Back to Journals » International Journal of Nanomedicine » Volume 13
Chitosan-based nanoformulated (–)-epigallocatechin-3-gallate (EGCG) modulates human keratinocyte-induced responses and alleviates imiquimod-induced murine psoriasiform dermatitis
Authors Chamcheu JC ![]() , Siddiqui IA, Adhami VM, Esnault S, Bharali DJ, Babatunde AS
, Siddiqui IA, Adhami VM, Esnault S, Bharali DJ, Babatunde AS ![]() , Adame S
, Adame S ![]() , Massey RJ, Wood GS, Longley BJ, Mousa SA
, Massey RJ, Wood GS, Longley BJ, Mousa SA ![]() , Mukhtar H
, Mukhtar H
Received 24 February 2018
Accepted for publication 19 April 2018
Published 20 July 2018 Volume 2018:13 Pages 4189—4206
DOI https://doi.org/10.2147/IJN.S165966
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Jean Christopher Chamcheu,1,2,* Imtiaz A Siddiqui,1,* Vaqar M Adhami,1,* Stephane Esnault,3 Dhruba J Bharali,4 Abiola S Babatunde,1,5 Stephanie Adame,1 Randall J Massey,6 Gary S Wood,1 B Jack Longley,1 Shaker A Mousa,4 Hasan Mukhtar1
1Department of Dermatology, School of Medicine and Public Health, University of Wisconsin, and the Middleton VA Medical Center, Madison, WI, USA; 2Department of Basic Pharmaceutical Sciences, School of Pharmacy, College of Health and Pharmaceutic Sciences, University of Louisiana at Monroe, Monroe, LA, USA; 3Department of Medicine, Division of Allergy, Pulmonary and Critical Care Medicine, The University of Wisconsin–Madison School of Medicine and Public Health, Madison, WI, USA; 4The Pharmaceutical Research Institute, Albany College of Pharmacy and Health Sciences, Albany, NY, USA; 5Department of Hematology, University of Ilorin, Ilorin, Nigeria; 6Electron Microscope Facility, Medical School Research Support Progs, School of Medicine and Public Health, University of Wisconsin, and the Middleton VAMedical Center, Madison, WI, USA
*These authors contributed equally to this work
Background: Psoriasis is a chronic and currently incurable inflammatory skin disease characterized by hyperproliferation, aberrant differentiation, and inflammation, leading to disrupted skin barrier function. The use of natural agents that can abrogate these effects could be useful for the treatment of psoriasis. Earlier studies have shown that treatment of keratinocytes and mouse skin with the green tea polyphenol (-)-epigallocatechin-3-gallate (EGCG) mitigated inflammation and increased the expression of caspase-14 while promoting epidermal differentiation and cornification. However, bioavailability issues have restricted the development of EGCG for the treatment of psoriasis.
Materials and methods: To overcome these limitations, we employed a chitosan-based polymeric nanoparticle formulation of EGCG (CHI-EGCG-NPs, hereafter termed nanoEGCG) suitable for topical delivery for treating psoriasis. We investigated and compared the efficacy of nanoEGCG versus native or free EGCG in vitro and in an in vivo imiquimod (IMQ)-induced murine psoriasis-like dermatitis model. The in vivo relevance and efficacy of nanoEGCG formulation (48 µg/mouse) were assessed in an IMQ-induced mouse psoriasis-like skin lesion model compared to free EGCG (1 mg/mouse).
Results: Like free EGCG, nanoEGCG treatment induced differentiation, and decreased proliferation and inflammatory responses in cultured keratinocytes, but with a 4-fold dose advantage. Topically applied nanoEGCG elicited a significant (p<0.01) amelioration of psoriasiform pathological markers in IMQ-induced mouse skin lesions, including reductions in ear and skin thickness, erythema and scales, proliferation (Ki-67), infiltratory immune cells (mast cells, neutrophils, macrophages, and CD4+ T cells), and angiogenesis (CD31). We also observed increases in the protein expression of caspase-14, early (keratin-10) and late (filaggrin and loricrin) markers of differentiation, and the activator protein-1 factor (JunB). Importantly, a significant modulation of several psoriasis-related inflammatory cytokines and chemokines was observed compared to the high dose of free EGCG (p<0.05). Taken together, topically applied nanoEGCG displayed a >20-fold dose advantage over free EGCG.
Conclusion: Based on these observations, our nanoEGCG formulation represents a promising drug-delivery strategy for treating psoriasis and possibly other inflammatory skin diseases.
Keywords: chitosan nanoparticles, topical delivery of chitosan nanoformulated EGCG, psoriasis-like skin inflammation, phytochemical treatment of psoriasis, normal human epidermal keratinocytes, differentiation, anti-inflammatory action
Erratum for this paper has been published.
Introduction
Psoriasis is a common, chronic, and currently incurable inflammatory skin disease that affects an estimated 125 million people worldwide, including ≈ 7.5 million in the USA alone.1–3 Even though the etiology of psoriasis is incompletely understood, it is known to be characterized by increased proliferation and aberrant differentiation of epidermal keratinocytes, as well as infiltration of immune cells and increased angiogenesis in the skin.4–7 There is evidence that inflammatory mediators produced by keratinocytes and infiltrating immune cells play a critical role in the immunopathogenesis of psoriasis.8–11 Despite advances in understanding psoriasis pathophysiology, curative treatments remain elusive, especially for mild to moderate cases for which powerful injectable biological therapies are not approved.12 Because inflamed psoriatic skin lesions in patients generally occur in visible skin sites, patients often experience significant emotional and social incapacities due to their condition. Standard of care for mild/moderate psoriasis mainly relies on palliative treatment involving psoralen and ultraviolet A therapy, topical treatment with small molecules, corticosteroids, retinoids, derivatives of vitamin D3, and/or concomitant immunomodulatory therapy.13–16 These therapies are limited by low efficacy and/or high relapse rates, high costs, and adverse side effects including relapse, as well as the chronic nature of these treatments, leading to widespread frustration and dissatisfaction among patients.17 These unmet challenges urgently necessitate the development of new mechanism-based treatments and drug delivery strategies. One such approach is to explore the use of non-toxic, natural botanicals that possess the ability to abrogate or mitigate multiple aspects of psoriasis.18
Earlier studies have shown that treatment of normal human epidermal keratinocytes (NHEKs) and mouse skin with (−)-epigallocatechin-3-gallate (EGCG), the main catechin found in green tea, induced the expression of critically important keratinocyte differentiation markers, including caspase-14 and p57/KIP2.19–21 Furthermore, studies in skin cancer cells, ultraviolet-irradiated human and IL-12 knockout mouse skin, as well as human vascular epithelial cells, demonstrated that EGCG promotes immunoregulatory/anti-neoplastic functions and anti-angiogenic effects.22–26 However, bioavailability issues have restricted the development and delivery of EGCG for the treatment of psoriasis. To overcome these limitations, we utilized an innovative chitosan-based nanocarrier encapsulating (−)-epigallocatechin-3-gallate (CHI-EGCG-NPs, hereafter referred to as nanoEGCG)27,28 as a novel topical delivery vehicle for the treatment of psoriasis. Chitosan is a naturally occurring N-deacetylated derivative of chitin composed of mainly glucosamine units. Its properties as an inert, biodegradable cationic polymeric nanoparticle have encouraged its utilization as a topical drug delivery vehicle for dermatological ailments.29 Another advantage is that at physiological pH, chitosan is positively charged as a result of the protonation of its primary amine groups, allowing for efficient encapsulation of negatively charged drugs via electrostatic interaction.29,30
In this study, the in vitro cytotoxicity, pro-differentiation and anti-inflammatory effects of chitosan-based nanoEGCG were first determined in vitro in NHEKs. We then examined the anti-psoriatic efficacy of topically applied nanoEGCG compared to free EGCG in an imiquimod (IMQ)-induced Balb/c mouse psoriasis-like skin inflammation model.
Materials and methods
Chemicals and reagents
EGCG, DMSO, sodium dodecyl sulfate, and MTT were procured from Sigma (St Louis, MO, USA). A 1,000-fold stock concentrate of EGCG was prepared in sterile endotoxin-free water, aliquoted, and stored at −20°C; other stock solutions were made in DMSO. Trypsin–EDTA and L-glutamine, conjugated secondary antibodies, and ProLong® Gold Antifade reagent containing DAPI were all obtained from Gibco BRL/Invitrogen (Carlsbad, CA, USA). IMQ cream 5% (Imiquad™) was purchased from Pattersson Veterinary Supply (Greeley, CO, USA). Human mix-matched 6-Plex and mouse 36-Plex ProcartaPlex™ multiplex bead-based immunoassay kits were purchased from eBioscience Affymetrix/eBioscience (Santa Clara, CA, USA). EpiLife growth medium and human keratinocyte growth supplement (HKGS) kit supplements were from Invitrogen Corp. CELLnTEC progenitor cell-culture medium (CnT-PR) was from ZenBio (Raleigh, NC, USA). A detailed list of primary antibodies used in this study is provided in Table S1. The Pierce BCA Protein Assay Kit and Invitrogen Novex precast Tris–glycine gels were purchased from Thermo Fisher Scientific (Rockford, IL, USA). The mini-protean precast Tris–glycine gel was from Bio-Rad Laboratories (Des Plaines, IL, USA). Ultrapure deionized water was used throughout all the experiments and all other reagents were of analytical grade or higher.
Preparation and characterization of nanoEGCG formulations
Preparation, optimization, and characterization of chitosan-based EGCG-loaded (nanoEGCG) or chitosan-based void (without EGCG) nanoparticles (CHI-Void-NPs) were essentially as previously described.27,28 Nanoparticles were then diluted in distilled water and further analyzed as detailed in the following subsections.
Size measurement with dynamic light scattering
The size distribution of CHI-Void-NPs and nanoEGCG in aqueous dispersion was determined using a Malvern Zetasizer (Malvern Instruments, Westborough, MA, USA). Two milliliters of nanoEGCG or CHI-Void NPs was placed in a 4-sided, clear, plastic cuvette and analyzed directly at 25°C. To prevent clumping (aggregation), the nanoparticle formulations were sonicated for several minutes, and the size of the nanoconjugates was found to be consistently less than 200 nm in diameter (Figure S1).
Zeta potential measurement
Zeta potentials of the chitosan-based nanoparticles encapsulating EGCG in aqueous dispersion were determined using the Malvern Zetasizer instrument. Approximately 1 mL of the nanoEGCG was placed in a disposable capillary zeta potential cell available from the Zetasizer Nano series. Measurements were performed at 25°C.
Size measurement with transmission electron microscopy (TEM)
The size and morphology of CHI-Void-NPs and nanoEGCG, as well as their localization and assessment of their ultrastructure in live cell cytoplasm, were determined with a JEOL JEM-100CX transmission electron microscope (JEOL, Peabody, MA, USA) as described previously.31 In brief, one drop of the nanoEGCG solution was mounted on a thin film of amorphous carbon deposited on a copper grid (300 mesh) then air dried, and the sample was examined directly. After 2 and 36 months, the size and morphology of the stored preparations were re-examined via negative staining using the drop method32 by placing a 2 μL drop of the sample on a Pioloform (Ted Pella, Redding, CA, USA) carbon-coated 300 mesh Cu grid (EMS, Hatfield, PA, USA). Samples were allowed to adsorb for 30 s, then the excess droplet was removed with the tip of a wedge-shaped filter paper. A 2 μL drop of Nano-W (Nanoprobes, Yaphank, NY, USA) was placed on the grid and the excess was again removed with fresh filter paper. After drying, the samples were placed in a Philips CM120 transmission electron microscope (FEI, Eindhoven, the Netherlands) at 80 kV, and photographed with an SIS MegaView III digital camera (EMSIS, Muenster, Germany). Size characterization analysis was performed using the SIS iTEM software (version 5.0) measurement feature calibrated to a grating replica on latex spheres (EMS).
Determination of encapsulation and loading efficiencies
The amount of EGCG bound to nanoparticles was determined by disintegrating the nanoparticles and measuring EGCG with ultraviolet–visible (UV-vis) spectroscopy. First, the amount of free EGCG (not encapsulated) was separated by filtering through a Millipore (Burlington, MA, USA) centrifugal device with a 3 kDa cut-off, assisted by centrifugation, at around 6,500 rpm for 30 min. A measured (known) amount of nanoEGCG particles was disintegrated by adding an acetic acid solution. The entire solution was passed through the Millipore filtration centrifugal device and centrifuged as before to separate the EGCG. The EGCG concentration of the centrifugate/filtrate was then determined using a Nanodrop 2000C UV-vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The encapsulation/entrapment and loading efficiencies were determined using the following formulae:
|
|
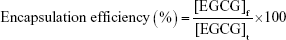
|
|
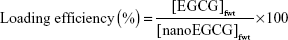
where [EGCG]f is the concentration of EGCG encapsulated in nanoEGCG and [EGCG]t is the total concentration of EGCG (meaning the total amount of EGCG added initially). [EGCG]fwt is the total weight of EGCG encapsulated in EGCG and [nanoEGCG]fwt is the total weight of the nanoparticles encapsulating EGCG (nanoEGCG). The results were used to determine the free EGCG equivalents for each batch of nanoEGCG particles.
Evaluation of storage stability, in vitro release kinetics, and cellular uptake of nanoparticles
To assess the stability of nanoEGCG, samples were added to 5 mL glass vials, sealed with plastic caps, and placed in an accelerated stability chamber at a temperature of 37°C±2°C with a relative humidity of 65%±5%. The in vitro release kinetics of EGCG from the nanoEGCG were investigated with a dialysis method with fresh release medium. In brief, 2.0 mL of a solution of nanoEGCG or free EGCG was dissolved in PBS solution, added to a dialysis bag (molecular weight cut-off=6–8 kDa), and dialyzed against 20 mL of release medium under gentle stirring with a paddle revolving at 100 rpm at a temperature of 37°C. At designated time intervals, 500 μL aliquots of external release medium were withdrawn, after which the dialysis buffer was replenished with an equal volume of fresh medium and returned to suspension. The withdrawn medium was centrifuged at 5,000× g for 5 min at 4°C, and the supernatant content of released EGCG in the supernatant was determined with high-performance liquid chromatography (HPLC) as in the Supplementary materials. The proportion as a percentage of cumulative EGCG release was plotted against dialysis time, and each experiment was executed in triplicate.
For the cellular uptake assay, NHEKs were cultured at 37°C under humidified air with 5% CO2 in serum-free CnT-PR growth medium supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and amphotericin (100 μg/mL) (Life Technologies, Carlsbad, CA, USA). NHEKs were harvested by trypsinization at optimal confluence and seeded into 6-well culture plates at a density of 5×104 cells/well. After 2 days, the cells were incubated with nanoEGCG at 0, 5, 10, 20, 40, and 80 μM (expressed as free EGCG equivalents) in growth medium for 12–24 h, followed by 2 washes in PBS to remove excess free EGCG or nanoEGCG. Cells were then disrupted by ultrasonication and centrifuged at 5,000× g for 6 min, and filtered supernatants were collected and analyzed with HPLC as described earlier27 to determine the cellular uptake of EGCG at each concentration of free EGCG equivalents.
Keratinocyte isolation, culture, treatment with 12-O-tetradecanoyl-phorbol-13-acetate (TPA) or recombinant human IL-22 (rhIL-22), and detection of secreted cytokines
Primary NHEKs used in this study were isolated from neonatal foreskin or adult skin biopsies and used to establish primary cultures as previously described,33 and were stored frozen until used. The samples were obtained from Meriter Hospital (Madison, WI, USA) following institutional guidelines under a University of Wisconsin–Madison-approved institutional review board protocol, and experiments were conducted with adherence to the principles of the Declaration of Helsinki. Cells were cultured in EpiLife medium supplemented with HKGS or in CnT-PR culture medium maintained at 37°C in a 5% CO2 humidified incubator.
Preparation of protein lysates and Western blotting
After treatments, cell or mouse skin lysates were prepared by homogenization. Protein concentrations were determined and Western blotting with various antisera was performed as previously described.34,35
In vivo anti-psoriatic activity studies
Ethical statement
The animal study in this research was carried out in accordance with the recommendations of the Guidelines for the Care and Use of Laboratory Animals. All protocols were approved by the University of Wisconsin–Madison School of Medicine and Public Health Animal Care and Use Committee.
Mouse IMQ-induced skin inflammation model and treatment with free EGCG and nanoEGCG
The in vivo efficacy studies were carried out using 6–8-week-old BALB/c mice (Harlan Laboratories, Madison, WI, USA). The mice were housed in cages at 25°C±2°C and 45% relative humidity, with a 12 h light/dark cycle, provided with food and water ad libitum, and maintained under pathogen-free conditions. Protocols for induction of psoriasis-like skin inflammation and treatment were essentially as previously described,34 with slight modifications as detailed in the Supplementary materials. Mice were shaved and divided into 4 groups of 5 mice (Figure S5). In brief, starting at day 3, control mice (group 1) were treated with Vaseline alone (local Walgreens pharmacy), while groups 2, 3, and 4 (a total of 15 mice) received a daily topical dose of 20 mg and 62.5 mg of 5% IMQ cream (Aldara) on the right ear and shaved back skin, respectively. Another group not included in this result was group 5, where mice were treated with CHI-Void-NPs alone (the results were similar to group 1 and we preferred to present group 1 as a common control because the effect of free EGCG is also compared). Group 2 (IMQ+) continued to receive topical IMQ cream alone for a total of 14 days (until day 17) to achieve the optimal chronic inflammation. Starting on day 9, mice of groups 3 and 4 were co-treated, in addition to IMQ, with free EGCG (group 3: 1 mg/cm2 shaved skin/right ear area) or nanoEGCG (group 4: 48 μg/cm2 shaved skin/right ear area). All EGCG treatments for both groups 3 (IMQ+free EGCG) and 4 (IMQ+nanoEGCG) were delivered in 100 μL of PBS, 7 times a week, 3 h before the daily application of IMQ cream, until day 17. Mice were monitored daily throughout the 14 day treatment period.
Inflammation severity scoring and measurement of ear and skin thickness
The degree of severity of erythema and inflammation, as well as the presence of scales and skin hardness on the ears and skin of treated mice, were scored using a semi-quantitative Psoriasis Area and Severity Index scoring system from 0 to 4 based on their physical appearance: 0=no skin abnormalities, 1=slight, 2=moderate, 3=marked, and 4=severe. In addition, ear skin thickening was assessed on alternate days using an electronic digital micrometer (Caliper; Fisher Scientific, Madison, WI, USA). On day 17, all mice were euthanized, and skin and spleen were harvested as described previously.34
Skin and spleen histology, immunohistochemistry, and analysis
Formalin-fixed, paraffin-embedded skin and spleen sections were processed for immunohistochemistry, mounted, and visualized with a Zeiss light microscope, and automated images were acquired with 20×/40× objectives as described earlier.34 Data acquisition and image analysis using Nuance™/Inform platform software (PerkinElmer, Boston, MA, USA) technology were as described previously.34,36 Isotype controls were used for immunostaining (proportion of brown and green/red pixels for antigen staining) with values averaged from at least 5 fields for each tissue/group. Other stainings were as detailed in Supplementary materials.
ProcartaPlex multiplex bead-based immunoassay for cytokine and chemokine profiling
The effects of nanoEGCG versus free EGCG interventions on inflammatory responses in either TPA-activated NHEKs or the IMQ-induced psoriasis-like skin inflammation mouse model were analyzed essentially as previously described,34 and as detailed in the Supplementary materials. Cytokine/chemokine values obtained with immunoassay were normalized against established referenced standards to allow for comparisons between the studied groups.
Statistical analysis
Statistical analyses were carried out with GraphPad Prism, version 6.1 (GraphPad Software, La Jolla, CA, USA). All quantitative data are expressed as mean±standard error of the mean (SEM) or SD, and significant differences between treatment groups were determined with the Student’s t-test or analysis of variance with Bonferroni post hoc testing. A p-value <0.05 was considered significant.
Results
Preparation and physical characterization of CHI-Void-NPs and nanoEGCG
We previously reported the synthesis and characterization of nanoEGCG formulations suitable for oral delivery.27 Here, chitosan nanoparticles encapsulating EGCG were synthesized by the gelation method. The size/size distribution and morphology of these nanoparticles were examined with dynamic light scattering (Figures 1A and S1A). As anticipated, the zeta potential of both CHI-Void-NPs (Figure S1B) and nanoEGCG (Figure 1B) was found to be positive as a result of chitosan having abundant free amine groups. Moreover, the size and morphology were further established with TEM, which showed that the size of the nanoparticles ranged between 80 nm and 225 nm in diameter, and most nanoparticles were more or less spherical (Figures 1C and S2). The encapsulation efficiency of the EGCG by the nanoparticles was found to be around 65%, while the loading efficiency was in the range of 8%–10% w/w. With the aid of UV-vis spectroscopy, the amount of EGCG encapsulated in the chitosan nanoparticles was determined (Figure 1D and E). The size, polydispersity factor/index, and zeta potentials of the CHI-Void-NPs and nanoEGCG are summarized in Figure 2A. Therefore, the positive charge is an indication of the superior bioadhesive properties of the nanoformulations, which is a prerequisite for topical delivery.
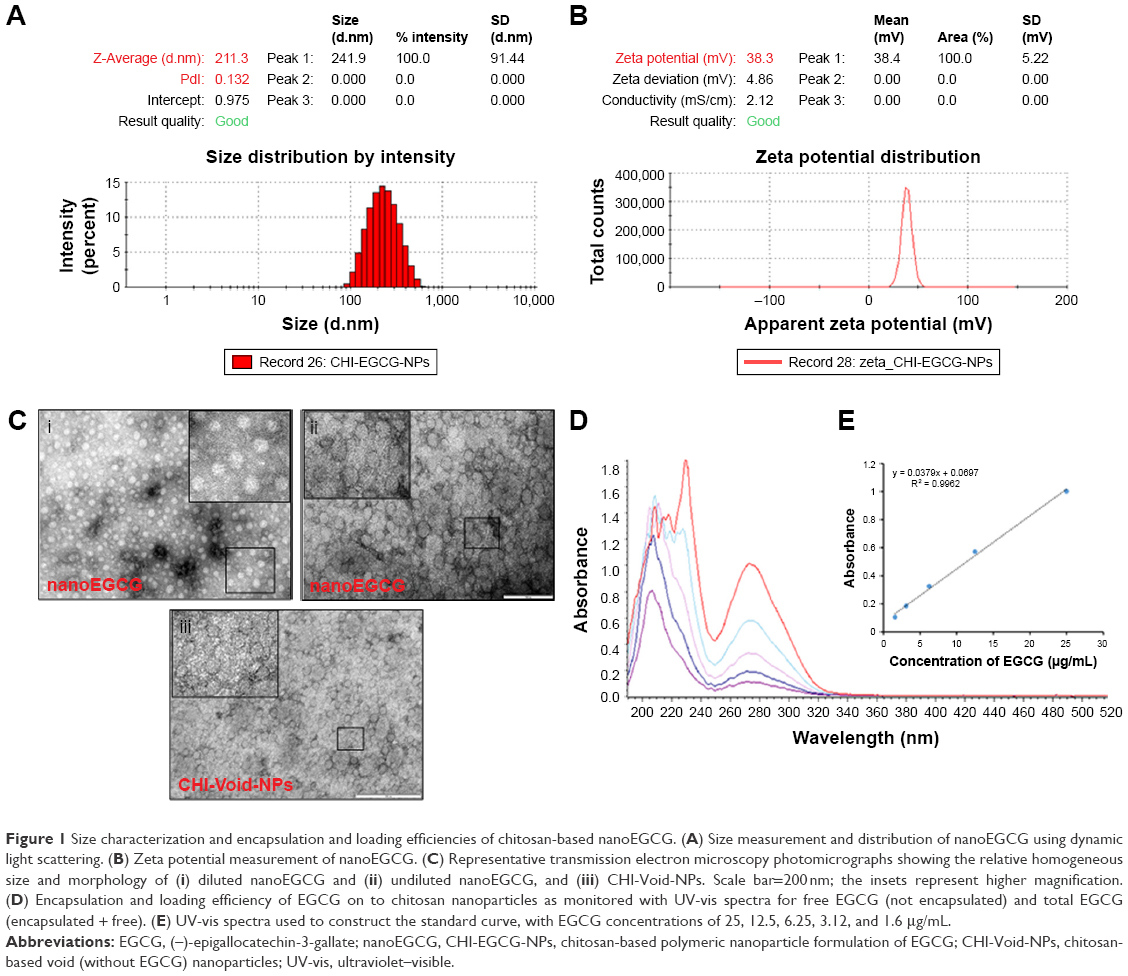 | Figure 1 Size characterization and encapsulation and loading efficiencies of chitosan-based nanoEGCG. (A) Size measurement and distribution of nanoEGCG using dynamic light scattering. (B) Zeta potential measurement of nanoEGCG. (C) Representative transmission electron microscopy photomicrographs showing the relative homogeneous size and morphology of (i) diluted nanoEGCG and (ii) undiluted nanoEGCG, and (iii) CHI-Void-NPs. Scale bar=200nm; the insets represent higher magnification. (D) Encapsulation and loading efficiency of EGCG on to chitosan nanoparticles as monitored with UV-vis spectra for free EGCG (not encapsulated) and total EGCG (encapsulated + free). (E) UV-vis spectra used to construct the standard curve, with EGCG concentrations of 25, 12.5, 6.25, 3.12, and 1.6 μg/mL. |
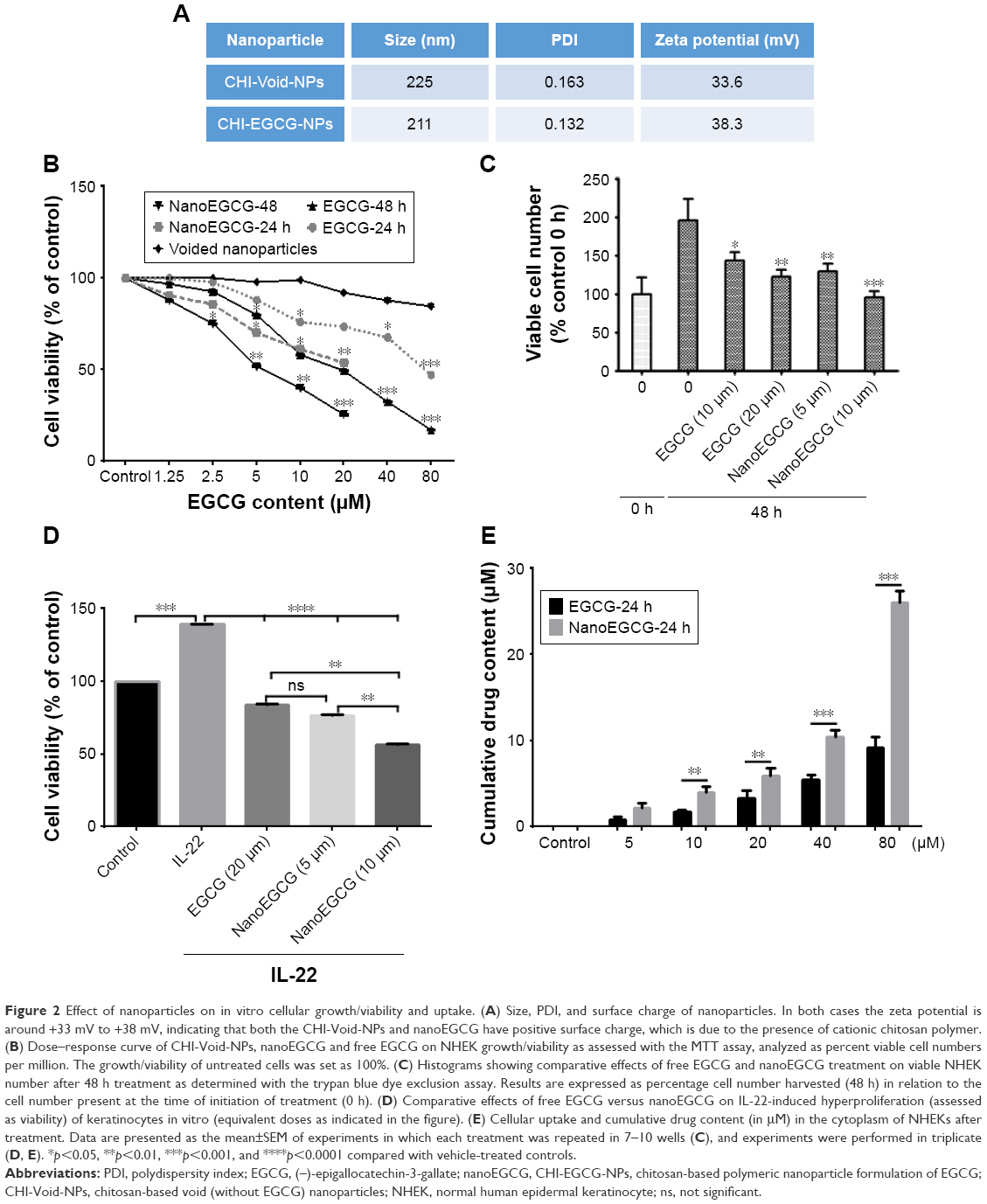 | Figure 2 Effect of nanoparticles on in vitro cellular growth/viability and uptake. (A) Size, PDI, and surface charge of nanoparticles. In both cases the zeta potential is around +33 mV to +38 mV, indicating that both the CHI-Void-NPs and nanoEGCG have positive surface charge, which is due to the presence of cationic chitosan polymer. (B) Dose–response curve of CHI-Void-NPs, nanoEGCG and free EGCG on NHEK growth/viability as assessed with the MTT assay, analyzed as percent viable cell numbers per million. The growth/viability of untreated cells was set as 100%. (C) Histograms showing comparative effects of free EGCG and nanoEGCG treatment on viable NHEK number after 48 h treatment as determined with the trypan blue dye exclusion assay. Results are expressed as percentage cell number harvested (48 h) in relation to the cell number present at the time of initiation of treatment (0 h). (D) Comparative effects of free EGCG versus nanoEGCG on IL-22-induced hyperproliferation (assessed as viability) of keratinocytes in vitro (equivalent doses as indicated in the figure). (E) Cellular uptake and cumulative drug content (in μM) in the cytoplasm of NHEKs after treatment. Data are presented as the mean±SEM of experiments in which each treatment was repeated in 7–10 wells (C), and experiments were performed in triplicate (D, E). *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 compared with vehicle-treated controls. |
As depicted in Figure S3, we observed similar in vitro release kinetics of free EGCG and nanoEGCG as reported earlier.27 In brief, a very rapid and complete dissolution of free EGCG was observed within 2 h, whereas a controlled release rate of EGCG from nanoEGCG peaked at about 50% within 6 h, approaching a maximal release of 100% after 24 h.
Biological characterization, in vitro uptake, and anti-proliferative effects of nanoEGCG in cultured normal and activated keratinocytes
We next determined the biological significance of nanoEGCG by examining its effects on cellular growth/viability and differentiation, as well as the uptake and accumulation into cultured NHEKs over 24–48 h, and compared these effects to those of free EGCG using spectroscopic data (Figure 1D) to calculate the relative doses of EGCG delivered by the nanoparticles. Employing MTT (Figure 2B) and trypan blue dye exclusion (Figure 2C) assays, we observed that whereas CHI-Void-NPs had a negligible effect on cell growth, nanoEGCG markedly and dose- and time-dependently inhibited cell growth/viability, with IC50 values for 24 h and 48 h of 37 μM and 5.2 μM, respectively, compared to 72.1 μM and 20.2 μM for free EGCG (Figure 2B and C). The IC50 values for nanoEGCG were consistently 2–4-fold lower than those for free EGCG with respect to inhibition of cell growth after 24 h and 48 h of treatment, respectively.
Hyperproliferation is a key feature of psoriatic keratinocytes, both in vivo and ex vivo,37 and IL-22-induced proliferation is related to psoriasis-like changes in vitro. We therefore sought to determine the ability of nanoEGCG to inhibit IL-22-induced keratinocyte proliferation.38 Pretreatment of NHEKs with or without nanoEGCG (5–10 μM) or 4-fold higher free EGCG (20 μM) followed by co-treatment with or without rhIL-22 (20 ng/mL) for a total of 48 h significantly inhibited IL-22-induced proliferation (Figure 2D). These data suggest that nanoEGCG can significantly reduce the hyperproliferative phenotype orchestrated by IL-22. Again, the doses of EGCG required to achieve a superior effect were 4-fold less than those of free EGCG. Next, we used liquid chromatography–mass spectrometry to determine the amount of EGCG that actually accumulates in cells over a 24 h period of treatment with nanoEGCG or free EGCG (5–80 μM). Our data indicated that uptake of nanoEGCG was approximately 3-fold higher than uptake of free EGCG (Figure 2E). Further assessments of the cellular uptake and localization of nanoparticles into cultured NHEKs using TEM directly demonstrated the uptake and localization of dense nanoparticle-containing vesicles inside the cell cytoplasm (Figure 3A–D). Therefore, for these and subsequent studies, we assumed a 4-fold advantage in effectiveness of nanoEGCG over free EGCG.
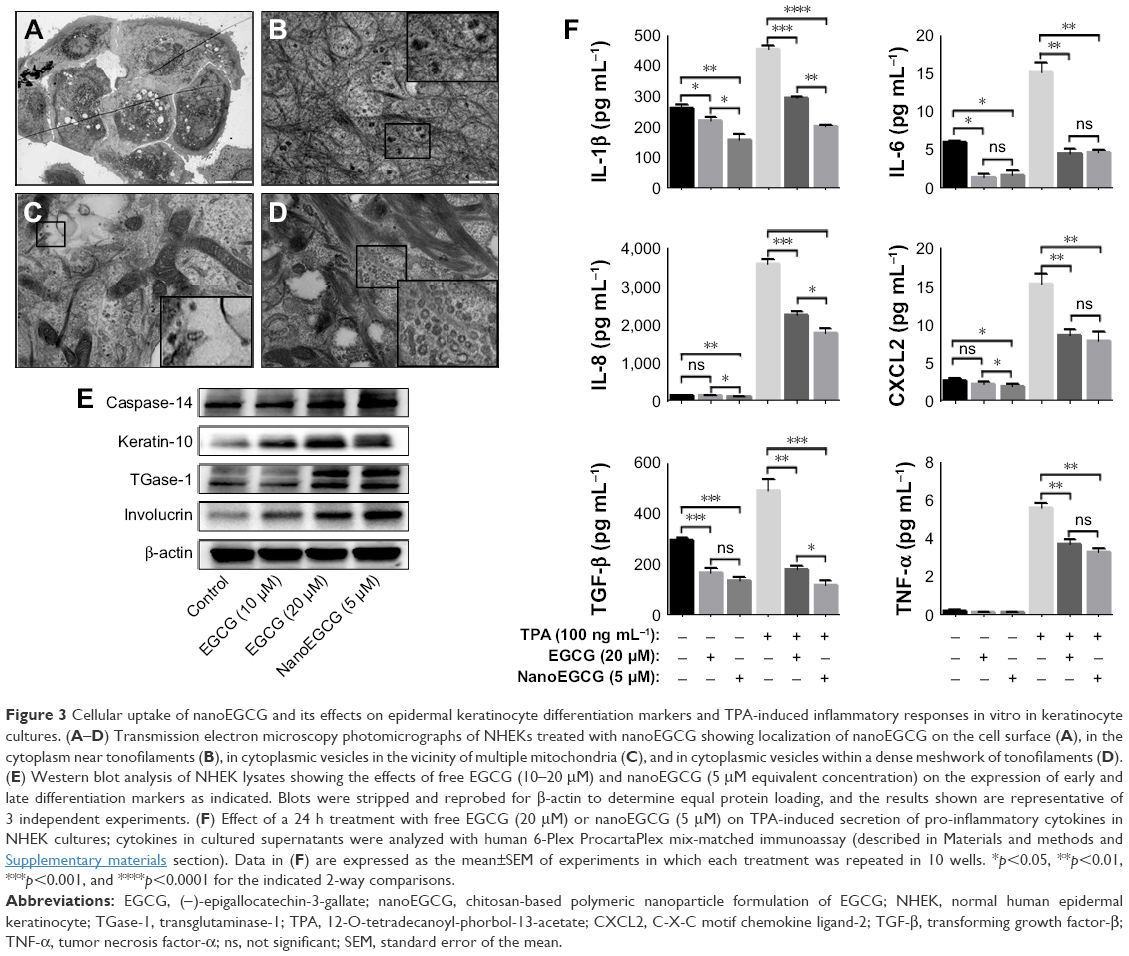 | Figure 3 Cellular uptake of nanoEGCG and its effects on epidermal keratinocyte differentiation markers and TPA-induced inflammatory responses in vitro in keratinocyte cultures. (A–D) Transmission electron microscopy photomicrographs of NHEKs treated with nanoEGCG showing localization of nanoEGCG on the cell surface (A), in the cytoplasm near tonofilaments (B), in cytoplasmic vesicles in the vicinity of multiple mitochondria (C), and in cytoplasmic vesicles within a dense meshwork of tonofilaments (D). (E) Western blot analysis of NHEK lysates showing the effects of free EGCG (10–20 μM) and nanoEGCG (5 μM equivalent concentration) on the expression of early and late differentiation markers as indicated. Blots were stripped and reprobed for β-actin to determine equal protein loading, and the results shown are representative of 3 independent experiments. (F) Effect of a 24 h treatment with free EGCG (20 μM) or nanoEGCG (5 μM) on TPA-induced secretion of pro-inflammatory cytokines in NHEK cultures; cytokines in cultured supernatants were analyzed with human 6-Plex ProcartaPlex mix-matched immunoassay (described in Materials and methods and Supplementary materials section). Data in (F) are expressed as the mean±SEM of experiments in which each treatment was repeated in 10 wells. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 for the indicated 2-way comparisons. |
NanoEGCG treatment induces expression of epidermal differentiation markers and inhibits TPA-induced inflammatory responses in cultured NHEKs
To determine whether nanoEGCG possesses pro-differentiative properties, NHEKs were treated with/without nanoEGCG (5–10 μM) or free EGCG (10–40 μM). The morphology and expression of early and late epidermal keratinocyte differentiation markers were examined, including caspase-14, keratin-10 (K10), transglutaminase-1, and involucrin, all of which are known to be deregulated in inflamed psoriatic lesions. Phase-contrast photomicrographs revealed morphological changes suggestive of differentiation (Figure S4, especially panel 4E). This was confirmed with Western blot analysis of each of these factors, which demonstrated that nanoEGCG (5 μM) induced the expression of all tested differentiation markers to equal or greater levels compared to a 4-fold higher dose of free EGCG (20 μM) (Figure 3E).
To determine whether nanoEGCG is capable of counteracting inflammation, we utilized an in vitro TPA-induced model of keratinocyte inflammation. NHEKs were pretreated with nanoEGCG (5 μM) or free EGCG (20 μM) before stimulation with TPA (100 ng/mL, 6 h) for a total of 24 h. Employing a mix-matched human ProcartaPlex multiplex immunoassay, analysis of a range of secreted inflammatory mediators revealed that both nanoEGCG and free EGCG significantly blunted TPA-induced secretion of IL-1β, IL-6, IL-8, tumor necrosis factor-α (TNF-α), transforming growth factor-β and C-X-C motif chemokine ligand-2 into culture media (Figure 3F). In addition, TPA-induced secretion of these mediators was particularly sensitive to nanoEGCG, with significant inhibition occurring at an equivalent concentration of 5 μM of nanoEGCG (Figure 3F), with effects in many cases superior to the effects of a 4-fold higher equivalent dose of free EGCG.
Topically applied nanoEGCG more significantly reduces IMQ-induced skin inflammation, while normalizing epidermal architecture and proliferation in lesioned mouse skin
To establish the in vivo anti-psoriatic effects of nanoEGCG, we employed the IMQ-induced murine psoriasis-like skin inflammation model39 using 4 treatment arms (Figure S5). At day 14 of the study, animals topically treated with IMQ cream displayed a psoriasis-like phenotype including prominent inflammation, scales, and thickened ears and trunk skin (Figure 4A–D and M–O). Ear thickness steadily increased by about 2–4-fold from days 3 to 6 after IMQ treatment and remained constant throughout the rest of the experiment (Figure 4O). Histological analyses of skin sections treated with IMQ alone (IMQ+, group 2) (Figure 4F and J) revealed acanthosis, epidermal rete-ridge projections (arrows in Figure 4F) and Munro’s microabscesses. In contrast, mice that received only control Vaseline cream (group 1) showed no skin abnormalities (Figure 4A and E). Topical application of free EGCG (1 mg/cm2 skin area; group 3) or nanoEGCG (48 μg/cm2 skin area; group 4) (Figure 4G– K and H–L) to lesioned skin co-treated with IMQ suppressed psoriasis-like symptoms. Indeed, treatment with nanoEGCG was even more successful in blocking erythema (Figure 4D and M), improving scale score (Figure 4N) and reducing ear and skin thickness (Figure 4O and P) to values similar to matched-control mice after 14 days of treatment. Further, lesioned skin treated with nanoEGCG (p<0.001) or free EGCG (p<0.01) exhibited significantly reduced histological psoriasis-like features (compare control IMQ(−) (Figure 4E), IMQ(+) (Figure 4F), IMQ(+)/EGCG (Figure 4G), and IMQ(+)/nanoEGCG (Figure 4H)-treated skin).
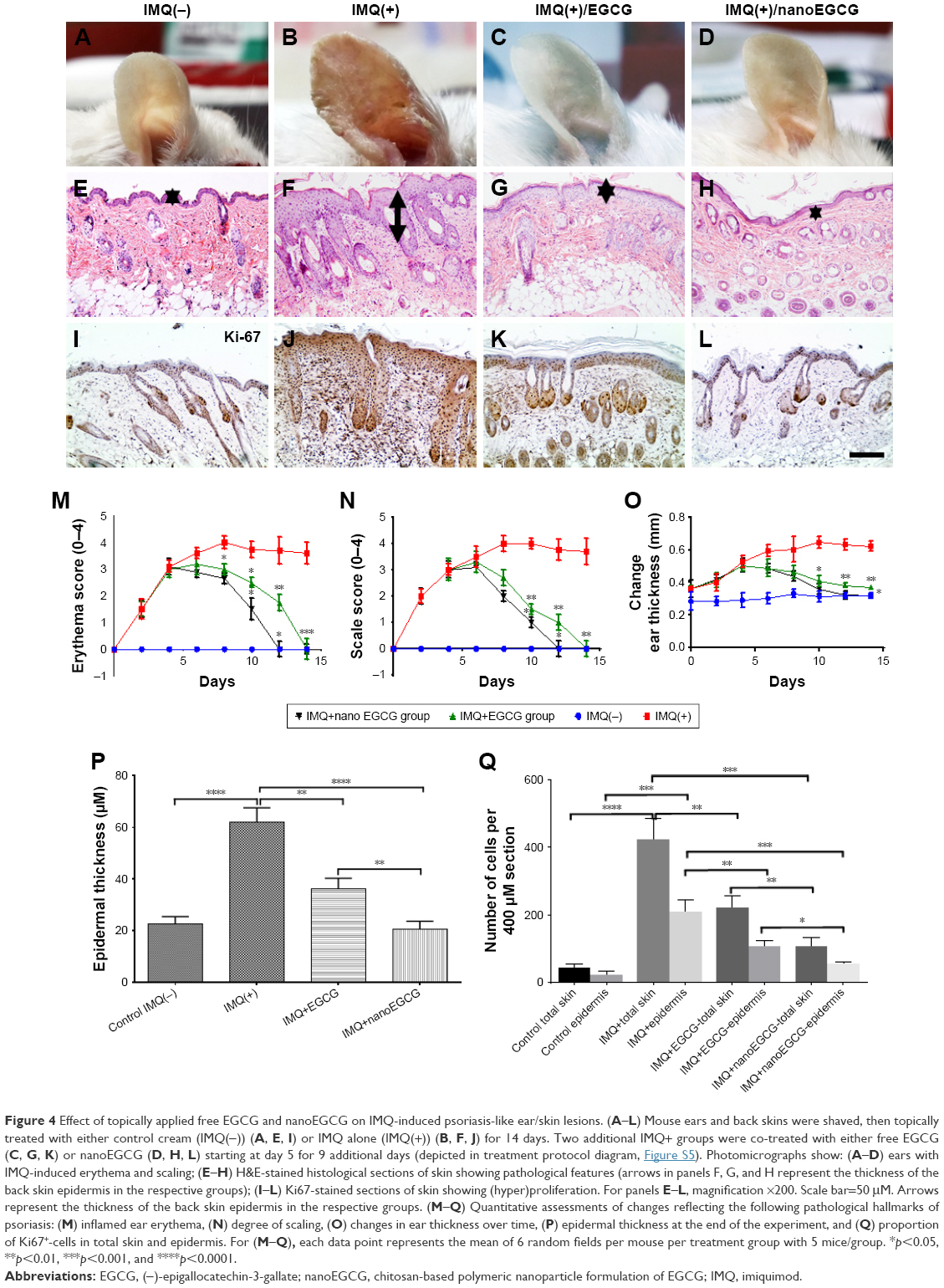 | Figure 4 Effect of topically applied free EGCG and nanoEGCG on IMQ-induced psoriasis-like ear/skin lesions. (A–L) Mouse ears and back skins were shaved, then topically treated with either control cream (IMQ(−)) (A, E, I) or IMQ alone (IMQ(+)) (B, F, J) for 14 days. Two additional IMQ+ groups were co-treated with either free EGCG (C, G, K) or nanoEGCG (D, H, L) starting at day 5 for 9 additional days (depicted in treatment protocol diagram, Figure S5). Photomicrographs show: (A–D) ears with IMQ-induced erythema and scaling; (E–H) H&E-stained histological sections of skin showing pathological features (arrows in panels F, G, and H represent the thickness of the back skin epidermis in the respective groups); (I–L) Ki67-stained sections of skin showing (hyper)proliferation. For panels E–L, magnification ×200. Scale bar=50 μM. Arrows represent the thickness of the back skin epidermis in the respective groups. (M–Q) Quantitative assessments of changes reflecting the following pathological hallmarks of psoriasis: (M) inflamed ear erythema, (N) degree of scaling, (O) changes in ear thickness over time, (P) epidermal thickness at the end of the experiment, and (Q) proportion of Ki67+-cells in total skin and epidermis. For (M–Q), each data point represents the mean of 6 random fields per mouse per treatment group with 5 mice/group. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. |
Because psoriasis is characterized by hyperproliferation of keratinocytes, we further determined the effect of nanoEGCG treatments on proliferation in vivo. Quantitative immunostaining analysis of Ki67+-epidermal keratinocytes/dermal cells revealed significantly reduced expression of Ki67 upon treatment with either free EGCG or nanoEGCG (p<0.01 for both treatments) (Figure 4K and L) compared to the IMQ(+) alone-treated group (Figure 4J) showing similarly lower expression values to the control IMQ(−) group (Figure 4I). These values were not as low as those from control mice, but were nevertheless markedly reduced compared to the robust increases in Ki67 expression in IMQ-treated skin (Figure 4Q).
Topical application of nanoEGCG inhibits leukocyte and lymphocyte infiltration in IMQ-induced back skin lesions
Psoriasis is characterized by increased dermal/epidermal infiltration of inflammatory cells.9,40–42 Using the Nuance imaging and Inform software analysis technology platforms with a 3-chromogen spectral library (Figure S6), we quantified selected markers of infiltrating immune cells by immunostaining in the skin of the 4 treatment groups. Markers were selected to visualize and quantitate mast cells (toluidine staining) (Figure 5A–D), neutrophils (neutrophil elastase staining) (Figure 5F–I), macrophages (F4/80 staining) (Figure 5K–N), and CD4+ T cells (red staining) (Figure 5P–S). Quantitative results are shown in Figure 5E, J, O, and T, respectively, along with the mean intensity in Figure 5U, and confirm that IMQ treatment alone causes infiltration of numerous inflammatory cells into the epidermis compared with control mice (compare Figure 5A, F, K, and P with Figure 5B, G, L, and Q). Treatment with either free EGCG (p<0.01 vs control) or nanoEGCG (p<0.0001 vs control) drastically reduced infiltration of all tested cell types, including CD4+ T lymphocytes (Figure 5P–T, red staining), with values for nanoEGCG-treated lesions reduced to levels almost as low as control skin. These results provide compelling evidence that topically applied EGCG, and especially nanoEGCG, inhibits and/or reverses inflammatory infiltrates typically associated with psoriasis pathophysiology, at least in the IMQ-induced mouse model of psoriasis.
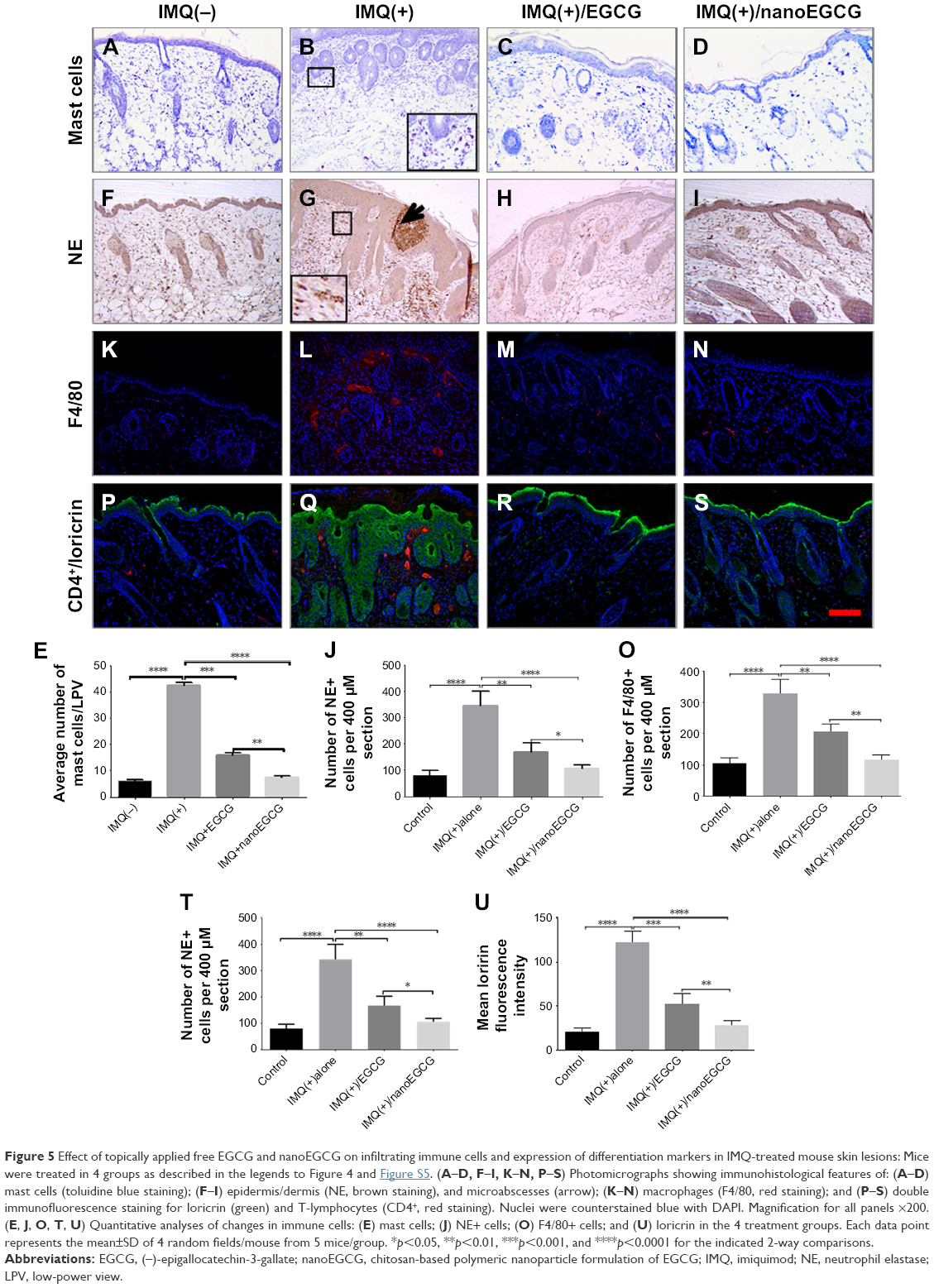 | Figure 5 Effect of topically applied free EGCG and nanoEGCG on infiltrating immune cells and expression of differentiation markers in IMQ-treated mouse skin lesions: Mice were treated in 4 groups as described in the legends to Figure 4 and Figure S5. (A–D, F–I, K–N, P–S) Photomicrographs showing immunohistological features of: (A–D) mast cells (toluidine blue staining); (F–I) epidermis/dermis (NE, brown staining), and microabscesses (arrow); (K–N) macrophages (F4/80, red staining); and (P–S) double immunofluorescence staining for loricrin (green) and T-lymphocytes (CD4+, red staining). Nuclei were counterstained blue with DAPI. Magnification for all panels ×200. (E, J, O, T, U) Quantitative analyses of changes in immune cells: (E) mast cells; (J) NE+ cells; (O) F4/80+ cells; and (U) loricrin in the 4 treatment groups. Each data point represents the mean±SD of 4 random fields/mouse from 5 mice/group. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 for the indicated 2-way comparisons. |
Topical application of nanoEGCG promotes epidermal differentiation and JunB expression in IMQ-induced skin lesions in mice
The expression of several differentiation-related proteins, including activator protein-1 (AP-1) factors, is altered in inflamed psoriatic lesions in both humans and mice.43
Loricrin is an insoluble precursor protein that is expressed in the granular layer of the epidermis and is a major component (70%–80%) of the cornified envelope.51 Loricrin expression occurs late in keratinocyte development and is typically down-regulated in lesioned and non-lesioned skin of psoriasis patients.48 However, unlike in human psoriasis, murine psoriasiform disease models overexpress loricrin in the entire epidermis43,52 (Figure 5Q and U, green fluorescence). We quantitatively examined the effect of free EGCG and nanoEGCG treatments on loricrin expression in this model. Immunostaining data revealed that both free EGCG and nanoEGCG treatments normalized loricrin expression to the granular layers, and nanoEGCG was 20-fold superior to free EGCG in reducing loricrin expression and could be reduced to levels comparable to those in the control group (Figure 5P–S and U).
Caspase-14 expression is down-regulated in parakeratotic psoriatic skin lesions compared to non-lesioned and normal skin in both humans and mice.43–45 Following morphometric and quantitative immunostaining analyses, we first observed that caspase-14 expression is indeed reduced in mouse skin treated with IMQ only (compare Figure 6A and B, brown color). Treatment of IMQ(+) lesions with free EGCG increased the expression of caspase-14 in the inflamed epidermis, and treatment with nanoEGCG was able to increase caspase-14 expression to levels qualitatively similar to the IMQ(−) control group (Figure 6A–D).
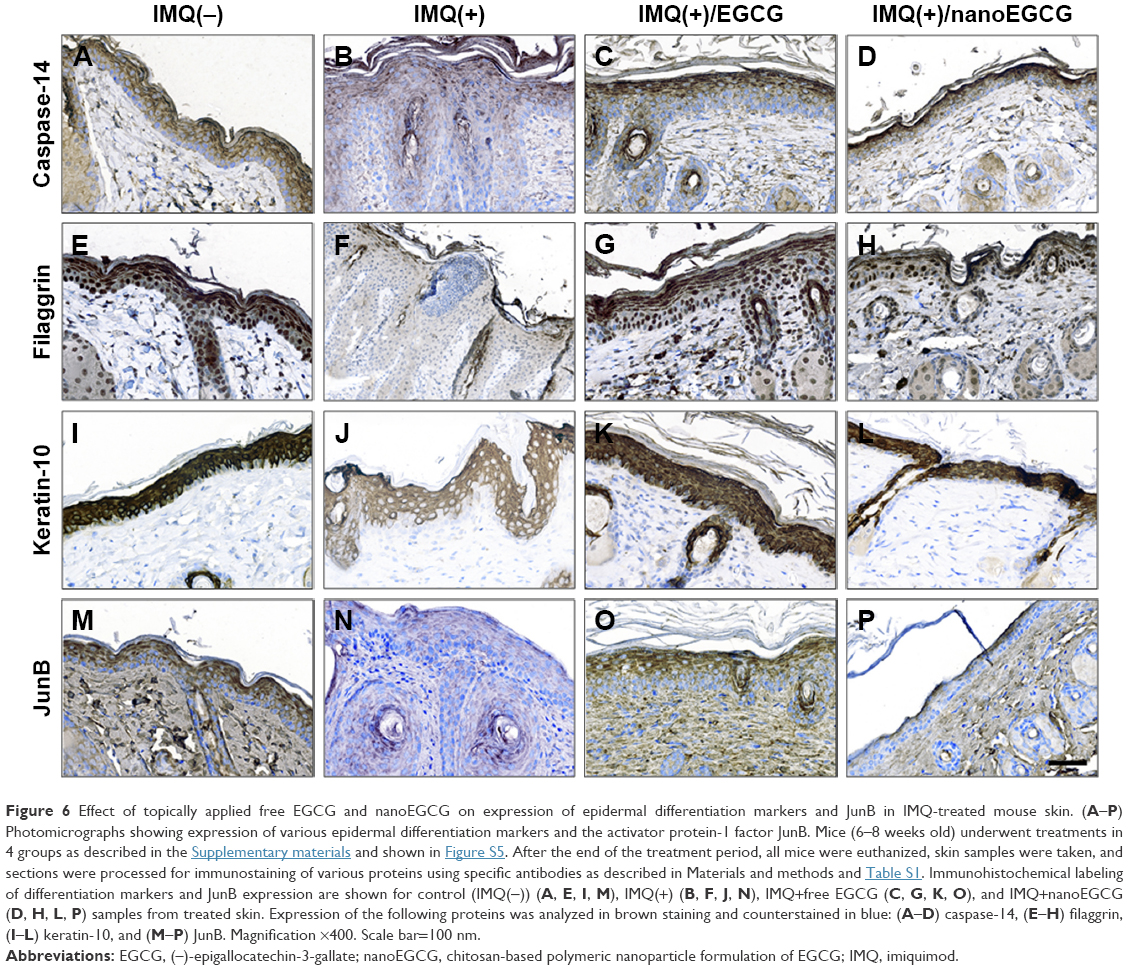 | Figure 6 Effect of topically applied free EGCG and nanoEGCG on expression of epidermal differentiation markers and JunB in IMQ-treated mouse skin. (A–P) Photomicrographs showing expression of various epidermal differentiation markers and the activator protein-1 factor JunB. Mice (6–8 weeks old) underwent treatments in 4 groups as described in the Supplementary materials and shown in Figure S5. After the end of the treatment period, all mice were euthanized, skin samples were taken, and sections were processed for immunostaining of various proteins using specific antibodies as described in Materials and methods and Table S1. Immunohistochemical labeling of differentiation markers and JunB expression are shown for control (IMQ(−)) (A, E, I, M), IMQ(+) (B, F, J, N), IMQ+free EGCG (C, G, K, O), and IMQ+nanoEGCG (D, H, L, P) samples from treated skin. Expression of the following proteins was analyzed in brown staining and counterstained in blue: (A–D) caspase-14, (E–H) filaggrin, (I–L) keratin-10, and (M–P) JunB. Magnification ×400. Scale bar=100 nm. |
Filaggrin, a major structural protein that is processed to its mature form by caspase-14, is involved in cornification.46,47 Approximately 5% of psoriasis patients harbor mutations in the filaggrin gene, and an estimated 80% exhibit depressed filaggrin expression in skin lesions.48,49 We previously reported a reduction in filaggrin expression in the IMQ-induced murine model of psoriasis43 and have confirmed this in the current study (compare Figure 6E and F, brown staining). We found that treatments with either nanoEGCG or free EGCG increased filaggrin expression in IMQ-induced skin lesions to values similar to control when compared to IMQ-alone-treated mouse skin (compare Figure 6E, G, and H to Figure 6F).
In normal epidermis, the most abundant structural proteins are keratins that form the intermediate filament cytoskeleton.50 Proliferative keratins-5 and -14 are expressed in the basal epidermal layers, while keratins-1 and -10 are expressed suprabasally during keratinocyte differentiation. Given that inflamed psoriatic skin displays aberrantly suppressed differentiation keratins (which is confirmed in our IMQ-induced mouse model) (compare Figure 6I and J), we investigated the effect of nanoEGCG treatment on protein expression of K10. We observed that treatment of IMQ-induced skin lesions with either free EGCG or nanoEGCG restored K10 expression in the upper granular layers (Figure 6K and L).
Abrogation of JunB, a member of the AP-1 signaling complex in keratinocytes, triggers release of inflammatory mediators that contribute to psoriasis immunopathogenesis by recruiting inflammatory cells to the skin.53 Because IMQ treatment down-regulates AP-1 transcription factor proteins43 (compare Figure 6M and N), we determined the effect of free EGCG and nanoEGCG on JunB expression and observed that treatment with both forms of EGCG restored epidermal expression (Figure 6M–P).
Topical application of EGCG/nanoEGCG alleviates tissue vascularization, and modulates IMQ-induced alteration of multiple immune mediators
An increased number of tortuous blood vessels (angiogenesis) was observed in both human psoriatic and IMQ-induced mouse skin lesions. Such blood vessels were visualized with CD31 immunofluorescence in the 4 treatment groups (Figure 7A–D; especially Figure 7B in the IMQ-only treatment group). Results from the 4 treatment groups reveal that treatment with either free EGCG or nanoEGCG strongly decreased tissue vascularization. Quantitation of results (Figure 7E) showed that nanoEGCG is superior to free EGCG, reducing the number of blood vessels per session to levels similar to control mice.
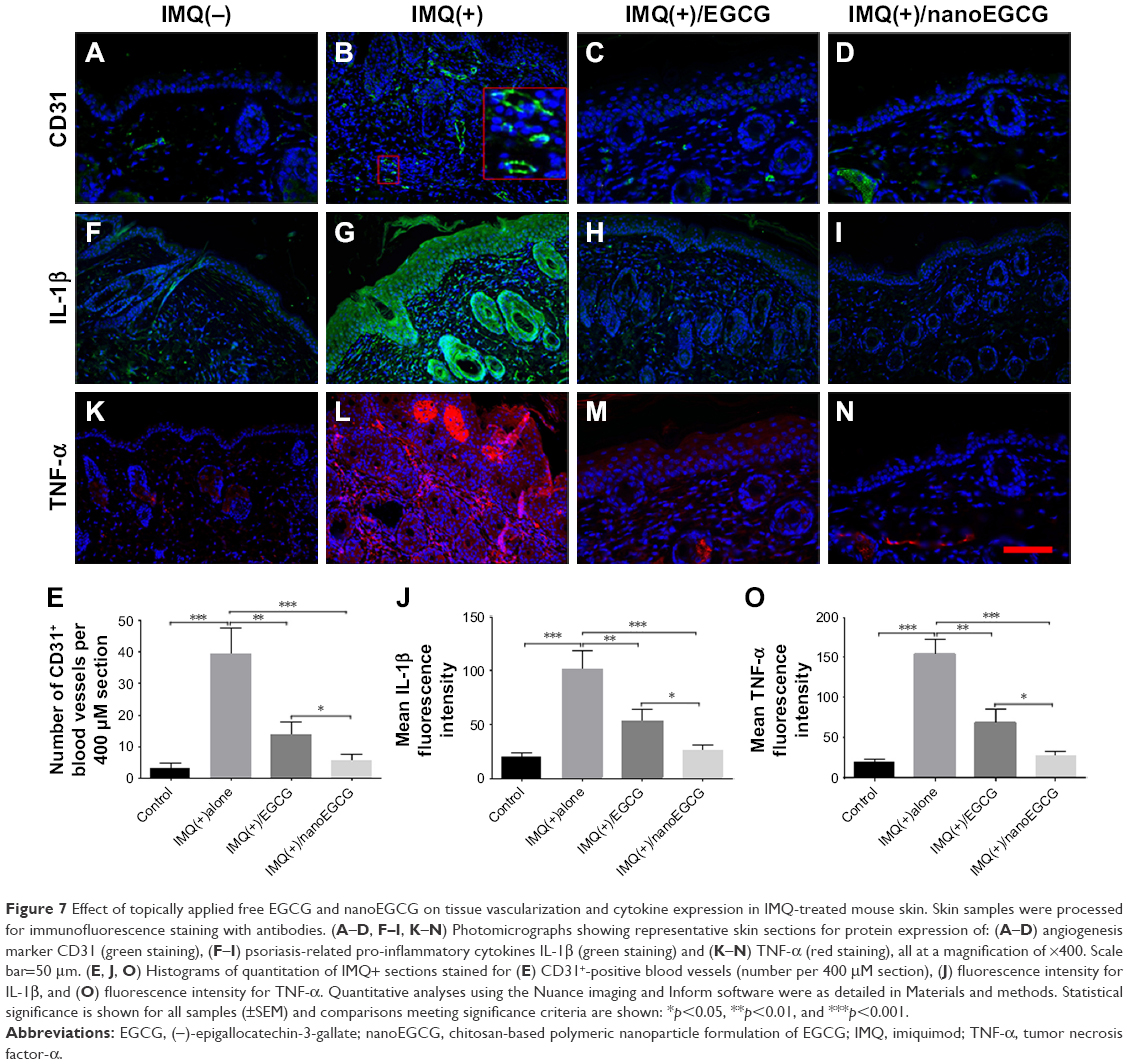 | Figure 7 Effect of topically applied free EGCG and nanoEGCG on tissue vascularization and cytokine expression in IMQ-treated mouse skin. Skin samples were processed for immunofluorescence staining with antibodies. (A–D, F–I, K–N) Photomicrographs showing representative skin sections for protein expression of: (A–D) angiogenesis marker CD31 (green staining), (F–I) psoriasis-related pro-inflammatory cytokines IL-1β (green staining) and (K–N) TNF-α (red staining), all at a magnification of ×400. Scale bar=50 μm. (E, J, O) Histograms of quantitation of IMQ+ sections stained for (E) CD31+-positive blood vessels (number per 400 μM section), (J) fluorescence intensity for IL-1β, and (O) fluorescence intensity for TNF-α. Quantitative analyses using the Nuance imaging and Inform software were as detailed in Materials and methods. Statistical significance is shown for all samples (±SEM) and comparisons meeting significance criteria are shown: *p<0.05, **p<0.01, and ***p<0.001. |
Pro-inflammatory cytokines IL-1β and TNF-α, produced either by keratinocytes or by immune cells, are believed to play a major role in psoriasis pathogenesis.2,10,54,55 Here, we observed that IMQ-induced lesions exhibited increased expression of IL-1β and TNF-α (Figure 7F–O), but that topical application of nanoEGCG (p<0.001) or free EGCG (p<0.01) significantly inhibited expression of both cytokines, with nanoEGCG reducing expression (measured by fluorescence intensities) to values similar to control mice (Figure 7F–J and K–O).
We also examined the expression of a panel of other Th1/Th17 cytokines and associated mediators, including interferon-γ, IL-17, IL-22, IL-23, and interferon-inducible protein-10 (a chemokine for Th1), which are reported to play important roles in human and murine psoriasiform immunopathogenesis.8,11,56,57 Using a 36-Plex ProcartaPlex-based multiplex immunoassay cytokine and chemokine profiling kit, we comparatively determined the effect of free EGCG and nanoEGCG treatments in our IMQ-induced mouse model. The expression levels of 4 mediators (IL-3, IL-9, epithelial neutrophil activating peptide-78 [ENA-78], and regulated on activation, normal T cell expressed and secreted [RANTES]) were not significantly altered by free EGCG treatment of IMQ-induced lesions (Figure S7). However, we found that treatment with nanoEGCG significantly decreased the expression levels of 31 out of the 36 factors tested, while free EGCG treatment decreased only 28 of the 36 immune mediators analyzed (Figure S8A and B), even though the equivalent EGCG concentration in free EGCG (1 mg) was approximately 21-fold more than that of nanoEGCG (48 μg). Furthermore, expression levels of 21 of the immune mediators that were inhibited by nanoEGCG were significantly less than by free EGCG (asterisks in Figures 8A and B and S8A and B). In addition, nanoEGCG was able to up-regulate the anti-inflammatory cytokine IL-10, potentially providing an additional benefit (Figure 8A, second row, extreme left graph). Finally, we also observed significantly reduced expression of chemokines such as macrophage inflammatory proteins (MIP-1α, MIP-1β, and MIP-2) by free EGCG treatments, with nanoEGCG again showing a superior ability to reduce expression of these factors (Figures 8B and S8B).
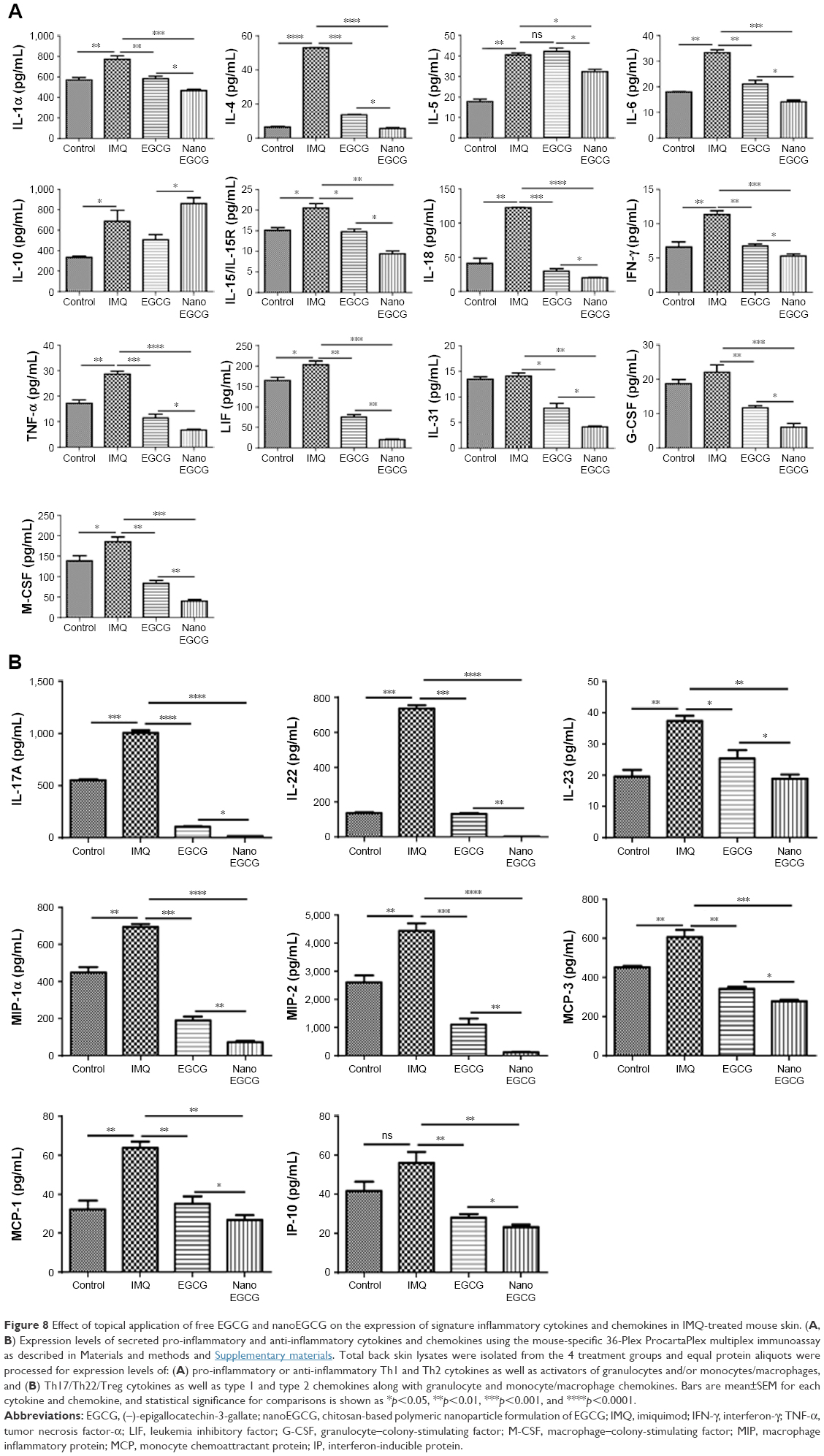 | Figure 8 Effect of topical application of free EGCG and nanoEGCG on the expression of signature inflammatory cytokines and chemokines in IMQ-treated mouse skin. (A, B) Expression levels of secreted pro-inflammatory and anti-inflammatory cytokines and chemokines using the mouse-specific 36-Plex ProcartaPlex multiplex immunoassay as described in Materials and methods and Supplementary materials. Total back skin lysates were isolated from the 4 treatment groups and equal protein aliquots were processed for expression levels of: (A) pro-inflammatory or anti-inflammatory Th1 and Th2 cytokines as well as activators of granulocytes and/or monocytes/macrophages, and (B) Th17/Th22/Treg cytokines as well as type 1 and type 2 chemokines along with granulocyte and monocyte/macrophage chemokines. Bars are mean±SEM for each cytokine and chemokine, and statistical significance for comparisons is shown as *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. |
Discussion and conclusion
This study describes the characterization and biological evaluation of a polymeric nanocarrier for delivery of green tea catechin EGCG as a novel therapeutic approach for the management of psoriasis. The efficacy of green tea or its constituent polyphenol EGCG is well documented in a variety of human disease models.58 Despite these preclinical proofs of concept, only limited clinical success has been reported,59 due at least in part to the low stability and bioavailability of EGCG in its free form.18 To circumvent these problems, we employed a nanoparticle formulation based on the biodegradable cationic polymer chitosan. A nanoformulation based on chitosan has several advantages over other known natural polymeric nanoparticles. Chitosan’s propensity to form hydrogels makes it an ideal carrier for natural and water-soluble drugs. In addition, chitosan polymers are able to release bound EGCG in a steady and sustained fashion, as we previously demonstrated in simulated gastric and intestinal fluids.27,28 This simulated behavior is very relevant because oral and topical deliveries are the preferred routes for therapeutic protocols in dermatological applications.
To assess whether nanoEGCG retains its mechanistic identity compared with free EGCG in a biologically relevant context, we examined the expression of several skin proteins known to be modulated by EGCG in keratinocytes in cell cultures and in an in vivo psoriasis-like mouse model system.
In the current study, our nanoEGCG formulation exhibited significantly improved cellular uptake, pro-differentiation, and anti-psoriatic activity compared to free EGCG in vitro and in vivo. We observed not only a steady release of nanoEGCG in cells, but also the inhibition of multiple TPA- and IL-22-induced proliferation and inflammatory responses. Using human epidermal keratinocytes, we observed that nanoEGCG can efficiently modulate key proteins related to keratinocyte differentiation, inflammation, and hyperplasia at a 4-fold lower equivalent dose compared with free EGCG. Thus, our nanoEGCG formulate may help to alleviate the reported problems with stability and/or bioavailability of EGCG in clinical tests.
We also used an in vivo mouse model of psoriasiform disease to test further the performance of our nanoEGCG formulation in reducing parameters of skin inflammation. Topical IMQ treatment produces a psoriasis-like dermatitis in both humans and mice,39,60 with lesions showing several parallels to genuine psoriatic lesions. The murine model therefore serves as an excellent preclinical model for testing the efficacy of promising therapeutics. Here, we demonstrate that topical application of nanoEGCG to skin lesions elicited by 5 days of pretreatment and 9 days of co-treatment with IMQ resulted in marked improvement in multiple aspects of psoriasiform disease in mice. NanoEGCG completely normalized the epidermal architecture and reduced inflammation via reduction and alleviation of psoriasiform phenotype. These effects of nanoEGCG were equal or superior to those obtained with over 20-fold higher doses of free EGCG. Further testing showed that nanoEGCG reduced the proportion of infiltrating immune cells as well as the expression of secreted Th1/Th2/Th17 inflammatory immune mediators and activators of mononuclear cells, all of which play important roles in human psoriasis pathophysiology.37 Quantitative analysis of the expression of 36 immune mediators in lesioned skin lysates, compared with treated and matched-control groups, confirmed the superiority of nanoEGCG over free EGCG in improving (31 out of 36) and, in several cases, almost completely normalizing the expression of the tested immunomodulatory markers. Taken together, our findings suggest that the nanoEGCG formulation is a novel and potentially potent approach for delivering EGCG, which possesses anti-psoriatic activity in vitro in cell culture and in vivo in IMQ-induced psoriasis-like disease in Balb/c mice.
The enhanced anti-proliferative, anti-inflammatory, and pro-differentiation effects of nanoEGCG indicate that it retains the mechanistic activity of free EGCG while improving its delivery and effectiveness. Our preclinical findings strongly suggest that nanoEGCG warrants further testing as a novel, effective addition to the currently available options for the topical treatment of psoriasis.
Acknowledgments
Part of this study was presented at the 2016 Annual Meeting of the Society for Investigative Dermatology in Scottsdale, AZ, USA.61 This research was supported in part by an National Institute of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases grant (R01 AR059742) (to HM), an American Skin Association Carson Research Scholar Award in psoriasis (to JCC), an American Cancer Society grant (MRSG-11-019-01-CNE) (to IAS), and an NIAMS core service resource University of Wisconsin-Skin Disease Research Center Pilot and Feasibility grant (P30 AR066524) (to GSW). The authors thank Maria Ines Chaves-Rodriguez, Elizabeth Kane, Anne Radtke, Nora Katib, Natalie Sholl, and Kayla Smith for their help with cell culture, immunohistochemistry, and Western blotting. We also thank Drs G Kerr Whitfield and Kelly A Keating, for helpful discussions and for critically reviewing this manuscript, and Dr Anna V Kiyanova for assistance with nanoparticle size characterization.
Author contributions
JCC designed and supervised the research, and wrote the first draft of the manuscript. DJB prepared and characterized the nanoparticle formulations. JCC, IAS, VMA, SE, DJB, ASB, SA, and RJM performed the experiments. JCC, VMA, SE, DJB, and RJM analyzed the data: JCC, VMA, and SE wrote the manuscript. All authors contributed toward data analysis, drafting, and critically revising the paper, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Stenderup K, Rosada C, Dam TN, Salerno E, Belinka BA, Kachlany SC. Resolution of psoriasis by a leukocyte-targeting bacterial protein in a humanized mouse model. J Invest Dermatol. 2011;131:2033–2039. | ||
Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. | ||
Gelfand JM, Dommasch ED, Shin DB, et al. The risk of stroke in patients with psoriasis. J Invest Dermatol. 2009;129:2411–2418. | ||
Hoffjan S, Stemmler S. On the role of the epidermal differentiation complex in ichthyosis vulgaris, atopic dermatitis and psoriasis. Br J Dermatol. 2007;157:441–449. | ||
Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–873. | ||
Segre JA. Epidermal barrier formation and recovery in skin disorders. J Clin Invest. 2006;116:1150–1158. | ||
Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385–422. | ||
Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339–1350. | ||
Wang H, Peters T, Kess D, et al. Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J Clin Invest. 2006;116:2105–2114. | ||
Rivas Bejarano JJ, Valdecantos WC. Psoriasis as autoinflammatory disease. Dermatol Clin. 2013;31:445–460. | ||
Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol. 2011;186:1495–1502. | ||
Crow JM. Therapeutics: Silencing psoriasis. Nature. 2012;492:S58–S59. | ||
Dubois Declercq S, Pouliot R. Promising new treatments for psoriasis. Scientific World Journal. 2013;2013:980419. | ||
Wong T, Hsu L, Liao W. Phototherapy in psoriasis: a review of mechanisms of action. J Cutan Med Surg. 2013;17:6–12. | ||
Villaseñor-Park J, Wheeler D, Grandinetti L. Psoriasis: evolving treatment for a complex disease. Cleve Clin J Med. 2012;79:413–423. | ||
Han R, Rostami-Yazdi M, Gerdes S, Mrowietz U. Triptolide in the treatment of psoriasis and other immune-mediated inflammatory diseases. Br J Clin Pharmacol. 2012;74:424–436. | ||
Lebwohl MG, Kavanaugh A, Armstrong AW, Van Voorhees AS. US Perspectives in the Management of Psoriasis and Psoriatic Arthritis: Patient and Physician Results from the Population-Based Multinational Assessment of Psoriasis and Psoriatic Arthritis (MAPP) Survey. Am J Clin Dermatol. 2016;17:87–97. | ||
Krupkova O, Ferguson SJ, Wuertz-Kozak K. Stability of (−)-epigallocatechin gallate and its activity in liquid formulations and delivery systems. J Nutr Biochem. 2016;37:1–12. | ||
Balasubramanian S, Eckert RL. Keratinocyte proliferation, differentiation, and apoptosis – differential mechanisms of regulation by curcumin, EGCG and apigenin. Toxicol Appl Pharmacol. 2007;224:214–219. | ||
Hsu S, Dickinson D, Borke J, et al. Green tea polyphenol induces caspase 14 in epidermal keratinocytes via MAPK pathways and reduces psoriasiform lesions in the flaky skin mouse model. Exp Dermatol. 2007;16:678–684. | ||
Hsu S, Yamamoto T, Borke J, et al. Green tea polyphenol-induced epidermal keratinocyte differentiation is associated with coordinated expression of p57/KIP2 and caspase 14. J Pharmacol Exp Ther. 2005;312:884–890. | ||
Bickers DR, Athar M. Novel approaches to chemoprevention of skin cancer. J Dermatol. 2000;27:691–695. | ||
Ellis LZ, Liu W, Luo Y, et al. Green tea polyphenol epigallocatechin-3-gallate suppresses melanoma growth by inhibiting inflammasome and IL-1β secretion. Biochem Biophys Res Commun. 2011;414:551–556. | ||
Katiyar SK, Afaq F, Perez A, Mukhtar H. Green tea polyphenol (−)-epigallocatechin-3-gallate treatment of human skin inhibits ultraviolet radiation-induced oxidative stress. Carcinogenesis. 2001;22:287–294. | ||
Meeran SM, Akhtar S, Katiyar SK. Inhibition of UVB-induced skin tumor development by drinking green tea polyphenols is mediated through DNA repair and subsequent inhibition of inflammation. J Invest Dermatol. 2009;129:1258–1270. | ||
Shankar S, Chen Q, Srivastava RK. Inhibition of PI3K/AKT and MEK/ERK pathways act synergistically to enhance antiangiogenic effects of EGCG through activation of FOXO transcription factor. J Mol Signal. 2008;3:7. | ||
Khan N, Bharali DJ, Adhami VM, et al. Oral administration of naturally occurring chitosan-based nanoformulated green tea polyphenol EGCG effectively inhibits prostate cancer cell growth in a xenograft model. Carcinogenesis. 2014;35:415–423. | ||
Siddiqui IA, Bharali DJ, Nihal M, et al. Excellent anti-proliferative and pro-apoptotic effects of (−)-epigallocatechin-3-gallate encapsulated in chitosan nanoparticles on human melanoma cell growth both in vitro and in vivo. Nanomedicine. 2014;10:1619–1626. | ||
Zhang Z, Tsai PC, Ramezanli T, Michniak-Kohn BB. Polymeric nanoparticles-based topical delivery systems for the treatment of dermatological diseases. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5:205–218. | ||
Kim DG, Jeong YI, Choi C, et al. Retinol-encapsulated low molecular water-soluble chitosan nanoparticles. Int J Pharm. 2006;319:130–138. | ||
Schrand AM, Schlager JJ, Dai L, Hussain SM. Preparation of cells for assessing ultrastructural localization of nanoparticles with transmission electron microscopy. Nat Protoc. 2010;5:744–757. | ||
Bozzola JJ, Russell LD. Electron microscopy. 2nd ed. Sudbury, MA: Jones & Bartlett; 1992. | ||
Chamcheu JC, Pal HC, Siddiqui IA, et al. Prodifferentiation, anti-inflammatory and antiproliferative effects of delphinidin, a dietary anthocyanidin, in a full-thickness three-dimensional reconstituted human skin model of psoriasis. Skin Pharmacol Physiol. 2015;28:177–188. | ||
Chamcheu JC, Chaves-Rodriquez MI, Adhami VM, et al. Upregulation of PI3K/AKT/mTOR, FABP5 and PPARβ/δ in Human Psoriasis and Imiquimod-induced Murine Psoriasiform Dermatitis Model. Acta Derm Venereol. 2016;96:854–856. | ||
Chamcheu JC, Navsaria H, Pihl-Lundin I, Liovic M, Vahlquist A, Törmä H. Chemical chaperones protect epidermolysis bullosa simplex keratinocytes from heat stress-induced keratin aggregation: involvement of heat shock proteins and MAP kinases. J Invest Dermatol. 2011;131:1684–1691. | ||
Huang W, Hennrick K, Drew S. A colorful future of quantitative pathology: validation of Vectra technology using chromogenic multiplexed immunohistochemistry and prostate tissue microarrays. Hum Pathol. 2013;44:29–38. | ||
Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–255. | ||
Mitra A, Raychaudhuri SK, Raychaudhuri SP. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine. 2012;60:38–42. | ||
van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182:5836–5845. | ||
Shukuya R, Hasegawa T, Niwa Y, Okuma K, Ikeda S. Granulocyte and monocyte adsorption apheresis for generalized pustular psoriasis. J Dermatol. 2011;38:1130–1134. | ||
Stratis A, Pasparakis M, Rupec RA, et al. Pathogenic role for skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation. J Clin Invest. 2006;116:2094–2104. | ||
Clark RA, Kupper TS. Misbehaving macrophages in the pathogenesis of psoriasis. J Clin Invest. 2006;116:2084–2087. | ||
Chamcheu JC, Adhami VM, Esnault S, et al. Dual Inhibition of PI3K/Akt and mTOR by the Dietary Antioxidant, Delphinidin, Ameliorates Psoriatic Features In Vitro and in an Imiquimod-Induced Psoriasis-Like Disease in Mice. Antioxid Redox Signal. 2017;26:49–69. | ||
Walsh DS, Borke JL, Singh BB, et al. Psoriasis is characterized by altered epidermal expression of caspase 14, a novel regulator of keratinocyte terminal differentiation and barrier formation. J Dermatol Sci. 2005;37:61–63. | ||
Lippens S, Kockx M, Denecker G, et al. Vitamin D3 induces caspase-14 expression in psoriatic lesions and enhances caspase-14 processing in organotypic skin cultures. Am J Pathol. 2004;165:833–841. | ||
Brown SJ, McLean WH. One remarkable molecule: filaggrin. J Invest Dermatol. 2012;132:751–762. | ||
Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122:1285–1294. | ||
Kim BE, Howell MD, Guttman-Yassky E, et al. TNF-α downregulates filaggrin and loricrin through c-Jun N-terminal kinase: role for TNF-α antagonists to improve skin barrier. J Invest Dermatol. 2011;131:1272–1279. | ||
Hüffmeier U, Traupe H, Oji V, et al. Loss-of-function variants of the filaggrin gene are not major susceptibility factors for psoriasis vulgaris or psoriatic arthritis in German patients. J Invest Dermatol. 2007;127:1367–1370. | ||
Chamcheu JC, Siddiqui IA, Syed DN, Adhami VM, Liovic M, Mukhtar H. Keratin gene mutations in disorders of human skin and its appendages. Arch Biochem Biophys. 2011;508:123–137. | ||
Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–340. | ||
Schonthaler HB, Huggenberger R, Wculek SK, Detmar M, Wagner EF. Systemic anti-VEGF treatment strongly reduces skin inflammation in a mouse model of psoriasis. Proc Natl Acad Sci U S A. 2009;106:21264–21269. | ||
Zenz R, Eferl R, Kenner L, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. | ||
Abramovits W, Rivas Bejarano JJ, Valdecantos WC. Role of interleukin 1 in atopic dermatitis. Dermatol Clin. 2013;31:437–444. | ||
Kunze A, Förster U, Oehrl S, Schmitz M, Schäkel K. Autocrine TNF-α and IL-1β prime 6-sulfo LacNAc+ dendritic cells for high-level production of IL-23. Exp Dermatol. 2017;26:314–316. | ||
Mabuchi T, Takekoshi T, Hwang ST. Epidermal CCR6+ γδ T cells are major producers of IL-22 and IL-17 in a murine model of psoriasiform dermatitis. J Immunol. 2011;187:5026–5031. | ||
Hedrick MN, Lonsdorf AS, Shirakawa AK, et al. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Invest. 2009;119:2317–2329. | ||
Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011;82:1807–1821. | ||
Chakrawarti L, Agrawal R, Dang S, Gupta S, Gabrani R. Therapeutic effects of EGCG: a patent review. Expert Opin Ther Pat. 2016;26:907–916. | ||
Gilliet M, Conrad C, Geiges M, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol. 2004;140:1490–1495. | ||
Chamcheu JC, Siddiqui IA, Adhami V, et al. Topical application of chitosan-based nanoformulated green tea polyphenol EGCG ameliorates imiquimod-induced psoriasis-like skin lesion in mice. J Invest Dermatol. 2016;136:S95. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.