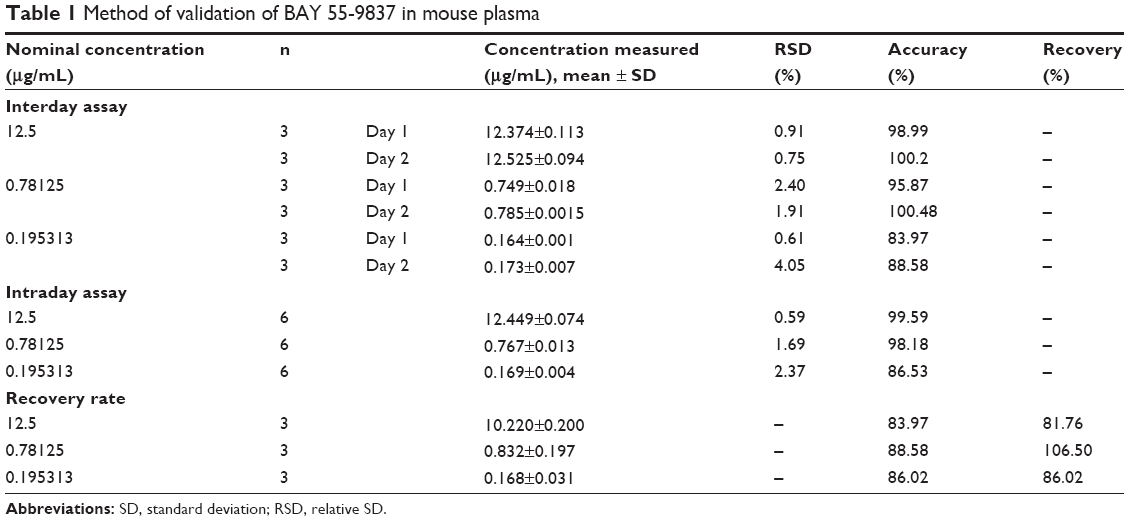 | Table 1 Method of validation of BAY 55-9837 in mouse plasma |
Pharmacokinetic studies
Figure 8 shows the plasma concentration–time profiles of BAY 55-9837 and BAY-CS-SeNPs. BAY 55-9837 exhibited a short life span, which is consistent with the previous study. However, when conjugated to CS-SeNPs, BAY 55-9837 was detectable for up to 70 hours. The corresponding pharmacokinetic parameters are summarized in Table 2. As shown, the half-life of BAY 55-9837 was approximately 0.32 hour, and was extended to approximately 20.81 hours following conjugation to CS-SeNPs. Additionally, the conjugation to CS-SeNPs significantly reduced the plasma clearance of BAY 55-9837 to a rate of 1.56 mL/h. Pharmacokinetic results also show that the area under the curve from time 0 to last sampling point of BAY-CS-SeNPs was 26.8-fold higher than that of BAY 55-9837. The remarkable improvement in pharmacokinetics (P<0.01) was most likely due to the increased apparent size of BAY 55-9837, indicating that the prolonged plasma persistence and decreased kidney clearance rate measured for BAY 55-9837 followed typical size-related effects.14 The prolonged in vivo half-life increased the acting time of BAY 55-9837, which would most likely improve insulin secretion.
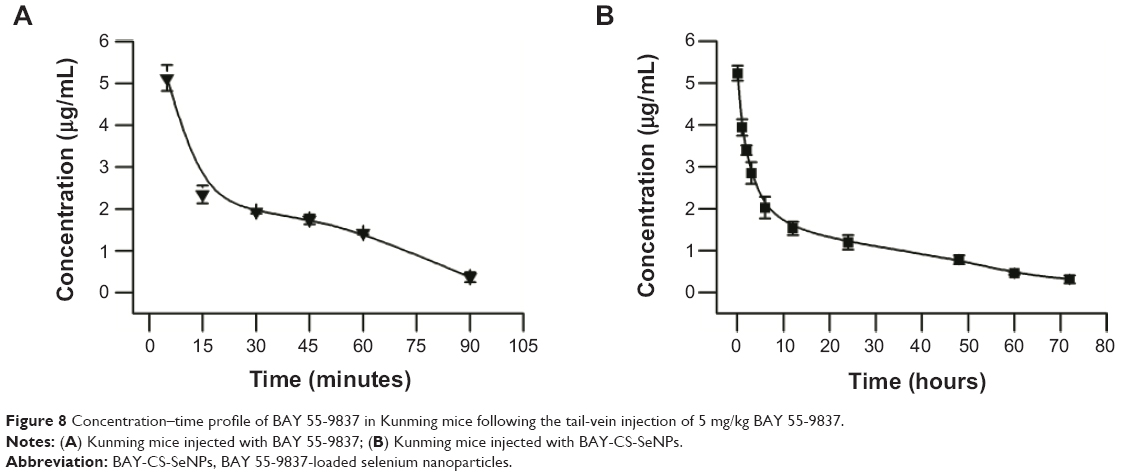 | Figure 8 Concentration–time profile of BAY 55-9837 in Kunming mice following the tail-vein injection of 5 mg/kg BAY 55-9837. |
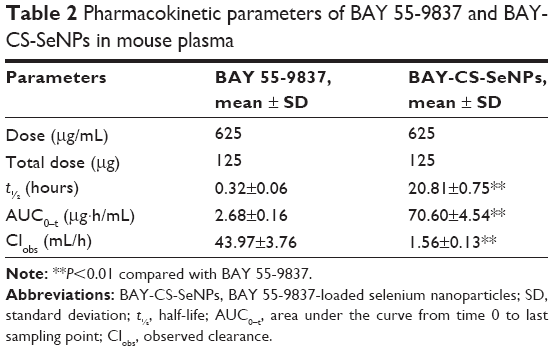 | Table 2 Pharmacokinetic parameters of BAY 55-9837 and BAY-CS-SeNPs in mouse plasma |
Conclusion
In summary, our work provides a simple method of extending the half-life of BAY 55-9837 in vivo by conjugating it to CS-SeNPs. BAY-CS-SeNPs possessed a desirable sustained-release profile and demonstrated high stability, with a diameter of approximately 200 nm in vitro. Furthermore, pharmacokinetic quantification revealed that after conjugation to CS-SeNPs, BAY 55-9837 exhibited a lower clearance rate, which resulted in a longer half-life. As illustrated in Figure 9, we believe that using nanocarrier conjugates can improve the half-life of low-molecular-weight therapeutics by increasing their apparent molecular size. Generally speaking, therapeutics with increased apparent molecular sizes possess lower rates of renal elimination. Furthermore, as carriers, SeNPs themselves may be beneficial in the treatment of T2DM, although a further understanding of the molecular mechanisms underlying this function is still in need of evaluation. The work presented here offers a new pathway forward toward improving the half-life of low-molecular-weight therapeutics in vivo by using nanotechnology.
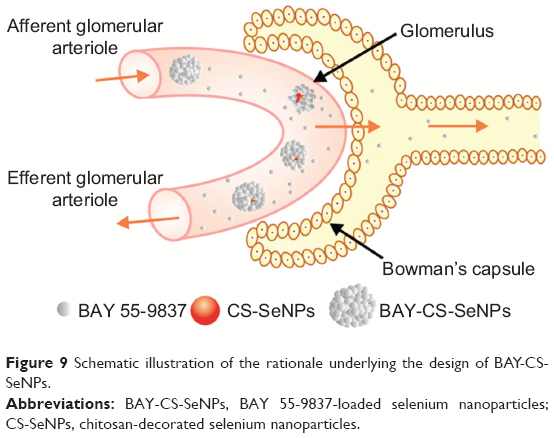 | Figure 9 Schematic illustration of the rationale underlying the design of BAY-CS-SeNPs. |
Acknowledgments
This work was supported by the grants from the NSFC (No 81373314), the Doctoral Program of Higher Education of China (No 20124401120012), the Natural Science Foundation of Guangdong Province (No S2012010008756), Special Fund for Subject Construction of Guangdong Province (No 2012KJCX0015), the Cooperation Project in Industry, Education and Research of Guangdong Province and Ministry of Education in China (No 2010B090400544), and the National Technical Innovation Fund for Medium and Small-Size Enterprise, China (No 11C26214413218). We also gratefully acknowledge financial support from the National 863 Project (2006AA02Z125), the Major Science and Technology Projects of Guangdong Province (2007B060310212), and the National Project for Significant New Drug Development of the Ministry of Science and Technology of China (2012ZX09202301). We would also like to thank Professor Tianfeng Chen from the Materials Chemistry Department of Jinan University for the FT-IR analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. | ||
Moller DE. New drug targets for type 2 diabetes and the metabolic syndrome. Nature. 2001;414:821–827. | ||
Walsh G. Biopharmaceutical benchmarks. Nat Biotechnol. 2000; 18:831–833. | ||
Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010; 28:917–924. | ||
Yung SL, Dela Cruz F, Hamren S, et al. Generation of highly selective VPAC2 receptor agonists by high throughput mutagenesis of vasoactive intestinal peptide and pituitary adenylate cyclase-activating peptide. J Biol Chem. 2003;278:10273–10281. | ||
Miyata A, Arimura A, Dahl RR, et al. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun. 1989;164:567–574. | ||
Miyata A, Jiang L, Dahl RD, et al. Isolation of a neuropeptide corresponding to the N-terminal 27 residues of the pituitary adenylate cyclase activating polypeptide with 38 residues (PACAP38). Biochem Biophys Res Commun. 1990;170:643–638. | ||
Winzell MS, Ahrén B. Role of VIP and PACAP in islet function. Peptides. 2007;28:1805–1813. | ||
Bertrand G, Puech R, Maisonnasse Y, Bockaert J, Loubatières-Mariani M. Comparative effects of PACAP and VIP on pancreatic endocrine secretions and vascular resistance in rat. Br J Pharmacol. 1996;117:764–770. | ||
Borboni P, Porzio O, Pierucci D, et al. Molecular and functional characterization of pituitary adenylate cyclase-activating polypeptide (PACAP-38)/vasoactive intestinal polypeptide receptors in pancreatic β-cells and effects of PACAP-38 on components of the insulin secretory system. Endocrinology. 1999;140:5530–5537. | ||
Tsutsumi M, Claus TH, Liang Y, et al. A potent and highly selective VPAC2 agonist enhances glucose-induced insulin release and glucose disposal a potential therapy for type 2 diabetes. Diabetes. 2002;51:1453–1460. | ||
Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem. 1993;214:829–835. | ||
Pan CQ, Li F, Tom I, et al. Engineering novel VPAC2-selective agonists with improved stability and glucose-lowering activity in vivo. J Pharmacol Exp Ther. 2007;320:900–906. | ||
Roberts M, Bentley M, Harris J. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2002;54:459–476. | ||
Cohen S, Yoshioka T, Lucarelli M, Hwang LH, Langer R. Controlled delivery systems for proteins based on poly(lactic/glycolic acid) microspheres. Pharm Res. 1991;8:713–720. | ||
Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J Biol Chem. 1977; 252:3582–3586. | ||
Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3:16–20. | ||
Liu W, Li X, Wong YS, et al. Selenium nanoparticles as a carrier of 5-fluorouracil to achieve anticancer synergism. ACS Nano. 2012;6: 6578–6591. | ||
Makhluf SBD, Abu-Mukh R, Rubinstein S, Breitbart H, Gedanken A. Modified PVA-Fe3O4 nanoparticles as protein carriers into sperm cells. Small. 2008;4:1453–1458. | ||
Singh Y, Gao D, Gu Z, et al. Influence of molecular size on the retention of polymeric nanocarrier diagnostic agents in breast ducts. Pharm Res. 2012;29:2377–2388. | ||
Lee C, Choi JS, Kim I, et al. Long-acting inhalable chitosan-coated poly(lactic-co-glycolic acid) nanoparticles containing hydrophobically modified exendin-4 for treating type 2 diabetes. Int J Nanomedicine. 2013;8:2975–2983. | ||
Rayman MP. The importance of selenium to human health. Lancet. 2000;356:233–241. | ||
Gao X, Zhang J, Zhang L. Hollow sphere selenium nanoparticles: their in-vitro anti hydroxyl radical effect. Adv Mater. 2002;14:290–293. | ||
Gates B, Mayers B, Cattle B, Xia Y. Synthesis and characterization of uniform nanowires of trigonal selenium. Adv Funct Mater. 2002; 12:219–227. | ||
Tran PA, Webster TJ. Selenium nanoparticles inhibit Staphylococcus aureus growth. Int J Nanomedicine. 2011;6:1553–1558. | ||
Li YH, Li XL, Wong YS, et al. The reversal of cisplatin-induced nephrotoxicity by selenium nanoparticles functionalized with 11-mercapto-1-undecanol by inhibition of ROS-mediated apoptosis. Biomaterials. 2011;32:9068–9076. | ||
Torres SK, Campos VL, León CG, et al. Biosynthesis of selenium nanoparticles by Pantoea agglomerans and their antioxidant activity. J Nanopart Res. 2012;14:1–9. | ||
Zhang J, Wang H, Yan X, Zhang L. Comparison of short-term toxicity between Nano-Se and selenite in mice. Life Sci. 2005;76:1099–1109. | ||
Wang H, Zhang J, Yu H. Elemental selenium at nano size possesses lower toxicity without compromising the fundamental effect on selenoenzymes: comparison with selenomethionine in mice. Free Radic Biol Med. 2007;42:1524–1533. | ||
Navarro-Alarcon M, López-G de la Serrana H, Perez-Valero V, López-Martínez C. Serum and urine selenium concentrations as indicators of body status in patients with diabetes mellitus. Sci Total Environ. 1999; 228:79–85. | ||
Kljai K, Runje R. Selenium and glycogen levels in diabetic patients. Biol Trace Elem Res. 2001;83:223–229. | ||
Zhang SY, Zhang J, Wang HY, Chen HY. Synthesis of selenium nanoparticles in the presence of polysaccharides. Mater Lett. 2004;58: 2590–2594. | ||
Yu B, Zhang Y, Zheng W, Fan C, Chen T. Positive surface charge enhances selective cellular uptake and anticancer efficacy of selenium nanoparticles. Inorg Chem. 2012;51:8956–8963. | ||
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99:306–314. | ||
Charrois GJ, Allen TM. Drug release rate influences the pharmacokinetics, biodistribution, therapeutic activity, and toxicity of pegylated liposomal doxorubicin formulations in murine breast cancer. Biochim Biophys Acta. 2004;1663:167–177. | ||
Imani M, Lahooti-Fard F, Taghizadeh SM, Takrousta M. Effect of adhesive layer thickness and drug loading on estradiol crystallization in a transdermal drug delivery system. AAPS Pharm Sci Tech. 2010; 11:1268–1275. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.