Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Chitosan-coated poly(lactic-co-glycolic) acid nanoparticles as an efficient delivery system for Newcastle disease virus DNA vaccine
Authors Zhao K ![]() , Zhang Y, Zhang Z, Shi C, Wang X, Wang X, Jin Z, Cui S, Chen G, Luo X
, Zhang Y, Zhang Z, Shi C, Wang X, Wang X, Jin Z, Cui S, Chen G, Luo X
Received 4 July 2014
Accepted for publication 24 July 2014
Published 30 September 2014 Volume 2014:9(1) Pages 4609—4619
DOI https://doi.org/10.2147/IJN.S70633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Kai Zhao,1,* Yang Zhang,1,2,* Xiaoyan Zhang,1,* Ci Shi,1,2 Xin Wang,1 Xiaohua Wang,1 Zheng Jin,3 Shangjin Cui2
1Laboratory of Microbiology, School of Life Science, Heilongjiang University, 2Division of Swine Infectious Diseases, State Key Laboratory of Veterinary Biotechnology, Harbin Veterinary Research Institute, 3Key Laboratory of Chemical Engineering Process and Technology for High-efficiency Conversion, Heilongjiang University, Harbin, People’s Republic of China
*These authors contributed equally to this work
Abstract: We determined the efficacy and safety of chitosan (CS)-coated poly(lactic-co-glycolic) acid (PLGA) nanoparticles (NPs) as a delivery system for a vaccine to protect chickens against Newcastle disease virus (NDV). The newly constructed vaccine contained DNA (the F gene) of NDV. The Newcastle disease virus (NDV) F gene deoxyribonucleic acid (DNA) plasmid (pFDNA)-CS/PLGA-NPs were spherical (diameter =699.1±5.21 nm [mean ± standard deviation]) and smooth, with an encapsulation efficiency of 98.1% and a Zeta potential of +6.35 mV. An in vitro release assay indicated that CS controlled the burst release of plasmid DNA, such that up to 67.4% of the entire quantity of plasmid DNA was steadily released from the pFDNA-CS/PLGA-NPs. An in vitro expression assay indicated that the expression of nanoparticles (NPs) was maintained in the NPs. In an immunization test with specific pathogen-free chickens, the pFDNA-CS/PLGA-NPs induced stronger cellular, humoral, and mucosal immune responses than the plasmid DNA vaccine alone. The pFDNA-CS/PLGA-NPs did not harm 293T cells in an in vitro assay and did not harm chickens in an in vivo assay. Overall, the results indicated that CS-coated PLGA NPs can serve as an efficient and safe mucosal immune delivery system for NDV DNA vaccine.
Keywords: mucosal immune delivery system, immune effect
Introduction
Virulent Newcastle disease virus (NDV) is the prototype of the paramixoviruses that cause Newcastle disease (ND). The virus, which causes high mortality among mature chickens and chicks, infects the respiratory tract, nerves, or intestines. NDV mainly expresses hemagglutinin-neuramindase and fusion (F) glycoproteins. In many countries, ND is a catastrophic problem for the poultry industry.1 Although the disease is difficult to control, vaccines are available. The efficacy of NDV vaccines depends on the induction of F glycoproteins.2 Traditional NDV vaccines include two types: inactivated vaccines and attenuated live vaccines.3 Both of these traditional types of vaccines have important limitations, including reversion to virulence and induction of respiratory pathological changes. In addition, the difficulty in differentiating between vaccinated chickens and naturally infected chickens complicates diagnosis.
In research by Robinson et al the injection of chickens with plasmid DNA provided a new way to protect chickens from lethal influenza viruses.4 In the same year, Fynan et al5 reported that the inclusion of gold-encapsulated plasmid particles in influenza virus vaccines reduced the requirement for DNA in the vaccines to 0.4 pg, which was 1/250th of the amount reported formerly. Reducing the DNA content was important because DNA-based vaccines can induce long-term cellular and humoral immune reactions in animals and humans.6–8
DNA vaccines have not been widely used for several other reasons. Some studies have shown that the vaccines, which are usually administered via intramuscular (IM) injection, can fail to reach the antigen-presenting cells and therefore fail to induce immune responses because of difficulty in crossing cell membranes.9–11 Sun et al reported that effective immunization of large animals required large amounts of DNA.12 Researchers have recently suggested several measures that could increase the efficacy of DNA vaccines. These measures include plasmid DNA optimization, improvement of delivery methods, the targeting of the antigen-presenting cells, and the use of immunologic adjuvants.13,14
Recent research has indicated that polymeric nanoparticles (NPs) can be used as potent adjuvants as part of a “nano” mucosal immune delivery system. NPs are biodegradable and biocompatible, have low toxicity, and protect the antigen or DNA from damage.15–17 Among all the polymers, polyesters based on polylactic acid, polyglycolic acid, and their copolymers, poly(lactic-co-glycolic) acids (PLGAs), have attracted the most attention and have been used as carriers for a wide range of vaccines.18–21
PLGA is authorized by the US Food and Drug Administration (FDA), and PLGA NPs (microspheres) have been thoroughly studied as a protein or DNA vaccine mucosal delivery system that protects the encapsulated protein or DNA vaccine from enzyme digestion and that extends the release time of the protein or DNA vaccine.22,23 A number of studies have reported improved antibody responses when antigens are orally administered in PLGA particles.24–27 Nevertheless, PLGA NPs have limited use in mucosal vaccination because of their poor mucoadhesivity and immunoenhancing ability. In recent years, chitosan (CS) has been used as a coating material for PLGA NPs because of its biological adhesive properties and ability to improve the immunological response to mucosal vaccination.28,29 By modifying the surface of PLGA NPs, CS provides the following advantages: 1) it decreases the burst release of the encapsulated protein or DNA; 2) it increases the stability of biological macromolecules; 3) it enhances the inversion of Zeta potential, and promotes cellular adhesion and retention of the delivery system at the target site; and 4) it offers the possibility of conjugating targeting ligands to free amino groups on its surface.30 Budhian et al reported that coating PLGA NPs with CS reduced the burst release of haloperidol from 70% to 36%.31 Tahara et al have also successfully developed gene delivery vectors using CS surface modification of PLGA NPs.32,33
In this study, we prepared CS-coated PLGA NPs containing the F gene plasmid DNA of NDV (pFDNA-CS/PLGA-NPs) and assessed the ability of the preparation to induce immune responses and protect specific pathogen-free (SPF) chickens from ND after intranasal (IN) administration. We also assessed the bioactivity and safety of the pFDNA-CS/PLGA-NPs with in vitro expression and cell cytotoxicity assays.
Materials and methods
Preparation of the pFDNA-CS/PLGA-NPs
The eukaryotic expression plasmid pVAX I-opti F DNA, which contains the F gene of NDV, was encapsulated in PLGA NPs (pFNDV-PLGA-NPs) by a water/oil/water double emulsion-solvent evaporation method.34 Previous research indicated that the optimal conditions for preparation of these NPs were 50 watts (w) for 30 seconds for the primary emulsion, 50 w for 60 seconds for the secondary emulsion, a DNA:PLGA ratio of 0.5:100, and combination of 40 mg/mL PLGA with 2.0% polyvinyl alcohol (PVA).35 Accordingly, pFDNA-CS/PLGA-NPs were prepared in three main steps. First, 40 mg of PLGA was dissolved in 1 mL of methylene chloride (oil phase), and 800 μg of the DNA solution was added with the primary emulsion. Second, 2 mL of 2% PVA and the secondary emulsion (W2) were added to the primary emulsion; the resulting compound emulsion (primary and secondary emulsions) was added in drops to the CS solutions, which contained 15 mL of 0.5% PVA, and the preparation was shaken at 500 r/min for 5 hours. Third, the CS/PLGA NPs containing pFDNA were recovered by centrifugation (4,500 r/min, 10 minutes, 4°C), washed three times with sterilized deionized water, centrifuged, and freeze-dried. The resulting NPs were referred to as pFDNA-CS/PLGA-NPs.
Characterization of the pFDNA-CS/PLGA-NPs
We determined the effects of CS concentration (0, 0.2, 0.4, 0.6, 0.8, and 1.0 mg/mL) and CS volume (0, 1.5, 3.0, 5.0, and 7.5 mL) on the characteristics of the pFDNA-CS/PLGA-NPs, respectively. Encapsulation efficiency (EE) was measured as previously described.3 The morphological and surface characteristics of the pFDNA-CS/PLGA-NPs were examined by JEM-200EX transmission electron microscopy (Hitachi Ltd., Tokyo, Japan). The particle size and Zeta potentials were measured using a Zetasizer 2000 from Malvern Instruments (Malvern, UK).
In vitro release of the pFDNA-CS/PLGA-NPs
An in vitro release assay was carried out to determine the release of plasmid DNA from the NPs.3 Briefly, samples were periodically collected (after 0, 6, 12, 24, 36, 48, 72, 96, 120, 144, 168, 192, 216, and 240 hours) and centrifuged at 10,000 r/min for 10 minutes at 4°C. The collected pFDNA-CS/PLGA-NPs were counted. All experiments were performed five times.
In vitro transfection and western blot analysis of the pFDNA-CS/PLGA-NPs
293T cells were grown in poly-lysine-treated 6-well plates and cultured at 37°C in a CO2 incubator. An in vitro transfection experiment was carried out according to the instructions from the Lipofectamine™ 2000 reagent kit (Invitrogen™; Life Technologies Corp, Carlsbad, CA, USA), using four groups: 1) the naked plasmid DNA group; 2) the pFDNA-CS/PLGA-NP transfected group; 3) the blank CS/PLGA-NP group; and 4) the negative cell control group. Western blot analysis was carried out as previously described.36 Briefly, after 72 hours of transfection, the 293T cells were collected and disrupted using radioimmunoprecipitation assay solution (50 mmol/L Tris-HCl [pH 8.5], 5 mmol/L 2-hydroxy-1-ethanethiol, 100 mmo1/L KCl, 1 mmol/L phenylmethanesulfonyl fluoride, and 1% Nonidet P-40 (octylphenoxypolyethoxyethanol). The lysate was centrifuged at 14,000 r/min at 4°C, and the supernatant was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis. Proteins were transferred to a nitrocellulose membrane (Amersham; GE Healthcare, Little Chalfont, UK) using a BioRad semidry unit. The membrane was washed with phosphate-buffered saline (PBS), blocked with 5% fat-free milk overnight, and then incubated with an NDV-positive serum at a 1:500 dilution for 1 hour. After the membrane was washed three times with PBS with Tween® (phosphate buffered saline with Tween-20), fluorescein isothiocyanate labeled goat anti-chicken secondary antibody was added at a dilution of 1:5,000 for 1 hour. The image was acquired with an Odyssey infrared imaging system (LI-COR Odyssey; LI-COR Biosciences Inc., Lincoln, NE, USA).
The safety of pFDNA-CS/PLGA-NPs
Cell Counting Kit-8 reagent was used to evaluate the in vitro cytotoxicity, and optical density at 450 nm (OD450) was measured to determine the survival rate of 293T cells. For the in vivo assay, 30 4-week-old SPF chickens from the Harbin Veterinary Research Institute Laboratory Animal Center were randomly assigned to two groups. The in vitro and in vivo cytotoxicity of the pFDNA-CS/PLGA-NPs were evaluated as previously described.36 Chickens in one group were immunized, by IN route, with 0.2 mL of the pFDNA-CS/PLGA-NPs, which contained a total of 200 μg of plasmid DNA. Chickens in the other group were immunized, by IM route, with 0.2 mL of the naked plasmid DNA. The chickens were continuously observed for 21 days, and any abnormalities were noted.
Immunization of SPF chickens
A total of 120 4-week-old SPF chickens were randomly assigned to six groups. Group 1 was treated with PBS buffer IM. Groups 2 and 3 were treated with blank CS/PLGA-NPs IM or IN, respectively. Group 4 was treated with 200 μg of naked plasmid DNA (0.1 mL) IM. Groups 5 and 6 were treated with 0.2 mL pFDNA-CS/PLGA-NPs (containing 200 μg of plasmid DNA) IM or IN, respectively. All treatments were repeated 14 days later.
Detection of immunoglobulin antibody in the serum of immunized chickens
Blood samples were collected from the wing veins of the six groups of chickens at 7, 14, 21, 28, 35, and 42 days after the first treatment. The serum was obtained by centrifugation at 3,000 r/min for 10 minutes at 4°C. Enzyme-linked immunosorbent assay (ELISA) was performed to assess the titers of the NDV-specific immunoglobulin G (IgG) in the sera, using a NDV IgG ELISA Kit (Rapidbio Co., Ltd., West Hills, CA, USA) according to the instruction manual.
Detection of IgA antibody in mucosa extracts of immunized chickens
Serum, tears, bile, and tracheal fluid were collected from two chickens from each of the six groups of treated (immunized) chickens, at 7, 14, 21, 28, 35, and 42 days after the first treatment. IgA antibody was evaluated with a NDV IgA ELISA Kit.
Lymphocyte proliferation in immunized chickens
To assess the cell-mediated immune responses of immunized chickens at 14, 28, and 42 days after the first treatment, we measured lymphocyte proliferation with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide colorimetric assay, as previously described.37 This was done with ten chickens from each of the six groups. The stimulation index (SI) was determined using the following formula:38
SI = OD570 T/OD570 C, | (1) |
where T is the mean value (number of OD at 570 nm) of the test group (pFDNA-CS/PLGA-NPs IN, pFDNA-CS/PLGA-NPs IM, pVAX I-opti F IM, blank CS/PLGA-NPs IN, or blank CS/PLGA-NPs IM), and OD570 C is the mean value of the control (PBS) group.
Protection against NDV strain F48E9
The ability of pFDNA-CS/PLGA-NPs to protect chickens against NDV strain F48E9 was determined. Strain F48E9 is highly virulent (mean death time ≤60 hours, intracerebral pathogenicity index >1.6). Protection was determined as previously described.3 Briefly, when the level of ND serum antibody of every immune group increased to 6.0 log 2, eight chickens were selected at random from the six groups and infected by IM route with 0.1 mL of the highly virulent NDV strain F48E9, for challenge studies with a viral titer of 104.5 egg infectious dose (EID)50/0.1 mL. Clinical signs of disease and mortality were monitored on a daily basis, and continuously observed for 14 days. The infected chickens and corresponding negative control chickens were euthanized, and the glandular stomach, duodenum, and bursa of Fabricius were collected for examination by histological staining.
Statistical analysis
Unless noted otherwise, all experiments were repeated three times, and each value was measured in triplicate. Data are presented as means ± standard deviation. Means were compared using one-sided Student’s t-tests. Differences were considered to be statistically significant at P<0.05.
Results
Preparation and characterization of the pFDNA-CS/PLGA-NPs
Based on EE, the optimal CS concentration and volume for preparation of pFDNA-CS/PLGA-NPs was 0.6 mg/mL (Table 1) and 1.5 mL (Table 2), respectively. With this concentration and volume, the mean EE was about 98.7%. The pFDNA-CS/PLGA-NPs were spheres with smooth surfaces. They did not aggregate, and they did not suffer from subsidence damage (Figure 1). The mean diameter was 699.1±5.21 nm, particle-size dispersity was 0.005 (Figure 2A), and Zeta potential was 6.35±2.75 mV (Figure 2B). Using the optimal CS concentration of 0.6 mg/mL and volume of 1.5 mL, we prepared and assessed five independent batches of pFDNA-CS/PLGA-NPs. The EE values did not significantly differ (P>0.05) among the five batches, indicating that the preparation procedure was reproducible and reliable.
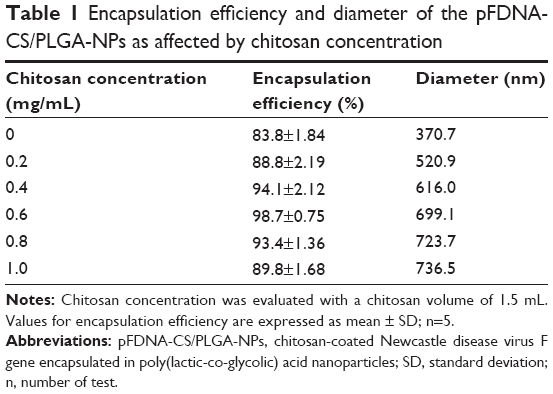 | Table 1 Encapsulation efficiency and diameter of the pFDNA-CS/PLGA-NPs as affected by chitosan concentration |
 | Table 2 Encapsulation efficiency of the pFDNA-CS/PLGA-NPs as affected by chitosan volume |
 | Figure 1 Transmission electron micrographs of the pFDNA-CS/PLGA-NPs at (A) 1,000× magnification and (B) 3,000× magnification. |
 | Figure 2 Size and Zeta potential of the pFDNA-CS/PLGA-NPs. |
In vitro release of the pFDNA-CS/PLGA-NPs
The plasmid DNA encapsulated in the CS/PLGA-NPs was gradually released throughout the assay, although the release was faster during the first 120 hours than during the subsequent 120 hours (Figure 3). The rate of release was relatively slow and sustained, apparently because the CS reduced desorption and diffusion.
 | Figure 3 In vitro release profiles of the plasmid DNA pVAX I-opti F from pFDNA-CS/PLGA-NPs. |
In vitro expression of the pFDNA-CS/PLGA-NPs
Specific fluorescence was detected in the naked plasmid DNA group (Figure 4A) and the pFDNA-CS/PLGA-NP transfected group (Figure 4B) but not in the blank CS/PLGA-NPs group (Figure 4C) or in the negative cell control group (Figure 4D). Expression of the antigen was further demonstrated by western blot (Figure 4E), which indicated that the expected 58 kDa antigen was expressed in 293T cells transfected with pFDNA-CS/PLGA-NPs and naked plasmid DNA but not in cells treated with the blank CS/PLGA-NPs or in the negative cell control group. These results proved that the pFDNA-CS/PLGA-NPs could express and protect the antigen in vitro.
 | Figure 4 In vitro expression of the pFDNA-CS/PLGA-NPs in 293T cells as indicated by indirect immunofluorescence analysis at ×40 (A–D) and western blotting (E). |
Safety of the pFDNA-CS/PLGA-NPs
The survival rate of 293T cells was 85.14%±8.13% when treated with pFDNA-CS/PLGA-NPs and was 84.72%±6.04% for nontreated control cells; the difference was not statistically significant (P>0.05). Cell morphology was similar for cells in the pFDNA-CS/PLGA-NPs and control groups. Chickens immunized with either the pFDNA-CS/PLGA-NPs or the naked plasmid DNA did not exhibit nervous signs, clinical symptoms, or necropsy lesions within the 3 weeks following treatment. These in vitro and in vivo results showed that the pFDNA-CS/PLGA-NPs were safe.
Immunization of SPF chickens with pFDNA-CS/PLGA-NPs
IgG antibody in serum
IgG antibody titers in sera peaked on day 28 for chickens immunized IM with naked plasmid DNA but peaked on day 35 for chickens immunized IM or IN with pFDNA-CS/PLGA-NPs; the titer on day 35 and day 42 was greater (P<0.05) with pFDNA-CS/PLGA-NPs than with naked plasmid DNA (Figure 5). IgG antibody titer did not increase in chickens treated with PBS alone or with blank CS/PLGA-NPs.
 | Figure 5 IgG antibody titers in the serum of SPF chickens immunized with PBS (IM), blank CS/PLGA-NPs (IM), blank CS/PLGA-NPs (IN), pVAX I-opti F (the naked plasmid DNA) (IM), pFDNA-CS/PLGA-NPs (IM), and pFDNA-CS/PLGA-NPs (IN). |
IgA antibody in mucosa extracts
The IgA antibody content was significantly higher (P<0.05) in the pFDNA-CS/PLGA-NPs IN group than in the other groups, for tears (Figure 6B), tracheal fluid (Figure 6C), and bile (Figure 6D). In serum, the IgA antibody content did not differ between the naked plasmid DNA IM group and the pFDNA-CS/PLGA-NPs IN group during day 7 to day 28 (Figure 6A) but was higher in the latter group on day 35 and day 42. These results indicated that the pFDNA-CS/PLGA-NPs induced a stronger and more sustained mucosal immune response than the naked plasmid DNA.
 | Figure 6 IgA antibody content in serum (A), tears (B), tracheal fluid (C), and bile (D) of SPF chickens immunized with PBS (IM), blank CS/PLGA-NPs (IM), blank CS/PLGA-NPs (IN), pVAX I-opti F (the naked plasmid DNA) (IM), pFDNA-CS/PLGA-NPs (IM), and pFDNA-CS/PLGA-NPs (IN). |
Lymphocyte proliferation
At 42 days after the first treatment, the SI was significantly higher (P<0.01) for chickens immunized with pFDNA-CS/PLGA-NPs (either IM or IN) than with the plasmid DNA or with blank CS/PLGA-NPs. The SI was significantly higher (P<0.05) with IN than with IM application of pFDNA-CS/PLGA-NPs. The results showed that the pFDNA-CS/PLGA-NPs significantly enhanced the immune function of T lymphocytes (Table 3).
 | Table 3 The stimulation index of T lymphocyte proliferation in SPF chickens after immunization |
Protection against NDV strain F48E9
Within 2 to 5 days, the highly virulent NDV strain F48E9 had killed all chickens treated with PBS or blank CS/PLGA-NPs (Table 4). In contrast, strain F48E9 killed only 12.5%, 0%, and 50% of the chickens treated with pFDNA-CS/PLGA-NPs IM, pFDNA-CS/PLGA-NPs IN, and the naked plasmid DNA, respectively (Table 4). Feeding, drinking, and other aspects of behavior were normal for chickens treated with pFDNA-CS/PLGA-NPs IM and IN but were somewhat abnormal for chickens treated with the naked plasmid DNA. Pathological and histopathological changes were not evident in chickens treated with pFDNA-CS/PLGA-NPs IN (Figure 7G), but small histopathological changes were evident in chickens treated with pFDNA-CS/PLGA-NPs IM (Figure 7F) and naked plasmid DNA (Figure 7E). Histopathological changes typical of ND were evident in chickens treated with PBS and blank CS/PLGA-NPs (Figure 7B–D). These findings demonstrated that treatment with the pFDNA-CS/PLGA-NPs IN induces an effective mucosal immune response and protects chickens against ND.
 | Table 4 Protection of the immunized SPF chickens after challenge with the highly virulent NDV strain F48E9 |
 | Figure 7 Histopathology of the glandular stomach, duodenum, and bursa of Fabricius of normal chickens (A) and of chickens challenged with the highly virulent NDV strain F48E9 after treatment with PBS IM (B); blank CS/PLGA-NPs IM (C); blank CS/PLGA-NPs IN (D); the naked plasmid DNA IM (E); pFDNA-CS/PLGA-NPs IM (F); or pFDNA-CS/PLGA-NPs IN (G). |
Discussion
Polymeric NPs have been widely used to deliver drugs because they can increase drug stability, are easily absorbed by cells, have good adhesive properties, are able to target specific organs, and can bind ligands on their surfaces.39 In particular, biodegradable NPs are available that deliver medicines to target sites and that are then degraded after passing the target site.40 We therefore determined whether biodegradable polymers could be used to deliver newly developed DNA vaccines. Zhao et al previously reported that pFNDV-PLGA-NPs that were prepared with a water/oil/water double emulsion-solvent evaporation method did not change the encapsulated plasmid DNA but promoted the sustained release of the plasmid DNA and induced stronger mucosal immune responses than for nonencapsulated plasmid DNA.35 Because other researchers showed that CS NPs could sustain release and stabilize the plasmid DNA,41 in the current study, we investigated whether CS could improve the performance of PLGA-NPs.
In our first experiments, we found that the EE was highest when NPs were prepared with a CS volume of 1.5 mL and a CS concentration of 0.6 mg/mL. The EE of NPs determines the effectiveness of the gene delivery and subsequent expression of encoded genes in vitro and in vivo.42 In this study, the use of 1.5 mL of CS at 0.6 mg/mL of CS produced an EE of 98.7%, which was higher than the previously reported EE for the pFNDV-PLGA-NPs without CS.35 When CS was used at this volume and concentration, the resulting pFDNA-CS/PLGA-NPs had an appropriate size and maintained the biological activity of the encapsulated plasmid DNA. In addition to not aggregating, the pFDNA-CS/PLGA-NPs had a core–shell structure, a diameter of 699.1±5.21 nm, good stability, a Zeta potential of 6.35±2.75 mV, and a polydispersity index of 0.005.
We also conducted an in vitro assay of the release of the DNA encapsulated in pFDNA-CS/PLGA-NPs. Hu et al43 showed that pH could influence the release of plasmid DNA from NPs and that NPs are more swollen at pH 7.0–7.4 than at pH 4.0. Thus, our assay used pFDNA-CS/PLGA-NPs that were swollen in PBS at pH 7.4. The results of the assay revealed a gradual and sustained release of DNA from the pFDNA-CS/PLGA-NPs, suggesting a strong physical interaction between the drug and polysaccharide layer on the NPs’ surface. The CS slightly reduced the DNA burst release effect in the first 24 hours, indicating that the CS layer surrounding the PLGA-NPs acts as a physical barrier that slows the release of DNA from the PLGA-NPs.
The cellular uptake of the NPs was visualized by indirect immunofluorescence. The in vitro cell toxicity and expression studies indicated that the pFDNA-CS/PLGA-NPs did not damage cells and maintained DNA bioactivity. The survival rate was higher than the previously reported 80.14% for pFNDV-PLGA-NPs without CS.35 In vivo cytotoxicity assay showed that the pFDNA-CS/PLGA-NPs did not have pathological effects on chickens. Overall, the results showed that the DNA in the NPs was not altered and that the NPs containing DNA and coated with CS would not harm chickens.
Analyses of IgG and IgA antibody responses revealed that IN immunization with pFDNA-CS/PLGA-NPs induced stronger responses than immunization with naked plasmid DNA. The IgA antibody contents of group pFDNA-CS/PLGA-NPs IN were much higher than the previously reported 800–1,000 ng/mL (P<0.05).35 IN immunization was very effective in eliciting mucosal and systemic immune responses.44,45 Our findings suggest that the nasal administration of pFDNA-CS/PLGA-NPs is effective in inducing the immune response against ND, probably because the CS coating changes the surface charge of the PLGA-NPs and prolongs the antigen contact time with mucosal surfaces.46
In summary, our comparison of the pFDNA-CS/PLGA-NPs and naked plasmid DNA showed that the immunogenicity and protective immunity can be improved by encapsulating the plasmid DNA into CS-coated PLGA NPs.
Although this study demonstrates the potential of CS-modified PLGA NPs as an efficient delivery system for NDV DNA vaccine in mucosal immunization, a number of challenges must be addressed. First, the trace amounts of initiator, toxic organics, and other impurities in the polymer must be removed before a vaccine is commercialized. Toxic solvent remaining in the natural polymer during NP preparation must also be removed. Second, the cost of preparation must be reduced. Finally, the controlled and targeted release of NPs must be improved. We suspect that these challenges can be met by technological advancements in the biomedical and material sciences.
Acknowledgments
We gratefully acknowledge the Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University), the Ministry of Education and Engineering Research Center of Agricultural Microbiology Technology, and the Ministry of Education for providing the facilities to carry out this work. This work was supported, in part, by the National Natural Science Foundation of the People’s Republic of China (grant number 31072119), the Key Project of the Chinese Ministry of Education (grant number 212048), the Program for New Century Excellent Talents in University (grant number NCET-12-0707), the Innovative Research Team for Agricultural Microbiology Fermentation Technology at Heilongjiang Provincial University (grant number 2012td009), the Changjiang Scholar Candidates Program for Provincial Universities in Heilongjiang (grant number 2014CJHB005), the Scientific and Technological Key Project of Heilongjiang Province (GC13B403), the Early Research and Development Cultivation Project of Scientific and Technological Achievements Industrialization for Provincial Universities in Heilongjiang (grant number 1253CGZH10), and the Innovation Foundation of Harbin (grant number 2013RFQXJ030).
Disclosure
The authors report no conflicts of interest in this work.
References
Meulemans G, Gonze M, Carlier MC, Petit P, Burny A, Long L. Protective effects of HN and F glycoprotein-specific monoclonal antibodies on experimental Newcastle disease. Avian Pathol. 1986;15(4):761–768. | ||
Arifin MA, Mel M, Abdul Karim MI, Ideris A. Production of Newcastle disease virus by Vero cells grown on cytodex 1 microcarriers in a 2-litre stirred tank bioreactor. J Biomed Biotechnol. 2010;2010:586363. | ||
Zhao K, Zhang Y, Zhang X, et al. Preparation and efficacy of Newcastle disease virus DNA vaccine encapsulated in chitosan nanoparticles. Int J Nanomedicine. 2014;9:389–402. | ||
Robinson HL, Hunt LA, Webster RG. Protection against a lethal influenza virus challenge by immunization with a haemagglutinin-expressing plasmid DNA. Vaccine. 1993;11(9):957–960. | ||
Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL. DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc Natl Acad Sci U S A. 1993;90(24):11478–11482. | ||
Donnelly JJ, Wahren B, Liu MA. DNA vaccines: progress and challenges. J Immunol. 2005;175(2):633–639. | ||
Hallengärd D, Haller BK, Petersson S, et al. Increased expression and immunogenicity of HIV-1 protease following inactivation of the enzymatic activity. Vaccine. 2011;29(4):839–848. | ||
Xu K, Ling ZY, Sun L, et al. Broad humoral and cellular immunity elicited by a bivalent DNA vaccine encoding HA and NP genes from an H5N1 virus. Viral Immunol. 2011;24(1):45–56. | ||
Pachuk CJ, McCallus DE, Weiner DB, Satishchandran C. DNA vaccines – challenges in delivery. Curr Opin Mol Ther. 2000;2(2):188–198. | ||
Robertson JS, Griffiths E. Assuring the quality, safety, and efficacy of DNA vaccines. Mol Biotechnol. 2001;17(2):143–149. | ||
Wu H, Dennis VA, Pillai SR, Singh SR. RSV fusion (F) protein DNA vaccine provides partial protection against viral infection. Virus Res. 2009;145(1):39–47. | ||
Sun J, Hou J, Li D, et al. Enhancement of HIV-1 DNA vaccine immunogenicity by BCG-PSN, a novel adjuvant. Vaccine. 2013;31(3):472–479. | ||
Manoj S, Babiuk LA, van Drunen Littel-van den Hurk S. Approaches to enhance the efficacy of DNA vaccines. Crit Rev Clin Lab Sci. 2004;41(1):1–39. | ||
Sun J, Li D, Hao Y, et al. Posttranscriptional regulatory elements enhance antigen expression and DNA vaccine efficacy. DNA Cell Biol. 2009;28(5):233–240. | ||
Csaba N, Garcia-Fuentes M, Alonso MJ. The performance of nanocarriers for transmucosal drug delivery. Expert Opin Drug Deliv. 2006;3(4):463–478. | ||
Moghimi SM, Kissel T. Particulate nanomedicines. Adv Drug Deliv Rev. 2006;58(14):1451–1455. | ||
Liang MT, Davies NM, Blanchfield JT, Toth I. Particulate systems as adjuvants and carriers for peptide and protein antigens. Curr Drug Deliv. 2006;3(4):379–388. | ||
Hsu SH, Chan SH, Chiang CM, Chen CC, Jiang CF. Peripheral nerve regeneration using a microporous polylactic acid asymmetric conduit in a rabbit long-gap sciatic nerve transection model. Biomaterials. 2011;32(15):3764–3775. | ||
Santo VE, Duarte ARC, Gomes ME, Mano JF, Reis RL. Hybrid 3D structure of poly(D,L-lactic acid) loaded with chitosan/chondroitin sulfate nanoparticles to be used as carriers for biomacromolecules in tissue engineering. J Supercrit Fluids. 2010;54(3):320–327. | ||
Mahmoudifar N, Doran PM. Chondrogenic differentiation of human adipose-derived stem cells in polyglycolic acid mesh scaffolds under dynamic culture conditions. Biomaterials. 2010;31(14):3858–3867. | ||
Thevenot PT, Nair AM, Shen J, Lotfi P, Ko CY, Tang L. The effect of incorporation of SDF-1alpha into PLGA scaffolds on stem cell recruitment and the inflammatory response. Biomaterials. 2010;31(14):3997–4008. | ||
Smith DJ, King WF, Barnes LA, Trantolo D, Wise DL, Taubman MA. Facilitated intranasal induction of mucosal and systemic immunity to mutans streptococcal glucosyltransferase peptide vaccines. Infect Immun. 2001;69(8):4767–4773. | ||
Peacock ZS, Barnes LA, King WF, et al. Influence of microparticle formulation on immunogenicity of SYI, a synthetic peptide derived from Streptococcus mutans GbpB. Oral Microbiol Immunol. 2005;20(1):60–64. | ||
Wierzbicki A, Kiszka I, Kaneko H, et al. Immunization strategies to augment oral vaccination with DNA and viral vectors expressing HIV envelope glycoprotein. Vaccine. 2002;20(9–10):1295–1307. | ||
Garinot M, Fiévez V, Pourcelle V, et al. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J Control Release. 2007;120(3):195–204. | ||
Allaoui-Attarki K, Fattal E, Pecquet S, et al. Mucosal immunogenicity elicited in mice by oral vaccination with phosphorylcholine encapsulated in poly(D,L-lactide-co-glycolide) microspheres. Vaccine. 1998;16(7):685–691. | ||
Rajkannan R, Dhanaraju MD, Gopinath D, Selvaraj D, Jayakumar R. Development of hepatitis B oral vaccine using B-cell epitope loaded PLG microparticles. Vaccine. 2006;24(24):5149–5157. | ||
Jabbal-Gill I, Fisher AN, Rappuoli R, Davis SS, Illum L. Stimulation of mucosal and systemic antibody responses against Bordetella pertussis filamentous haemagglutinin and recombinant pertussis toxin after nasal administration with chitosan in mice. Vaccine. 1998;16(20):2039–2046. | ||
Kawashima Y, Yamamoto H, Takeuchi H, Kuno Y. Mucoadhesive DL-lactide/glycolide copolymer nanospheres coated with chitosan to improve oral delivery of elcatonin. Pharm Dev Technol. 2000;5(1):77–85. | ||
Chronopoulou L, Massimi M, Giardi MF, et al. Chitosan-coated PLGA nanoparticles: a sustained drug release strategy for cell cultures. Colloids Surf B Biointerfaces. 2013;103:310–317. | ||
Budhian A, Siegel SJ, Winey KI. Controlling the in vitro release profiles for a system of haloperidol-loaded PLGA nanoparticles. Int J Pharm. 2008;346(1–2):151–159. | ||
Tahara K, Yamamoto H, Kawashima Y. Cellular uptake mechanisms and intracellular distributions of polysorbate 80-modified poly(D,L-lactide-co-glycolide) nanospheres for gene delivery. Eur J Pharm Biopharm. 2010;75(2):218–224. | ||
Tahara K, Sakai T, Yamamoto H, Takeuchi H, Hirashima N, Kawashima Y. Improvements in transfection efficiency with chitosan modified poly(DL-lactide-co-glycolide) nanospheres prepared by the emulsion solvent diffusion method, for gene delivery. Chem Pharm Bull (Tokyo). 2011;59(3):298–301. | ||
Zhao K, Li GX, Jin YY, et al. Preparation and immunological effectiveness of a Swine influenza DNA vaccine encapsulated in PLGA microspheres. J Microencapsul. 2010;27(2):178–186. | ||
Zhao K, Li W, Huang T, et al. Preparation and efficacy of Newcastle disease virus DNA vaccine encapsulated in PLGA nanoparticles. PLoS One. 2013;8(12):e82648. | ||
Zhao K, Shi X, Zhao Y, et al. Preparation and immunological effectiveness of a swine influenza DNA vaccine encapsulated in chitosan nanoparticles. Vaccine. 2011;29(47):8549–8556. | ||
Zeng W, Wang Y, Shi X, et al. Optimization of codon usage of F gene enhanced efficacy of Newcastle disease virus DNA vaccine. Chin J of Anim Infect Dis. 2009;17(2):8–16. | ||
Zhao F, Wu Y, Zhang X, et al. Enhanced immune response and protective efficacy of a Treponema pallidum Tp92 DNA vaccine vectored by chitosan nanoparticles and adjuvanted with IL-2. Hum Vaccin. 2011;7(10):1083–1089. | ||
Löbenberg R, Araujo L, Kreuter J. Body distribution of azidothymidine bound to nanoparticles after oral administration. Eur J Pharm Biopharm. 1997;44(2):127–132. | ||
Belbella A, Vauthier C, Fessi H, Devissaguet JP, Puisieux F. In vitro degradation of nanospheres from poly(D,L-lactides) of different molecular weights and polydispersities. Int J Pharm. 1996;129(1–2):95–102. | ||
Kawashima Y, Handa T, Kasai A, Takenaka H, Lin SY, Ando Y. Novel method for the preparation of controlled-release theophylline granules coated with a polyelectrolyte complex of sodium polyphosphate-chitosan. J Pharm Sci. 1985;74(3):264–268. | ||
Boyoglu S, Vig K, Pillai S, et al. Enhanced delivery and expression of a nanoencapsulated DNA vaccine vector for respiratory syncytial virus. Nanomedicine. 2009;5(4):463–472. | ||
Hu Y, Jiang X, Ding Y, Ge H, Yuan Y, Yang C. Synthesis and characterization of chitosan-poly(acrylic acid) nanoparticles. Biomaterials. 2002;23(15):3193–3201. | ||
Xu J, Dai W, Wang Z, Chen B, Li Z, Fan X. Intranasal vaccination with chitosan-DNA nanoparticles expressing pneumococcal surface antigen a protects mice against nasopharyngeal colonization by Streptococcus pneumoniae. Clin Vaccine Immunol. 2011;18(1):75–81. | ||
Cripps AW, Kyd JM. Comparison of mucosal and parenteral immunisation in two animal models of pneumococcal infection: otitis media and acute pneumonia. Vaccine. 2007;25(13):2471–2477. | ||
Yamamoto H, Kuno Y, Sugimoto S, Takeuchi H, Kawashima Y. Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions. J Control Release. 2005;102(2):373–381. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.