Back to Journals » International Journal of Nanomedicine » Volume 15
Chitosan-Coated PLGA Nanoparticles for Enhanced Ocular Anti-Inflammatory Efficacy of Atorvastatin Calcium
Authors Arafa MG ![]() , Girgis GNS
, Girgis GNS ![]() , El-Dahan MS
, El-Dahan MS ![]()
Received 4 November 2019
Accepted for publication 15 January 2020
Published 28 February 2020 Volume 2020:15 Pages 1335—1347
DOI https://doi.org/10.2147/IJN.S237314
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Mona G Arafa,1,2 Germeen NS Girgis,3 Marwa S El-Dahan3
1Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, The British University in Egypt (BUE), El-Sherouk City, Cairo, 11837, Egypt; 2Chemotherapeutic Unit, Mansoura University Hospitals, Mansoura 35516, Egypt; 3Department of Pharmaceutics, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt
Correspondence: Mona G Arafa Suez Desert Road, El Sherouk City, Cairo Governorate 11837, Egypt
Tel +20 1005055557
Fax +20 226300010
Email [email protected]
Background: Atorvastatin calcium (AT) is an ocular anti-inflammatory with limited bioavailability when taken orally due to its low solubility in low pH and extensive first-pass effect. To overcome these problems, AT was entrapped in polymeric nanoparticles (NPs) to improve surface properties and sustained release, in addition to achieving site-specific action.
Methods: AT was entrapped in chitosan (CS)-coated polylactic-co-glycolic acid (PLGA) NPs to form AT-PLGA-CS-NPs (F1). F1 and free AT were embedded in thermosensitive Pluronic® 127-hydroxypropyl methylcellulose (HPMC) to form thermosensitive gels (F2) and (F3) while F4 is AT suspension in water. F1 was assessed for size, surface charge, polydispersity index (PDI), and morphology. F2 and F3 were examined for gelation temperature, gel strength, pH, and viscosity. In vitro release of the four formulations was also investigated. The ocular irritancy and anti-inflammatory efficacy of formulations against prostaglandin E1-(PGE1) induced ocular inflammation in rabbits were investigated by counting the polymorphonuclear leukocytes (PMNs) and protein migrated in tears.
Results: Oval F1 of 80.0– 190.0± 21.6 nm exhibited a PDI of 0.331 and zeta potential of 17.4± 5.62 mV with a positive surface charge. F2 and F3 gelation temperatures were 35.17± 0.22°C and 36.93± 0.31°C, viscosity 12,243± 0.64 and 9759± 0.22 cP, gel strength 15.56± 0.6 and 12.45± 0.1 s, and pHs of 7.4± 0.02 and 7.4± 0.1, respectively. In vitro release of F1, F2, F3, and F4 were 48.21± 0.31, 26.48± 0.5, 84.76± 0.11, and 100% after 24 hrs, respectively. All formulations were non-irritant. F2 significantly inhibited lid closure up to 3 h, PMN counts and proteins in tear fluids up to 5 h compared to other formulations.
Conclusion: AT-PLGA-CS-NP thermosensitive gels proved to be successful ocular anti-inflammatory drug delivery systems.
Keywords: atorvastatin calcium, nanoparticles, PLGA, chitosan, ocular, thermosensitive gel
Introduction
Statins such as simvastatin, lovastatin, and atorvastatin calcium (AT) are 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. These compounds effectively lower blood cholesterol levels and hence reduce mortality risks in patients with cardiovascular disease.1 Recently statins were shown to have both immunomodulatory effects2–4 and anti-inflammatory properties.5,6 However, it is unclear if these drugs can be topically applied. A topical effect of AT on the healing process of patients with pressure ulcers was demonstrated.7 Moreover, some statin derivatives showed efficacy in treating intraocular inflammation; orally administrated AT has pharmacological effects on autoimmune uveoretinitis (EAU) induced experimentally in rats.8 Drug delivery to targeted eye tissues is a major challenge faced by formulation scientists. Orally administered statins may have side effects on the liver and may cause myopathy. As a result, topical delivery may be a reasonable alternative with several merits compared with oral administration, including localized and prolonged effects on treated eye tissue and lower dose frequency, which could reduce many drug adverse effects.9
In the past two decades, ophthalmic research has moved toward the formulation of novel and safe drug delivery systems to counteract eye barriers and maintain drug levels in target tissues. Anterior segment drug delivery dosage forms are produced by modifying traditional topical formulations such as solutions, suspensions, emulsions, gels, and ointments. Novel advanced ocular delivery systems have been extensively investigated, such as in-situ forming gel,10 nanocarrier systems,11 inserts,12 and vesicular systems.13
The major dilemma encountered while developing new formulations of ocular drug with poor solubility is limited bioavailability due to low dissolution rate and consequent poor permeability. Other factors such as nasolacrimal drainage can necessitate recurrent drug administration. This reduces patient compliance and increases systemic side effects. There was a growing need to develop new approaches that control particle size, enhance drug dissolution rate, and accordingly improve its bioavailability.14 Encapsulating drug in biodegradable, biocompatible, natural or synthetic polymeric nanoparticles (NPs) is one such approach. Polymers like poly lactic-co-glycolic acid (PLGA) and chitosan (CS) are often used to overcome the aforementioned problems, and they also help achieve mucoadhesive ocular controlled drug delivery. Mucoadhesiveness in ocular drug delivery is a robust approach that prolongs drug retention time and increases the membrane permeability and intracellular uptake of drugs. This could be attained by establishing a bond between the negatively charged cornea or sclera and positively charged ocular system. For example, cationic CS is a biodegradable polymer with positively charged amino groups.15,16
Based on the above considerations, the present study aimed to enhance ocular bioavailability of AT by preparing and evaluating CS-coated PLGA NPs entrapping AT (AT-PLGA-CS-NPs, F1). We also developed a topical AT-PLGA-CS-NPs thermosensitive gel (AT-PLGA-CS-NPs-PF127-HPMC, F2) as an efficient ocular anti-inflammatory targeting drug delivery system. Its efficacy against inflammation of rabbit’s eye induced by prostaglandin E1 (PGE1) instillation was assessed by quantifying polymorphonuclear leukocytes (PMNs) and protein levels in tear fluid.
Materials and Methods
Materials and Reagents
AT was kindly supplied by Epico Company,(10th of Ramadan city, Cairo, Egypt). Pluronic®F127 (PF127) purified poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), CAS Number: 9003-11-6, was purchased from Sigma-Aldrich (St. Louis, MO, USA). Polyvinyl alcohol (PVA, CAS: 9002-89-5, MW:89, 000–98,000, 99% hydrolyzed) were purchased from Sigma-Aldrich. Hydroxypropyl methylcellulose (HPMC, USP grade, CAS: 9004-65-3, 2600–5600 cP), PLGA (lactic acid: glycolic acid, 50:50) was purchased from Sigma-Aldrich. CS 100,000:300,000 was purchased from Sigma-Aldrich. Monopotassium dihydrogen phosphate and anhydrous dibasic sodium phosphate were purchased from Adwic, Al Nasr Pharmaceutical Chemicals Co. (Qalyubia, Egypt). Spectra/por dialysis cellophane membranes (12,000–14,000 MW cut off) were used for filtration. PGE1 (Alprostadil) was obtained from Pfizer Limited, United Kingdom. Protein dye reagent kits were from Spinreact (Girona, Spain; Pyrogallol red Colorimetric, # 1001024, S.A./S.A.U. Ctra. Santa Coloma).
Methodology
Preparation of AT-PLGA-CS-NPs
The NP entrapping drug (F1) was prepared by single emulsion and solvent evaporation technique. In brief, 30 mg drug was dissolved in 5 mL dichloromethane (DCM) containing 100 mg PLGA. CS and PVA solutions were prepared by solubilizing 0.5 g CS in (1% acetic acid solution) at room temperature followed the addition of 0.5 g PVA. The organic phase was drop-wise injected in 15 mL aqueous stabilizer solution. The injection procedure and reaction process were carried out under constant magnetic stirring (Branson Sonifier Model S-450D, Branson, Danbury, CT, USA) (1500 rpm) at room temperature until the solvent vaporized. The suspension was freeze centrifuged (2–16KL, Sigma Laborzentrifugen GmbH, Osterode am Harz, Germany) and the obtained NPs were washed with distilled water and kept in the refrigerator at °C for further studies.17–19
Characterization of AT-PLGA-CS-NPs
Particle size, particle distribution, and the surface charge of formed NPs were quantized on a Zetasizer Nano ZS90 (Malvern Instruments Ltd., Malvern, UK). AT-PLGA-CS-NP morphology was inspected by transmission electron microscopy (TEM; H-600, Hitachi, Tokyo, Japan).20,21
Entrapment Efficiency (EE) and Drug Loading of AT-PLGA-CN-NPs
Five milliliters of acetonitrile were added to 20 mg of the sediments, and the mixture was sonicated (Elmasonic S60 H, Elma Hans Schmidbauer GmbH, Singen, Germany) for 5 min. An additional 5 mL acetonitrile was added to 0.1 mL of each suspension and sonicated again for 10 min until clear solutions were obtained, and then concentrations of entrapped AT were determined spectrophotometrically at 241 using an ultraviolet spectrophotometer (V-630, Jasco, Tokyo, Japan) against a blank of the corresponding plain PLGA-CS-NPs dispersion treated in the same way.
EE% = entrapped drug concentration/initial drug concentration *100 (Equation 1) was used to calculate the EE, while drug loading was determined as DL% = ED/weight of NPs* 100 (Equation 2).22,23
Preparation of AT-PLGA-CS-NP-PF127-HPMC Thermosensitive Gel
The thermosensitive gelling property is one of the important features of Pluronic®F127 (PF127), as a result; the cold method was employed to formulate the gel.24 In brief, a calculated amount of PF127 was gradually added to half the certain volume of distilled water containing an accurately weighed amount of HPMC at 4°C with continuous stirring. The calculated amount of F1 (equivalent to 0.3 g drug) was added to the PF127 and HPMC solution to form F2 under stirring to attain an homogenous mixture, and at that time, the final volume was attained with the rest of water. To obtain Pluronic®F127 and HPMC solution containing 0.3 g drug (F3), the preparation was continued as described in Table 1. The mixture was stored at 4°C until a mucilaginous lucent gel was attained.
 |
Table 1 Formulae Composition of Thermosensitive Gels |
Evaluation of AT-PLGA-CS-NP Thermosensitive Gels
Determination of Drug Contents
AT contents were determined for all formed gels, with an anticipated range of 100 ± 10%. In brief, 10 mL of phosphate-buffered saline (PBS) was mixed with 100 mg of each thermosensitive gel and then diluted. The absorbance of each solution was measured at 241 nm spectrophotometrically in triplicate.
Gelation Temperature Measurement of the AT-PLGA-CS-NP Thermosensitive Gels
A test tube containing 2 mL of each thermosensitive gel was wrapped in a thin aluminum sheet and placed in a 4°C water bath. Gelation temperature was assessed when the temperature of the water bath was elevated by 1°C. The temperature was elevated constantly with leaning the tube over 90 degrees until immobilization of the gel was observed. The sol–gel transition temperature was the lowest temperature at which gel immobility was attained.25
Gel Strength Measurement of AT-PLGA-CS-NP Thermosensitive Gels
In a 100-mL graduated cylinder, sufficient amount of each medicated gel was added and gelled at 35–36°C depending on its specific gelation temperature.26 A weight of 5 g was located on every gel formula. Gel strength was defined as the time (s) needed for the weight to run down 5 cm.27
Viscosity Determination of AT-PLGA-CS-NP Thermosensitive Gels
A rotary viscometer HAAKE RV3 with digital circulating water bath (Haake, KarlsruheGoerzallee 249, Germany) was used to measure the viscosities of the prepared thermosensitive gels.28 One gram of each gel was straightened out on the lower flat piece of the viscometer and Roto Cone. A 32 rpm angular velocity was employed to determine the viscosity of every sample at 35–36 ± 0.5°C depending on its specific gelation temperature. The angular velocity was increased, and the process was duplicated to calculate the viscosity.24
pH Determination of AT-PLGA-CS-NP Thermosensitive Gels
The pH values of thermosensitive gels were determined in triplicate using a digital pH meter (CA 92634, Beckman Instruments, Fullerton, CA, USA), and the mean values were calculated.
In vitro Release Study of AT-PLGA-CS-NPs and AT-PLGA-CS-NP Thermosensitive Gels
The diffusion cell method using PBS, pH 7.4, at 37±0.5°C was employed using a digital shaking water bath (WSB-18, Dahan Scientific Co. Ltd., Beijing, China). In brief, 3.3 g of each gel formula with the same amount of polymeric NPs entrapping AT and 3.3 mL drug dispersion F429 (equivalent to 10 mg AT) was spread on the membrane, and 1.5 mL of PBS was added to the F1, F2, and F3 tubes. For each sample, and at pre-set timepoints of 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, and 24 hrs, a certain volume was taken and substituted with equal volumes of newly prepared PBS at the same temperature.24 The amounts of AT released from all formulations were investigated spectrophotometrically at 241 nm.22
Kinetic Study
Zero-order, first-order, and Higuchi diffusion models of linear regression and the Korsmeyer–Peppas model were used to study the in vitro release of AT.20
In vivo Anti-Inflammatory Assessment of AT-PLGA-CS-NP Thermosensitive Gels
Albino rabbits (2–2.5 kg) were obtained from Egyptian farms and kept under standard laboratory conditions. All animal work was conducted in accordance with the guidelines for the care and use of laboratory animals and was approved by the ethical committee of faculty of pharmacy of the British University in Egypt.30
Ocular Irritancy Test
The ocular irritancy test was performed for all tested formulations using a previously reported method.31 Briefly, a volume of 100 µL of each preparation was instilled twice a day for a period of 1 week into the lower lid of the rabbits’ eyes and observed for any signs of irritation such as eye swelling, hyperemia, or rise in the rate of tear formation after predetermined time intervals of 1, 24, 48, and 72 hrs and 1 week after application to examine the potential ocular irritancy of the tested formulations.
Each formulation was tested employing albino rabbits divided into four groups (n=4 each). In each group, the tested preparations were instilled in the right eyes while the left eyes were received PBS. Group I received AT-PLGA-CS-NPs (F1), group II received AT-PLGA-CS-NPs in thermosensitive gel (F2), group III received free AT in thermosensitive gel (F3), and group IV received AT suspended in water (control group, F4).
Anti-Inflammatory Study
The prostaglandin E1 (PGE1) solution was instilled in rabbit eyes to model ocular inflammation. The animals used for this study were 16 albino rabbits divided into 4 groups of 4 each. Animals were maintained in an institutional animal household and given unrestricted access to food and water.32
For each group of rabbits, the right eyes were treated with 100 µL of AT-PLGA-CS-NPs (F1 in case of Group I), AT-PLGA-CS-NPs in thermosensitive gel (F2 for Group II), free AT in thermosensitive gel (F3 for Group III), and AT suspended in double-distilled water (F4 for Group IV). Left rabbit eyes served as control and were instilled with equal volumes of PBS (pH 7.4).
Ten minutes after formulae application, 50 µL of PGE1 solution (1 µg/mL normal saline) was instilled in both eyes. Then, all eyes were examined for inflammation characteristics. Lid closure was scored as follows: 0, fully open; 1, one-third closed; 2, two-thirds closed; and 3, fully closed eye. PMN counts and protein concentration in tear fluid were also measured.33
To determine PMN counts and protein concentrations in tears following PGE1 instillation, at each time interval (1, 2, 3, 4, and 5 hrs), 100 µL of normal saline was dropped in the lower lid of the rabbit eye, mixed quickly, and then after 10 (s); 50 µL of the tear fluid was taken out for subsequent analysis.
PMN Quantification
Tear fluid PMN counts were measured for all eyes treated with either the formulation or vehicle. This was performed by diluting tear fluid with Turke’s fluid reagent consisting of 1.5% glacial acetic acid and 1% gentian violet in double-distilled water. PMNs were counted using a Neubauer hemocytometer (PRECICOLOR HBG, Lützellinden, Germany).34
Protein Estimation
The protein concentrations in eye tear fluid samples were determined using the pyrogallol-red colorimetric method according to the manufacturer's instructions.35
Statistical Analysis
One-way analysis of variance followed by Tukey-Kramer multiple comparisons statistical analysis were applied36 using GraphPad Prism-5 software (GraphPad Software Inc., San Diego, CA, USA).
Results
AT-PLGA-CS-NP Characterization
The average size of AT-PLGA-CS-NPs was 192.0±21.6 nm, which was in agreement with the TEM results showing a PDI of 0.331 (Figure 1A). The zeta potential of the AT-PLGA-CS-NPs indicated a positive surface charge with a mean zeta potential of (17.4±5.62 mV) (Figure 1B). The present results are in accordance with many reports.15,37
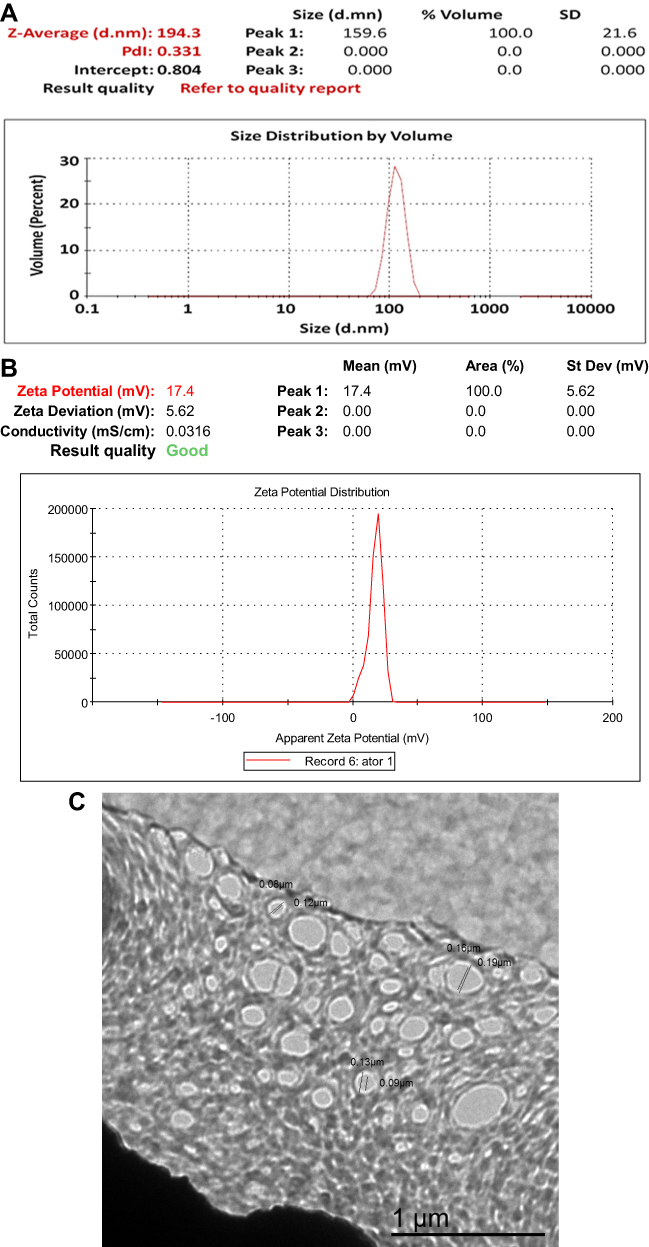 |
Figure 1 Particle size and particle size distribution (A), zeta potential (B), and transmission electron microscopy (C) of F1 AT-PLGA-CS-NPs. |
TEM analysis was carried out to identify the size and shapes of the formed AT-PLGA-CS-NPs. The results confirmed the formation of oval to spherical NPs of 80.0–190.0±21.6 nm in size without aggregation (Figure 1C). These results are in agreement with previous studies.14,38
EE and Drug Loading of AT-PLGA-CS-NPs
The results showed an EE and DL of 83.9 ±4.1% 10 ±0.6%, respectively.
Evaluation of the AT-PLGA-CS-NP Thermosensitive Gels
Determination of Drug Contents
The drug contents of F2 and F3 in gels were 95.2 ± 1.1% and 97.6 ± 0.7%, respectively.
Gelation Temperatures of AT-PLGA-CS-NP Thermosensitive Gels
The results in Table 2 showed that F2 and F3 were liquid at 4°C and demonstrated gelling temperatures of 35.1±0.22 and 36.93±0.31°C, respectively, by steadily increasing the temperature (Figure 2A).
 |
Table 2 Comparative Effect of AT-NP Thermosensitive Gel and Other Drug Formulations on PGE1-Induced Rabbit Eye Lid Closure |
 |
Figure 2 Gelation temperature (A), gel strength (B), and viscosity profile at 32 rpm (C) of F2 (AT-PLGA-CS-NPs-PF127-HPMC thermosensitive gel). |
Gel Strength Measurement of the AT-PLGA-CS-NP Thermosensitive Gels
While the gel strength increased as a function of time, the gelation temperature of the gel system decreased. The results showed that F2 and F3 exhibited gel strengths of 15.56±0.6 and 12.45±0.1 s, and shear thinning occurred between 35.17±0.22 and 36.93±0.31°C, respectively. As shown in Figure 2B, the gel strength of F2 was higher than that of F3, as the weight (5 g) took more time to fold away.
Viscosity Determinations of the AT-PLGA-CS-Ns Thermosensitive Gels
The viscosities in Figure 2C were 12,243± 0.64 and 9759± 0.22 cP for F2 and F3, respectively. The process was operated for 10 runs at 32 rpm. The viscosities of gelling systems increased in the presence of polymeric NPs.
pH Determination of AT-PLGA-CS-NP Thermosensitive Gels
The respective pH values of F2 and F3 were 7.4±0.02 and 7.4±0.1.
In vitro Release of AT-PLGA-CS-NP Dispersion from Thermosensitive Gels
The percentages of AT released from all formulations were studied (Figure 3A). Formula F4 showed the highest percent of drug released followed by F3, F1, and F2. The release percentages after 24 hrs were 100, 84.76 ±0.11, 48.21 ±0.31, and 26.48 ±0.5%, respectively.
 |
Figure 3 Release of AT from F1, F2, F3, and F4 in phosphate buffer (pH 7.4) at 37°C (A). Zero model of F1 (B), non-Fickian model of F1 (C), zero model of F2 (D), non-Fickian model of F2 (E), zero model of F3 (F), and non-Fickian model of F3 (G). |
According to this result, incorporation of NPs into thermosensitive gel (F2) resulted in significantly (p<0.05) slower AT release compared with the other formulae (F1, F3, and F4) at 10 and 24 h.
Kinetic Study
Correlations of release data for F1, F2, and F3 are explained by a zero-order model with regression coefficients (R2) of 0.9972, 0.9811, and 0.9917, respectively, at 35–36±0.5°C (Figure 3B, D, and F). The Korsmeyer–Peppas equation was also employed, and the release exponents “n” were 0.616, 0.619, and 0.5309 for F1, F2, and F3, respectively (Figure 3C, E, and G). This result demonstrated that the drug release rate was mainly controlled by anomalous or non-Fickian transport.
Ocular Irritancy Test
During the entire study time, no inflammation signs such as eye redness, swelling, or increase in tearing were recorded after instillation of the studied preparations in comparison to PBS. This indicated that all the investigated formulae were non irritant and safe to the eye.
In vivo Ocular Anti-Inflammatory Study
Topical instillation of PGE1 induces ocular inflammation including increased numbers of PMNs, higher protein concentration in tears, and eyelid closure.39 Hence, these characteristics were used to assess the anti-inflammatory effect of AT formulations. Figure 4 shows the gross appearance of inflamed rabbit eyes after 1 hr (Figure 4A) and after 5 hrs treatment with F2 (Figure 4B). The results shown in Table 2 show that lid closure was obvious for up to 3 hrs, after which point it declined. The lid closure score was found to be higher in all control eyes compared to drug-treated eyes. In addition, the lid closure scores of eyes treated with AT-PLGA-CS-NP thermosensitive gel was much lower compared to that observed with other formulations, indicating a higher inflammation inhibitory effect of AT-PLGA-CS-NPs over the other formulations. F2 achieved a statistically significant difference (p < 0.05) in lid closure score after 3 hrs with respect to other formulations.
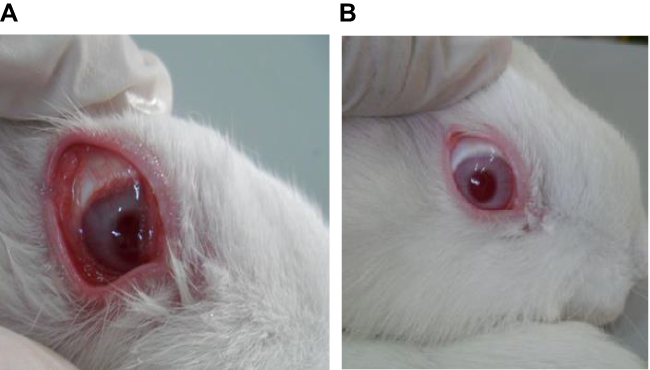 |
Figure 4 Gross appearance of an inflamed rabbit eye after 1 hr (A), and an eye treated with F2 formula 5 hrs earlier (B). |
The results shown in Figure 5A and B indicated that both PMN counts and protein levels in tear fluid increased up to 3 hrs and afterward decreased with all AT ophthalmic formulations and PBS. For NP formulations (F1 and F2), the PMN counts and protein % were lower than for F3 and F4 throughout the 5 hrs study.
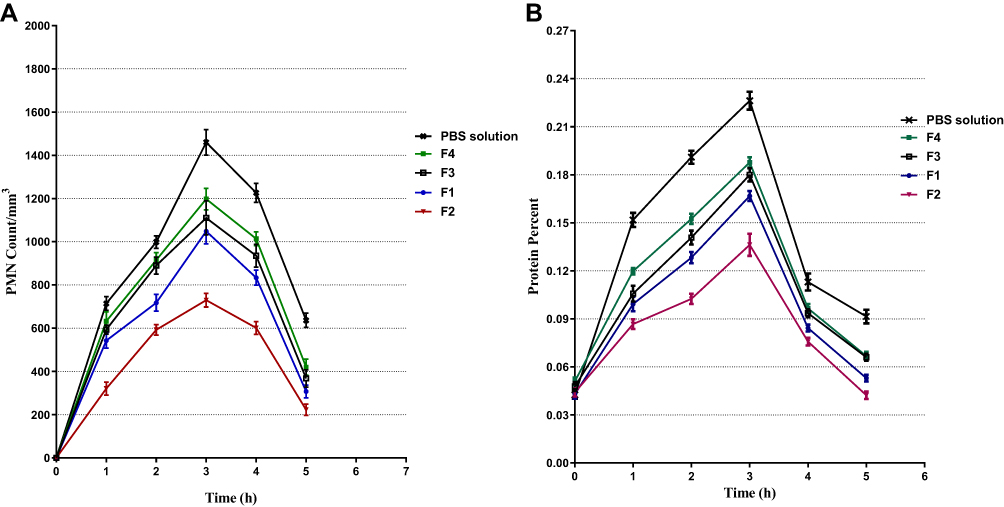 |
Figure 5 Effect of AT formulations against prostaglandin E1–induced polymorphonuclear leukocytes (PMN) (A) and protein (B) in tears of rabbits. |
Discussion
The small particle size of formed NPs may be attributed to the fact that a stabilizer favors smaller particle size because it tends to form a boundary layer around droplets, which prevents favors non-aggregated dispersion. Also, the volume of DCM reduced the mean particle size due to decreased viscosity of the organic phase and increased polymer dilution. Finally, stirring also provides shear force to reduce particle size. The small PDI value stipulated the homogenous dissemination of formed NPs. Positive zeta potentials were detected for F3 due to the presence of CS film coating PLGA-NPs,40 enhancing the stability of formed system. The TEM results corroborated those of the particle size study.
EE% and DL% are the dominating parameters underlying the efficiency of the selected nanocarrier system. Because of their poor aqueous solubility and high binding to PLGA polymer; hydrophobic drugs are liable to remain in the oil phase. In the presence of PLGA, the organic phase becomes more viscous, preventing drug molecules from moving toward the water phase, consequently more drug is trapped and loaded inside the polymeric system.41 EE also increased in the presence of CS,37 which is in accordance with previous findings.42 The presence of stabilizers PVA and CS increases the aqueous phase’s viscosity, which eliminates the loading of AT halfway between the organic and aqueous phases, leading to reasonable loading.43
The content of drug was homogeneous in hydrogels. Two main factors are responsible for the low gelling temperature of the PF127-HPMC mixture. The first is the presence of an oxygen atom in PF127 that can form a bond with the water proton when PF127 and HPMC are dissolved. The second factor is the abundant OH groups in HPMC, which cross-link with the propylene oxide chain of PF127. In other words, PF127 has the ability to form links with both water and HPMC so stuff gel formation requires less heat energy.44,45 When the temperature of thermosensitive PF127-HPMC systems is raised, it breaks down the hydrogen bonds between PF127 and HPMC molecules, causing polymers to aggregate and form micelles. Upon continuous heating, the sizes and numbers of micelles increase, and a stiff gel is produced.
The rheology values as a function of temperature regarding F2 (Figure 2A–C) showed that the addition of NPs to gel could enhance its mechanical features; accordingly, its viscosity value and gel strength were higher than that of F3. Consequently, F2 showed lower temperature for gel formation, higher gel strength, and higher viscosity than F3.46
F1, F2, and F3 showed sustained release compared to normal drug suspension (F4). Notably, there was no burst effect, and constant drug release was achieved for F1.47 This was due to several reasons. Firstly, the physical barrier of CS that coated PLGA NPs limited the diffusion/erosion process of drug from NPs.48 Another reason is the tendency of CS to hydrate, limitedly swell when it comes in contact with high-pH dissolution media of a high pH, then form a gelatinous mass that slows drug diffusion.49,50
F3 also showed sustained release due to the formation of a three-dimensional physical network of rigid gel. Mixing HPMC with water slows gel dissolution. Another reason is the swelling effect of PF127, which is always in a controlled manner to delay drug diffusion. Therefore, we achieved a controlled and sustained AT release rate for F3.
F2 showed the slowest release profile because of the aforementioned effect of NPs in addition to PF127-HPMC gel in hindering and controlling drug release. We also observed that gel dissolution was retarded up to 24 hrs at pH 7.4 because of the higher hydrophilicity of PF127 when mixed with water. This hydrophilicity opposed gel formation under high temperature 35–36±0.5°C.51
The data obtained from dissolution studies of all formulations were fitted into various release kinetics models, and the regression coefficients were calculated. From these results, we concluded that the release of AT from F1 follows a zero-order, diffusion, and swelling mechanism.38 More than one mechanism exerts a dominant effect on the release pattern of the drug from biodegradable polymeric systems; for example, diffusion-controlled release from the matrix of polymers is followed by readsorption, then the drug is desorbed from the surface of formed NPs, leading to degradation and dissolution/erosion of the polymeric network.52 Nevertheless, drug release from the formed NPs is a combination of drug diffusion and surface erosion; CS forms a coat that acts as a physical barrier that swells or erodes, consequently restraining the release of drug from NPs. Swelling and erosion occur due to the ability of CS to absorb water, resulting in weakening the bonds between CS and PLGA followed by erosion of the NP surface, resulting in drug diffusion.53
AT release from F2 is also explained by zero-order modeling. This may be because the gel system continues to swell in a controlled manner, simultaneous with prompt polymer dissolution. This result indicated a two-phase drug release process. First, AT was released from the nanoparticles into the network structure of the colloidal gel; then, the released drug diffused through the micro-pipes of the gel network and was finally transported to the dissolution medium. It seems that the second phase may be the rate-limiting step of this proposed theory. In addition, the gel matrix could reduce the sudden initial burst of drug release associated with temperature-sensitive gels.54–56
In vitro release of AT from F3 also followed zero-order release kinetics. A similar effect was noted in several studies57–60 that reported a robust correspondence between the percent of gel dissolved versus percent of drug released in a specific time span that indicates a gel dissolution-controlled release mechanism. Polymer (PF127-HPMC) dissolution could be explained well with this theory, in which drug–polymer–solvent systems demonstrate release of drug that may be diffusion- or dissolution-controlled, compared to slow diffusion of the drug over the micro pipes of gel networks and adsorption of the drug onto nanoparticles. Blending with polymeric gelling agents enables a sustained release pattern of AT.
All formulations followed non-Fickian anomalous diffusion-controlled mechanism since values of n≥0.5, indicating diffusion-controlled and swelling-controlled drug release.15,16,38
The in vivo results may be due to the unparalleled properties of CS that formed the nanoparticles,61 including bioadhesivity and penetration enhancement. CS carries a positive charge, so the bioadhesive property is due to its interaction with the negatively charged mucin on the eye surface.61,62 The penetration-enhancing property of CS enables it to open the tight junctions between cells and enhance cell membrane permeability.63 The percent inhibition of PMN migration by nanoparticles in a thermosensitive gel formulation (F2) was much higher than observed with other formulations.
The in vivo results conclusively demonstrate the effectiveness of nanoparticles in targeted, sustained drug release. The enhanced anti-inflammatory efficacy of gel-containing nanoparticles may be due to two factors. The first is that nanoparticles act as drug reservoirs for continuous drug release.62 The second is the high viscosity and bioadhesiveness characteristics of the gel matrix containing PF127/HPMC. These properties enhance the residence time of the formulations with the ocular surface, which improves their pharmacological anti-inflammatory effects.64 Similar observations were reported in several studies.65 Similarly, nanoparticle formulation treatment led to statistically significant reductions (p < 0.05) in PMN count and protein content after 3 hrs, suggesting an excellent potential of gel-formulating nanoparticles in anti-inflammatory applications.
Conclusions
CS-coated PLGA nanoparticles incorporated in thermosensitive and bioadhesive systems were successfully developed to achieve ocular drug targeting of highly entrapped AT calcium and sustain its delivery over a long period of time.
The prepared formulation was non-irritant and prolonged drug retention at the corneal site. Furthermore, AT calcium nanoparticles possessed high anti-inflammatory activity compared to the other formulations. AT-PLGA-CS-NPs thermosensitive gel is an efficient ocular anti-inflammatory targeting agent for controlled drug delivery.
Ethics Approval
All animal work was conducted in accordance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals and was approved by the Ethical Committee of Faculty of Pharmacy, The British University in Egypt.
Author Contributions
We declare that this work was done by the authors named in this article, and all liabilities pertaining to claims relating to the content of this article will be borne by the authors. All authors conceived, designed, and conducted the study, and wrote, reviewed, and approved the manuscript including figures and tables. All authors contributed towards data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Funding
This work was funded by the authors named in this article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Veillard N, Mach F. Statins: the new aspirin? Cell Mol Life Sci. 2002;59(11):1771–1786. doi:10.1007/PL00012505
2. Vollmer T, Key L, Durkalski V, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363(9421):1607–1608. doi:10.1016/S0140-6736(04)16205-3
3. McCarey DW, McInnes IB, Madhok R, et al. Trial of Atorvastatin in Rheumatoid Arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet. 2004;363(9426):2015–2021. doi:10.1016/S0140-6736(04)16449-0
4. Mach F. Statins as immunomodulatory agents. Circulation. 2004;109(21suppl 1):
5. Kwak BR, Mulhaupt F, Mach F. Atherosclerosis: anti-inflammatory and immunomodulatory activities of statins. Autoimmun Rev. 2003;2(6):332–338. doi:10.1016/S1568-9972(03)00049-1
6. Schieffer B, Drexler H. Role of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors, angiotensin-converting enzyme inhibitors, cyclooxygenase-2 inhibitors, and aspirin in anti-inflammatory and immunomodulatory treatment of cardiovascular diseases. Am J Cardiol. 2003;91(12):12–18. doi:10.1016/S0002-9149(03)00429-6
7. Farsaei S, Khalili H, Farboud ES, Karimzadeh I, Beigmohammadi MT. Efficacy of topical atorvastatin for the treatment of pressure ulcers: a randomized clinical trial. Pharmacotherapy. 2014;34(1):19–27. doi:10.1002/phar.2013.34.issue-1
8. Kohno H, Sakai T, Saito S, Okano K, Kitahara K. Treatment of experimental autoimmune uveoretinitis with atorvastatin and lovastatin. Exp Eye Res. 2007;84(3):569–576. doi:10.1016/j.exer.2006.11.011
9. Farsaei S, Khalili H, Farboud ES. Potential role of statins on wound healing: review of the literature. Int Wound J. 2012;9(3):238–247. doi:10.1111/iwj.2012.9.issue-3
10. Abraham S, Furtado S, Bharath S, Basavaraj B, Deveswaran R, Madhavan V. Sustained ophthalmic delivery of ofloxacin from an ion-activated in situ gelling system. Pak J Pharm Sci. 2009;22:2.
11. Maurice DM. Prolonged-action drops. Int Ophthalmol Clin. 1993;33(4):81–91. doi:10.1097/00004397-199303340-00009
12. Nadkarni SR, Yalkowsky SH. Controlled delivery of pilocarpine. 1. In vitro characterization of Gelfoam® matrices. Pharm Res. 1993;10(1):109–112. doi:10.1023/A:1018985332376
13. Davies NM, Farr SJ, Hadgraft J, Kellaway IW. Evaluation of mucoadhesive polymers in ocular drug delivery. II. Polymer-coated vesicles. Pharm Res. 1992;9(9):1137–1144. doi:10.1023/A:1015891419676
14. Li Z, Tao W, Zhang D, et al. The studies of PLGA nanoparticles loading atorvastatin calcium for oral administration in vitro and in vivo. Asian J Pharm Sci. 2017;12(3):285–291. doi:10.1016/j.ajps.2016.08.006
15. Khan N, Khanna K, Bhatnagar A, Ahmad FJ, Ali A. Chitosan coated PLGA nanoparticles amplify the ocular hypotensive effect of forskolin: statistical design, characterization and in vivo studies. Int J Biol Macromol. 2018;116:648–663. doi:10.1016/j.ijbiomac.2018.04.122
16. Bathool A, Vishakante GD, Khan MS, Shivakumar H. Development and characterization of atorvastatin calcium loaded chitosan nanoparticles for sustain drug delivery. Adv Mat Lett. 2012;3(6):466–470. doi:10.5185/amlett
17. Taghavi S, Ramezani M, Alibolandi M, Abnous K, Taghdisi SM. Chitosan-modified PLGA nanoparticles tagged with 5TR1 aptamer for in vivo tumor-targeted drug delivery. Cancer Lett. 2017;400:1–8. doi:10.1016/j.canlet.2017.04.008
18. Van de Ven H, Paulussen C, Feijens P, et al. PLGA nanoparticles and nanosuspensions with amphotericin B: potent in vitro and in vivo alternatives to fungizone and AmBisome. J Control Release. 2012;161(3):795–803. doi:10.1016/j.jconrel.2012.05.037
19. Arafa MG, Mousa HA, Afifi NN. Preparation of PLGA-chitosan based nanocarriers for enhancing antibacterial effect of ciprofloxacin in root canal infection. Drug Deliv. 2020;27(1):26–39. doi:10.1080/10717544.2019.1701140
20. Arafa MG, Ayoub BM. Nano-vesicles of salbutamol sulphate in metered dose inhalers: formulation, characterization and in vitro evaluation. Int J App Pharm. 2017;9(6):100–105. doi:10.22159/ijap.2017v9i6.22448
21. Song XR, Cai Z, Zheng Y, et al. Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur J Pharm Sci. 2009;37(3–4):300–305. doi:10.1016/j.ejps.2009.02.018
22. Arafa MG, Ayoub BM. DOE optimization of nano-based carrier of pregabalin as hydrogel: new therapeutic & chemometric approaches for controlled drug delivery systems. Sci Rep. 2017;7:41503. doi:10.1038/srep41503
23. Song X, Zhao Y, Wu W, et al. PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: systematic study of particle size and drug entrapment efficiency. Int J Pharm. 2008;350(1–2):320–329. doi:10.1016/j.ijpharm.2007.08.034
24. Arafa MG, El-Kased RF, Elmazar M. Thermoresponsive gels containing gold nanoparticles as smart antibacterial and wound healing agents. Sci Rep. 2018;8(1):13674. doi:10.1038/s41598-018-31895-4
25. Varshosaz J, Tavakoli N, Saidian S. Development and physical characterization of a periodontal bioadhesive gel of metronidazole. Drug Deliv. 2002;9(2):127–133. doi:10.1080/10426500290095601
26. Bhandwalkar MJ, Avachat AM. Thermoreversible nasal in situ gel of venlafaxine hydrochloride: formulation, characterization, and pharmacodynamic evaluation. AAPS PharmSciTech. 2013;14(1):101–110. doi:10.1208/s12249-012-9893-1
27. Arafa MG, Ghalwash D, El-Kersh DM, Elmazar M. Propolis-based niosomes as oromuco-adhesive films: a randomized clinical trial of a therapeutic drug delivery platform for the treatment of oral recurrent aphthous ulcers. Sci Rep. 2018;8(1):18056. doi:10.1038/s41598-018-37157-7
28. Liu Z, Li J, Nie S, Liu H, Ding P, Pan W. Study of an alginate/HPMC-based in situ gelling ophthalmic delivery system for gatifloxacin. Int J Pharm. 2006;315(1):12–17. doi:10.1016/j.ijpharm.2006.01.029
29. Ayoub BM, Mowaka S, Safar MM, et al. Repositioning of Omarigliptin as a once-weekly intranasal Anti-parkinsonian Agent. Sci Rep. 2018;8(1):8959. doi:10.1038/s41598-018-27395-0
30. Arafa MG, Ayoub BM. Bioavailability study of niosomal salbutamol sulfate in metered dose inhaler: controlled pulmonary drug delivery. J Aerosol Med Pulm Drug Deliv. 2018;31(2):114–115. doi:10.1089/jamp.2017.1448
31. Abraham MH, Hassanisadi M, Jalali-Heravi M, Ghafourian T, Cain WS, Cometto-Muñiz JE. Draize rabbit eye test compatibility with eye irritation thresholds in humans: a quantitative structure-activity relationship analysis. Toxicol Sci. 2003;76(2):384–391. doi:10.1093/toxsci/kfg242
32. Mathurm M, Gilhotra RM. Glycerogelatin-based ocular inserts of aceclofenac: physicochemical, drug release studies and efficacy against prostaglandin E2-induced ocular inflammation. Drug Deliv. 2011;18(1):54–64. doi:10.3109/10717544.2010.509366
33. Katara R, Sachdeva S, Majumdar DK. Enhancement of ocular efficacy of aceclofenac using biodegradable PLGA nanoparticles: formulation and characterization. Drug Deliv Transl Res. 2017;7(5):632–641. doi:10.1007/s13346-017-0416-1
34. Sood R. Medical Laboratory Technology: Methods and Interpretations. New Delhi: Jaypee Brothers; 1999:169–177.
35. Orsonneau J, Douet P, Massoubre C, Lustenberger P, Bernard S. An improved pyrogallol red-molybdate method for determining total urinary protein. Clin Chem. 1989;35(11):2233–2236. doi:10.1093/clinchem/35.11.2233
36. De Muth JE. Basic Statistics and Pharmaceutical Statistical Applications. Chapman and Hall/CRC; 2014.
37. Ali J, Bhatnagar A, Kumar N, Ali A. Chitosan nanoparticles amplify the ocular hypotensive effect of cateolol in rabbits. Int J Biol Macromol. 2014;65:479–491. doi:10.1016/j.ijbiomac.2014.02.002
38. Ahmed AB, Konwar R, Sengupta R. Atorvastatin calcium loaded chitosan nanoparticles: in vitro evaluation and in vivo pharmacokinetic studies in rabbits. Braz J Pharm Sci. 2015;51(2):467–477. doi:10.1590/S1984-82502015000200024
39. Srinivasan BD, Kulkarni PS. The role of arachidonic acid metabolites in the mediation of the polymorphonuclear leukocyte response following corneal injury. Invest Ophthalmol Vis Sci. 1980;19(9):1087–1093.
40. Galindo-Rodriguez S, Allemann E, Fessi H, Doelker E. Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification-diffusion, and nanoprecipitation methods. Pharm Res. 2004;21(8):1428–1439. doi:10.1023/B:PHAM.0000036917.75634.be
41. Sharma N, Madan P, Lin S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study. Asian J Pharm Sci. 2016;11(3):404–416. doi:10.1016/j.ajps.2015.09.004
42. Gryparis E, Mattheolabakis G, Bikiaris D, Avgoustakis K. Effect of conditions of preparation on the size and encapsulation properties of PLGA-mPEG nanoparticles of cisplatin. Drug Deliv. 2007;14(6):371–380. doi:10.1080/10717540701202937
43. Ilium L. Chitosan and its use as a pharmaceutical excipient. Pharm Res. 1998;15(9):1326–1331. doi:10.1023/A:1011929016601
44. Gupta S, Samanta MK, Raichur AM. Dual-drug delivery system based on in situ gel-forming nanosuspension of forskolin to enhance antiglaucoma efficacy. AAPS Pharm Sci Tech. 2010;11(1):322–335. doi:10.1208/s12249-010-9388-x
45. Mohanambal E, Arun K, Sathali AHA. Formulation and evaluation of pH-triggered in situ gelling system of levofloxacin. Indian J Pharm Educ Res. 2011;45(1):58–64.
46. Turabee MH, Jeong TH, Ramalingam P, Kang JH, Ko YT. N, N, N-trimethyl chitosan embedded in situ Pluronic F127 hydrogel for the treatment of brain tumor. Carbohydr Polym. 2018;203.
47. Durán-Lobato M, Martín-Banderas L, Gonçalves LM, Fernández-Arévalo M, Almeida AJ. Comparative study of chitosan-and PEG-coated lipid and PLGA nanoparticles as oral delivery systems for cannabinoids. J Nanoparticle Res. 2015;17(2):61. doi:10.1007/s11051-015-2875-y
48. Vandervoort J, Yoncheva K, Ludwig A. Influence of the homogenisation procedure on the physicochemical properties of PLGA nanoparticles. Chem Pharm Bull. 2004;52(11):1273–1279. doi:10.1248/cpb.52.1273
49. Wang W, Luo C, Shao S, Zhou S. Chitosan hollow nanospheres fabricated from biodegradable poly-D, L-lactide-poly (ethylene glycol) nanoparticle templates. Eur J Pharm Biopharm. 2010;76(3):376–383. doi:10.1016/j.ejpb.2010.08.009
50. Ayoub BM, Mowaka S, Arafa MG. Factorial design optimization of micelle enhanced synchronous spectrofluorimetric assay of omarigliptin: applied to content uniformity testing and in vitro drug release. Luminescence. 2018;33(4):797–805. doi:10.1002/bio.3479
51. Klouda L, Mikos AG. Thermoresponsive hydrogels in biomedical applications. Eur J Pharm Biopharm. 2008;68(1):34–45. doi:10.1016/j.ejpb.2007.02.025
52. Warsi MH, Anwar M, Garg V, et al. Dorzolamide-loaded PLGA/vitamin E TPGS nanoparticles for glaucoma therapy: pharmacoscintigraphy study and evaluation of extended ocular hypotensive effect in rabbits. Colloids Surf B Biointerfaces. 2014;122:423–431. doi:10.1016/j.colsurfb.2014.07.004
53. Mohammed MA, Syeda J, Wasan KM, Wasan EK. An overview of chitosan nanoparticles and its application in non-parenteral drug delivery. Pharmaceutics. 2017;9(4):53. doi:10.3390/pharmaceutics9040053
54. Tao SL, Desai TA. Microfabricated drug delivery systems: from particles to pores. Adv Drug Deliv Rev. 2003;55(3):315–328. doi:10.1016/S0169-409X(02)00227-2
55. Wang Q, Wang J, Lu Q, Detamore MS, Berkland C. Injectable PLGA based colloidal gels for zero-order dexamethasone release in cranial defects. Biomaterials. 2010;31(18):4980–4986. doi:10.1016/j.biomaterials.2010.02.052
56. Zhu S, Tian H, Dou M, Huang G. Chitosan temperature-sensitive gels: reduce the burst release of microspheres containing lornoxicam and enhance drug targeting. MOJ Bioequiv Availab. 2018;5(1):00076.
57. Akash MSH, Rehman K, Sun H, Chen S. Assessment of release kinetics, stability and polymer interaction of poloxamer 407-based thermosensitive gel of interleukin-1 receptor antagonist. Pharm Dev Technol. 2014;19(3):278–284. doi:10.3109/10837450.2013.775158
58. Barichello JM, Morishita M, Takayama K, Nagai T. Absorption of insulin from Pluronic F-127 gels following subcutaneous administration in rats. Int J Pharm. 1999;184(2):189–198. doi:10.1016/S0378-5173(99)00119-2
59. Nie S, Hsiao WW, Pan W, Yang Z. Thermoreversible Pluronic® F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: in vitro drug release, cell cytotoxicity, and uptake studies. Int J Nanomedicine. 2011;6:151. doi:10.2147/IJN.S25646
60. Liu Y, Lu W-L, Wang J-C, et al. Controlled delivery of recombinant hirudin based on thermo-sensitive Pluronic® F127 hydrogel for subcutaneous administration: in vitro and in vivo characterization. J Control Release. 2007;117(3):387–395. doi:10.1016/j.jconrel.2006.11.024
61. Alonso MJ, Sánchez A. The potential of chitosan in ocular drug delivery. J Pharm Pharmacol. 2003;55(11):1451–1463. doi:10.1211/0022357022476
62. De Campos AM, Sánchez A, Alonso M. Chitosan nanoparticles: a new vehicle for the improvement of the delivery of drugs to the ocular surface. Application to cyclosporin A. Int J Pharm. 2001;224(1–2):159–168. doi:10.1016/S0378-5173(01)00760-8
63. de la Fuente M, Raviña M, Paolicelli P, Sanchez A, Seijo B, Alonso MJ. Chitosan-based nanostructures: a delivery platform for ocular therapeutics. Adv Drug Deliv Rev. 2010;62(1):100–117. doi:10.1016/j.addr.2009.11.026
64. Budai L, Hajdú M, Budai M, et al. Gels and liposomes in optimized ocular drug delivery: studies on ciprofloxacin formulations. Int J Pharm. 2007;343(1–2):34–40. doi:10.1016/j.ijpharm.2007.04.013
65. Gupta AK, Madan S, Majumdar D, Maitra A. Ketorolac entrapped in polymeric micelles: preparation, characterisation and ocular anti-inflammatory studies. Int J Pharm. 2000;209(1–2):1–14. doi:10.1016/S0378-5173(00)00508-1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.