Back to Journals » Hepatic Medicine: Evidence and Research » Volume 16
Chimeric Livers: Interspecies Blastocyst Complementation and Xenotransplantation for End-Stage Liver Disease
Received 17 September 2023
Accepted for publication 10 February 2024
Published 16 February 2024 Volume 2024:16 Pages 11—29
DOI https://doi.org/10.2147/HMER.S440697
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Madelyn J Blake,1 Clifford J Steer2
1Department of Medicine, University of Minnesota Medical School, Minneapolis, MN, USA; 2Departments of Medicine, and Genetics, Cell Biology and Development, University of Minnesota Medical School, Minneapolis, MN, USA
Correspondence: Madelyn J Blake, Department of Medicine, University of Minnesota Medical School, 420 Delaware Street SE, MMC 195, Minneapolis, MN, 55455, USA, Email [email protected] Clifford J Steer, Department of Medicine, University of Minnesota Medical School, 420 Delaware Street SE, MMC 36, Minneapolis, MN, 55455, USA, Email [email protected]
Abstract: Orthotopic liver transplantation (OLT) currently serves as the sole definitive treatment for thousands of patients suffering from end-stage liver disease; and the existing supply of donor livers for OLT is drastically outpaced by the increasing demand. To alleviate this significant gap in treatment, several experimental approaches have been devised with the aim of either offering interim support to patients waiting on the transplant list or bioengineering complete livers for OLT by infusing them with fresh hepatic cells. Recently, interspecies blastocyst complementation has emerged as a promising method for generating complete organs in utero over a short timeframe. When coupled with gene editing technology, it has brought about a potentially revolutionary transformation in regenerative medicine. Blastocyst complementation harbors notable potential for generating complete human livers in large animals, which could be used for xenotransplantation in humans, addressing the scarcity of livers for OLT. Nevertheless, substantial experimental and ethical challenges still need to be overcome to produce human livers in larger domestic animals like pigs. This review compiles the current understanding of interspecies blastocyst complementation and outlines future possibilities for liver xenotransplantation in humans.
Keywords: end-stage liver disease, interspecies blastocyst complementation, blastocyst complementation, chimeras, xenotransplantation
Introduction
End-stage liver disease (ESLD) is analogous to advanced liver disease, decompensated cirrhosis, and liver failure due to the irreversible nature of the aforementioned conditions.1–4 In 2021, mortality attributed to ESLD ranked as ninth out of all causes of death in the United States and was responsible for more than 55,000 deaths.5 As such, the condition remains a substantial burden for modern medicine. However, in direct contrast to other leading causes of mortality, orthotopic liver transplantation (OLT) is an effective therapeutic and even curative option for those with ESLD. Despite OLT being the second most frequently used transplantation procedure across the globe, its efficacy in alleviating the significant burden posed by ESLD is blunted by a substantial lack of liver donors, and an incredibly high demand for the procedure.3 Across all organ donation, the odds are equally dismal. In 2021, the total number of transplant candidates in the United States waiting for an organ was over 100,000, and it is estimated that each year, only a third of all patients waiting on a transplant list will receive an organ.6 In response to these glaring challenges, several innovative approaches including blastocyst complementation and xenotransplantation of chimeric livers, have been proposed as viable alternatives to overcome the treatment challenges presented for patients with ESLD.7,8
These methods contrast with other noteworthy solutions for the treatment of ESLD, namely hepatocyte grafts and transplantation, as well as extensive bioengineering of discarded livers.9–11 While the latter approaches possess potential for the treatment of ESLD, it is necessary to acknowledge that their efficacy is largely dependent upon several factors. These include reliable sources of primary human hepatocytes (PHH), optimal cell culture techniques for their propagation in vitro, and sustained viability and strict maintenance of phenotypic characteristics within cultured hepatocytes in addition to effective protocols for the hepatic differentiation. Unfortunately, these obstacles are also in the setting of a well-characterized tendency of PHHs to de-differentiate and die off rapidly in vitro.12
Several novel cell culture techniques have been developed in response to the difficulties posed by generating large quantities of suitable PHHs for transplantation in vivo, including the application of positively-charged surfaces, double gels, albumin-coated surfaces, varying extracellular matrix compositions, various 3D spheroids and bioreactors, microencapsulation techniques, and liver-on-chip microdevices.13–21 Nevertheless, the task of securing a high volume of PHHs and ensuring consistent reproducibility of results while utilizing commercially available PHHs are lofty challenges for the realization of these methods as effective treatment options for patients with ESLD. Thus, with these obstacles in mind, induced pluripotent stem cell (iPSC) derived hepatocytes have become increasingly preferred to PHHs but even iPSCs are plagued by several issues, including safety, blunted hepatic maturation in both in vitro and in vivo settings, and lacking engraftment into the hepatic parenchyma following implantation.22–24
It is for these reasons that blastocyst complementation and xenotransplantation have become particularly appealing avenues for the future of ESLD treatment. In the setting of hepatic medicine, blastocyst complementation is a biological technique that could potentially generate a whole, human liver in utero of an animal, such as a pig, by microinjecting stem cells into a blastocyst-stage, dysorganogenetic embryo that possesses functionally normal pluripotent cells.25,26 The efficacy of this technique is reliant upon the dysorganogenetic status of the embryo, which is achieved by knocking out pivotal gene(s) that direct the biogenesis of an organ at the blastocyst stage of development through the use of genome editing, before PSCs are injected into the knockout blastocysts to complement the deleted gene(s).27,28 Despite the relatively recent emergence of this technology, blastocyst complementation has already been used to generate numerous organs in animal models, including, the heart and vascular system, whole lungs, kidneys, pancreas, salivary glands, parathyroid glands, and liver.7,25,29–32 In addition to possessing the potential to generate a large variety of analogous cells, tissues, and organs, cells produced through blastocyst complementation are also immunocompatible, reducing the risk of rejection in the recipient species following transplantation.33,34
The potential of generating human-scale, chimeric organs using large animal fetuses and human pluripotent stem cells (PSCs) through blastocyst complementation is abundantly clear. Commonly, porcine models are preferred for growing chimeric organs for xenotransplantation due to myriad physiological, anatomical, and genetic characteristics shared between pigs and humans. With reference to the liver specifically, porcine models would be an ideal match because their livers are similar to human livers in size, segmentation and arterial vasculature.35–38 As such, chimeric human livers generated in pigs are well suited to serve as a potential treatment option for ESLD through OLT. The application of blastocyst complementation and xenotransplantation for ESLD has only grown with timely advances in the field. Recent studies utilizing an apancreatic pig embryo phenotype have demonstrated that it was possible through blastocyst complementation to form an entire pancreas from normal heterologous PSCs. Moreover, there has been noteworthy success in generating both pig-human and pig-primate chimeras. Together, there is an increasing amount of data to suggest the future reality of pig-human liver chimeras as a viable therapeutic option for patients requiring transplantation.7,39
Early Hepatogenesis
The liver is the largest solid internal organ in the human body, and it is also one of the most complex. It is principally formed from the anterior primitive streak and the endoderm germ layer that originates from within a gastrulating embryo. While the liver is composed of a multitude of different cell types, hepatocytes and cholangiocytes, commonly known as bile duct cells, are the only cells to possess embryonic endoderm-derived origin. Other cells are formed from embryonic mesoderm such as Kupffer cells (bone marrow derived), epithelial cells, hepatic sinusoidal cells, and stellate cells. Most notably, hepatocytes make up approximately 80% of the liver cell mass, and about 50% of the cellular population. Moreover, the liver possesses a distinct tissue architecture, with lobules that receive arterial blood flow from the hepatic artery and portal vein.37–39
Hepatic biogenesis, initially identified and characterized using mouse embryonic studies, is tightly regulated, and conserved throughout evolution as per numerous mammal, bird, and amphibian studies. During elementary stages of hepatogenesis, the endoderm can be detected around E7.5 in a mouse embryo, equivalent to about the third week of gestation in humans.40 At this stage, embryonic stem cells (ESCs) undergo a migration process towards the anterior portion of the embryo before forming the definitive endoderm by segregating from the mesoderm. A cylindrical structure is then formed by the definitive endoderm, which rotates along the anterior and posterior axis of the embryo.
The definitive embryo is segregated into three distinct regions: foregut, midgut, and hindgut. The formation of the liver diverticulum, also known as the liver bud, is initiated by the ventral portion of the foregut and diverse signaling from the surrounding mesodermal embryonic tissue at an E8.5.41 In contrast, development of the liver outgrowth and primordium is initiated by adjacent cardiac mesodermal embryonic tissue.42 Following the formation of the outgrowth and primordium, the liver bud begins to thicken and subsequently transitions to multiple layers of pseudostratified epithelium that facilitate the synthesis of hepatoblasts. By E9, it has been well established by tissue explant studies that endothelial cells then surround the newly developed hepatic endoderm and bind the mesenchymal domain that the liver bud then grows into as it continues to develop.43,44 At this point in the developmental process, the liver’s endodermal cells are now polarized, and their stroma further evokes developmental signals.
By E10.5, a migration of hepatoblasts into the surrounding mesenchyme has occurred, and the cystic primordium has formed as a result. Following this, the liver and intrahepatic biliary tree develops from the cranial portion of the liver diverticulum and the intrahepatic biliary tree is derived from the caudal portion.45 At around E13.5 when hepatoblasts have undergone specification and migration to the septum transversum mesenchyme surrounding the liver bud, they proliferate and then differentiate into hepatocytes and cholangiocytes.46 Through the use of experimental studies involving tissue explants, animal models, and primary culture systems utilizing stem cells, several integral cellular and molecular signaling pathways have been implicated in the tight regulation of early hepatogenesis.47–52
Hhex and the Beginnings of Liver Formation
Hhex (also referred to as proline-rich homeodomain gene (Prh), Hex in rodents, and Xhex in Xenopus) is a homeobox gene. It was initially discovered in studies involving chicken embryos where it was found to be widely expressed in several areas including, vascular endothelial cells, thyroid gland primordia, liver, lung, and hematopoietic cells.53,54 Following initial identification, several Hhex homologs were discovered in the dorsal endomesoderm within gastrula-stage Xenopus embryos, primitive endoderm of mouse embryos, and differentiation of F9 cells into visceral endoderm cells within rodent embryos at 15 days-old.55 It is well established that Hhex is tightly conserved across many multicellular organisms, including mice, humans, and pigs.56
Through the use of in situ hybridization studies, it has been demonstrated that expression of Hhex occurs within the blastocyst’s primitive endoderm as early as E4.5 and by E5.5, and is confined to a collection of visceral endodermal cells localized at the distal end of the developing embryo.57 At E7.5, Hhex’s expression is limited to the visceral endoderm, but it begins to increase in the chorion region of the ectoplacental cavity.58 Following this at E8.5, localization of Hhex to the anterior portion of the foregut endodermal tissue, which is principally involved in the formation of the liver diverticulum - a region vital for primary hepatogenesis - occurs.59,60 Expression of Hhex remains elevated in both the thyroid primordium and hepatic anlage from E9.5 to E16.5, although the latter half mainly involves expression in the thyroid, pancreas, liver, lung, and thymus.59,61 By E18.5, the expression of Hhex is significantly reduced in the lungs but persists in both the bile ducts and thyroid.61 As such, these findings demonstrate that Hhex is consistently expressed throughout various stages of development and cements its vital role in hepatogenesis.
Given that Hhex is expressed very early on in hepatogenesis, it is one of the most basic molecular markers of the anterior endoderm within the developing embryo and alongside albumin, its consistent expression is suggestive of committed hepatoblasts.57,61 The principal means by which Hhex fosters endoderm organogenesis is in supporting a transition to pseudostratified epithelium. In the elementary stages of hepatogenesis, around E8, Hhex is regulated by FGF and BMP signaling pathways.62 Moreover, several studies have shown that Hhex expression is transactivated by GATA4 and HNF3β through direct binding to its promoter region. It has also been shown that elimination of Hhex completely blunts hepatogenesis in mouse models, and Hhex knockout mice are able to promote hepatic specification, but liver bud morphogenesis fails. This ultimately results in the formation of hepatic structures devoid of parenchymal cell components.57,63 However, some of the earliest genes specific to the liver (actin, albumin, transthyretin) remain activated in the ventral endoderm of Hhex knockout mice at E8.5.64
The importance of Hhex was further cemented by mouse studies utilizing Hhex-null embryos, which showed that the endoderm was able to initiate the expression of hepatic genes at E8.5, but expansion of the liver bud was blunted, despite survival of hepatic endodermal cells with detectable expression of genes within the midgut epithelium, such as Shh.43,64,65 It is, however, significant to note that in these mice embryos, there was hepatoblast migration into surrounding mesenchymal tissue and with corresponding arrest of hepatogenesis at E9.5, resulting in embryonic lethality.58
It has also been well documented in the literature that in early hepatoblasts, conditional ablation of Hhex expression significantly disrupts the formation of intrahepatic bile ducts and the differentiation of hepatoblasts into hepatocytes (Figure 1).66 Although complete Hhex knockouts are undoubtedly lethal, studies involving conditional Hhex knockouts generated by means of transgenic, vav-Cre mice were, in fact, viable. Furthermore, these studies illustrated that Hhex expression is vital at multiple stages for the proper differentiation of both hematopoietic stem cells and hematopoietic progenitor cells.67 When examining embryoid bodies obtained from complete Hhex knockouts, ESCs possessed deficits in the formation of hematopoietic and granulocyte-monocyte cell colonies. In contrast, null Hhex mutants were unable to develop the hepatopancreatic duct system and possessed noticeable abnormalities in cardiac and vascular development.58,68–70
 |
Figure 1 Phenotypes of wild-type: WT and HHEX knockout: KO embryos. (A) E18 pig embryos; (B) E9.5 and E10.5 mouse embryos and Hhex KO. |
Further evaluation of the molecular marker involved functional studies of the HHEX protein and ultimately revealed that HHEX could act as either a transcriptional activator or repressor.67–69 When HHEX served as a transcriptional activator, it was found to promote HNF1ɑ expression, a transcription factor principally involved in liver differentiation.71 Moreover, when serving as a transcriptional repressor, HHEX was involved in the specification of anterior embryonic structures through direct suppression of genes responding to Spemann organizing signals, such as Chordin and Goosecoid signals that are vital for anterior patterning.72 As such, these data collectively highlight the extreme importance of Hhex not only for regulation of the initiation of hepatogenesis, but morphogenesis of the intrahepatic bile duct and hepatoblast differentiation as well.
The Application of Blastocyst Complementation for Hepatogenesis
Blastocyst complementation has attracted substantial attention in recent years for its potential to serve as an alternative avenue for organ regeneration. The technique is centered around the generation of cells and even whole organs of one animal species, the donor, through the use of another animal species in utero, the recipient.73–76 When compared to other contemporary methods for organ regeneration, such as the previously discussed implementation of PHH transplantation for ESLD, blastocyst complementation has the benefit of requiring remarkably few PSCs, and most of the inductive cues required for hepatogenesis are already present within the developing blastocyst.77–79 In reference to blastocyst complementation for hepatogenesis, genetic modification of the recipient species’ blastocysts occurs such that key gene(s), such as the aforementioned Hhex, required for hepatogenesis and liver bud development are knocked out, and subsequently complemented with a microinjection of PSCs from the donor species – often ESCs or iPSCs (Figure 2). Following this, the complemented blastocysts are then directly implanted into the uterus of a pseudo-pregnant animal of the recipient species.80,81 At the completion of the gestation period, the complemented offspring born from the recipient species possess chimeric livers entirely derived from the PSCs injected from the donor species. The resulting humanized livers would then be transplanted into a human patient with ESLD (Figure 3).
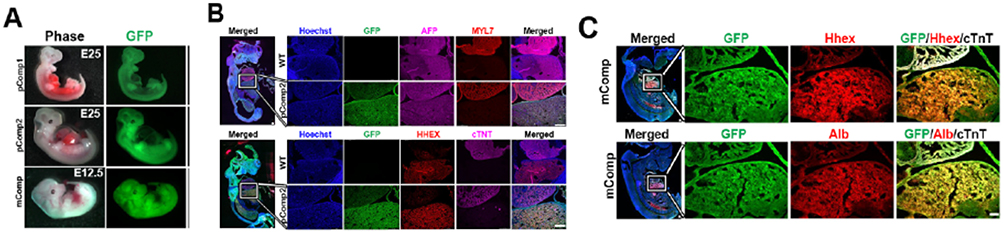 |
Figure 2 Mouse and pig complemented embryos. (A) Whole embryo images showing GFP expression and the developing liver. IHC images of the pig (B) and mouse (C) complemented embryos showing the liver-heart area in the insets in the magnifications. mComp: mouse complemented embryo; pComp1: pig complemented embryo 1; pComp2: pic complemented embryo 2; MYL7: myosin light chain 7; cTNT: cardiac troponin T (white signal in mouse); ALB: albumin. |
 |
Figure 3 Visual representation of blastocyst complementation for ESLD in humans. |
The feasibility of blastocyst complementation was first demonstrated in 1993 when mouse-derived blastocysts from a Rag2 knockout strain without mature lymphocytes were microinjected with mouse ESCs.82 The chimeric offspring produced from the Rag2 knockout mice were found to possess mature B and T lineages entirely derived from the donor ESCs. Subsequently, wild-type murine ESCs were microinjected into blastocysts derived from a mutant mouse strain known as aphakia which possesses the affliction of being unable to develop an ocular lens. This resulted in the creation of allogenic chimeras that possessed normal ocular lenses secondary to complementation.75 These promising results spurred further complementation studies responsible for the creation of a chimeric kidney, pancreatic islets, and thymus in mice, and an exogenic pancreas was even developed in pigs.25,83–85 Furthermore, advancements in gene editing also promoted additional investigation in the field by enabling efficient development of robust interspecies chimeric organs. In particular, the CRISPR/Cas9 and TALEN gene editing techniques have become the primary methods for blastocyst modification due to their groundbreaking efficiency in genetic modification and broad accessibility.86,87
The aforementioned successes with chimeric organs produced within organisms of the same species laid the foundation for additional experimental exploration involving interspecies, rather than intraspecies, chimeras. One of the first endeavors that was fruitful in producing an interspecies chimeric organ was centered upon the inactivation of a gene required for the development of the pancreas in mice, known as Pdx1. Subsequent microinjection of PSCs from wild-type rats into mouse blastomeres resulted in interspecies chimeric offspring possessing rat-derived pancreases.88 Following this triumph, interspecies blastocyst complementation was applied for the synthesis of several additional chimeric organs, including the growth of rat-derived kidneys and thymuses in mice.85,89 With this in mind, several studies began investigating whether human PSCs could be used to generate chimeric organs using interspecies blastocyst complementation, including that of livers for OLT. However, the utilization of interspecies blastocyst complementation for the purposes of xenotransplantation would require several steps. Principally, mutant blastocysts with knockout modifications for the expression of integral gene(s) involved in liver formation would need to be generated. As previously mentioned, the gene Hhex is considered to be one of the most vital genes for early hepatogenesis - particularly with regard to the synthesis of hepatocytes and cholangiocytes, morphogenesis of bile ducts, and hepatoblast differentiation.43,58–60 As such, it is notably apparent that the creation of mutant blastocysts lacking Hhex possesses substantial potential for interspecies blastocyst complementation aimed at producing human chimeric organs for xenotransplantation.
A pioneering study detailing the compensation of Hhex+ cells in hepatogenesis was that of Bort et. al. The study was centered upon elucidation of the precise mechanism underlying the emergence of progenitor cells from the epidermal epithelium and subsequent initiation of organogenesis. The experimental design involved developing chimeras of E9.5 and microinjecting Hhex knockout ESCs into Hhex+ and GFP+ blastocysts with corresponding evaluation of which cells were contributing to the formation of the liver bud. It was found that the Hhex knockout ESCs were substantially involved in the formation of dorsal and posterior embryonic structures, with no significant contribution to the development of the liver bud.43 These findings provided a rationale for the previous observations from chimeric Hhex knockout mice embryos, where the elimination of Hhex consistently resulted in embryonic mortality. With this in mind, there was a query regarding whether the microinjection of Hhex+ PSCs into a Hhex knockout embryo would allow for entirely chimeric hepatogenesis. Clearly, given the aforementioned similarities between human and pig livers, investigation of this hypothesis in porcine models was ideal, and similar to mice, Hhex expression is also integral to hepatogenesis in pigs.
A study led by Matsunari et. al utilized the TALEN gene editing technique in fetal porcine fibroblasts to create mutations in exon 1 of the Hhex gene sequence and then conducted somatic cell nuclear transfer with the mutated fibroblasts as donors for compensation of Hhex knockout embryos. In embryos lacking Hhex expression, no successful liver development was observed, in turn illustrating inadequate hepatogenesis in pigs lacking Hhex expression and mirroring previous observations in mice. Collectively, these findings also cemented that Hhex knockout blastocysts could generate whole livers following intraspecies blastocyst complementation. However, despite the promising nature of these findings, it is important to acknowledge a major shortfall in that out of the 95 complemented blastocysts, only 3 produced chimeric fetuses with normal liver development and function akin to wild-type, non-complemented, pig fetuses.25 Nevertheless, the promising nature of the study’s findings remains, and there is clearly further investigation required to examine whether pig-derived PSCs could produce a whole liver in Hhex knockout pigs, similar to that of results previously described by Bort et. al.43
Feasibility and Ethical Challenges for Interspecies Blastocyst Complementation
Relevant Questions and Concerns
When comparing interspecies blastocyst complementation to other noteworthy techniques discussed in reference to alleviating the scarcity of organs for OLT, it has the clear advantage of being able to construct an entire liver in utero. The large-scale implementation of human PSCs and interspecies blastocyst complementation in pigs for the generation of chimeric human livers could transform the current landscape of regenerative medicine and OLT. Moreover, with contemporary advances in personalized medicine, the PSCs used for generation of a chimeric liver through blastocyst complementation could even be derived from the patients themselves. Nevertheless, while chimeric organs and their potential for xenotransplantation present a wealth of opportunity to advance contemporary medical knowledge and practices, the path towards generating chimeric organs in domestic livestock remains riddled with challenges. As such, there are several questions that must be answered through scientific experimentation and inquiry before sufficient progress can be made for interspecies blastocyst complementation and xenotransplantation in hepatic medicine. These include, (i) how and to what extent do the injected human PSCs contribute to liver-specific vascular development and organization; (ii) in chimeric livers, which hepatic cell types do the xenogenic injected stem cells produce; and (iii) is it possible for human PSC-derived liver cells to cooperate with donor-derived liver cells to permit proper hepatic function?
These pertinent questions have arisen secondary to a hum of anxiety surrounding the interaction between liver cells of xenogenic origin, and those derived from the host. It is well established that the more plentiful, host-derived liver cells would contribute more robustly to vascular development of the liver and other associated tissue, so there is no guarantee that human cells would function properly in a pig. However, it is not just the human cells that may be of concern. There is also the thought that host-derived endothelial cells remain in the chimeric livers and thus, could trigger an immune response following xenotransplantation. With this in mind, there has been considerable effort to address and alleviate these immune concerns. This has included the modification of xenoantigens on pig vascular endothelial cells, such as glycosylated epitopes. Thus, if the aforementioned challenges could be rectified in an efficient manner, the application of interspecies blastocyst complementation and xenotransplantation harbors notable potential for the treatment of ESLD and other aspects of regenerative medicine.
Feasibility and Potential for Large-Scale Development of Chimeric Livers
Interspecies blastocyst complementation possesses a unique advantage with its rapid turnaround time for the growth of chimeric organs, which equates to the recipient animal’s gestation period. In pigs, the gestation period is approximately 114 days, and a sow can produce up to two litters per year, with an average of 16 piglets per litter.25,90 As such, the quantity of chimeric livers necessary for hepatic xenotransplantation could be accomplished in an efficient manner. Moreover, it is also important to note that the HHEX protein plays an instrumental role in organ development as a whole, and therefore other organs such as the gallbladder, lung, and pancreas could also be produced simultaneously alongside the liver in human-pig chimeras with blastocysts containing the corresponding knockouts.60,61,91–93 As an example, if the CRISPR-cas9 system was utilized to create a triple knockout porcine blastocyst with elimination of Sal1, Pdx1, and Hhex genes, human PSCs could be injected to produce a human-pig chimeric kidneys, pancreas, and liver, all within the same pig.
Interspecies blastocyst complementation also possesses an advantage through its application to personalized medicine. If patient-derived PSCs were used for interspecies blastocyst complementation, the resulting chimeric liver would be genetically and immunologically specific to that patient (Figure 4). As such, xenotransplantation of patient-specific livers could be a major step towards preventing any adverse immune response, such as xenograft rejection, following the procedure. Nevertheless, as previously discussed, the chimeric liver, regardless of whether patient-derived cells were used for complementation, still receives much of its vascular development from recipient-derived cells and therefore, immunosuppressive therapy may still be required for proper function of the chimeric organ.77 It is also pertinent to discuss the potential that chimeric livers have for the treatment of ESLD outside of xenotransplantation. As example, innovative drugs and therapies could be applied to human-pig chimeric livers to assess efficacy before clinical studies. In addition, human-pig chimeric hepatocytes could be isolated and used for cell therapy treatments in patients with ESLD, such as a liver assist device. For example, Takebe et. al reported success with the development of functional IPSC-derived liver buds in vitro that were then transplanted into a drug-induced human liver failure model. Following transplantation, the liver bud transplants connected to the host vasculature and became functional within 48 hours, underscoring the potential of chimerism for ESLD outside of whole organ generation.94 Simply stated, there are many potential avenues for the application of human-pig chimeric livers in the contemporary treatment of ESLD.
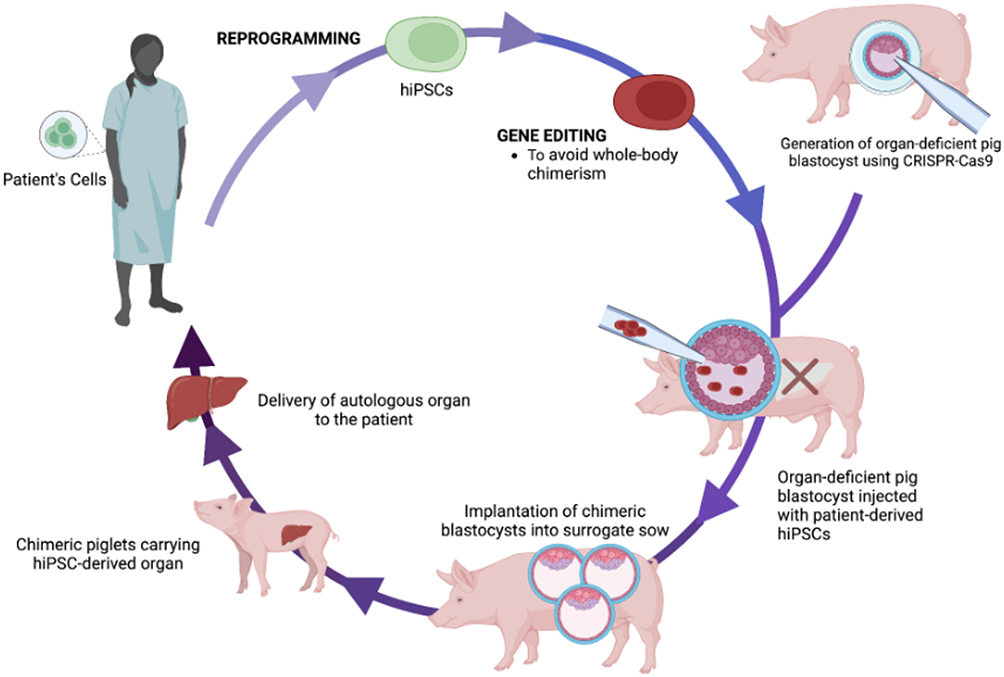 |
Figure 4 Schematic representation of proposed workflow for generation of personalized chimeric livers as a treatment for ESLD through isolation and subsequent utilization of patient-derived hiPSCs for interspecies blastocyst complementation. |
Ensuring Necessary Engraftment and Pluripotency for Efficient Complementation
At present, there are several challenges to overcome with interspecies blastocyst complementation, including the environmental niche and pluripotency of PSCs. Several complementation experiments involving mouse and rat blastocysts with both human iPSCs and ESCs have had only intermittent success with producing chimeras.85 These pitfalls in achieving consistent chimera production have been mirrored by similar results from experiments involving mouse embryos and human PSCs.73,95 At present, it is posited that these difficulties are principally due to growth and differentiation issues following injection. A recent study utilizing naive human PSCs for blastocyst complementation in non-human primates reported that PSCs exhibited extremely low colonization in utero due to an immediate stall in the G1 phase of cell cycle and premature differentiation following injection. This lack of chimeric competence existed for the human PSCs regardless of the host species.96 However, this problem appears to persist beyond rodent and human cells. For experiments involving pigs, robust engraftment of naive human PSCs has been observed in pre-implantation porcine and bovine blastocysts but only contributed minimally to post-implantation embryos. In fact, when assessing the cellular composition of post-implantation pig embryos following blastocyst complementation with human PSCs, approximately 1 out of every 100,000 cells were that of human origin and the more human cells contained within the chimeric embryo, the less developed the embryo was overall.36 In a separate study involving monkey-pig chimeras, over 4000 pig embryos underwent blastocyst complementation with monkey-derived ESCs, and 10 piglets made it to term, with only two of those piglets being chimeras. Tragically, all 10 piglets then died within one week of birth due to unknown medical issues.39
These studies collectively highlight the difficulty of the challenges associated with the growth of chimeric organs. However, the solution to these pervasive problems may be within reach. A unique type of stem cell known as a region-specific PSC can be isolated from mouse embryos and generated in human and primate PSCs through the modulation of cell culture parameters. These stem cells have had notable success with engraftment into gastrulation-stage mouse embryos.97 Thus, current issues surrounding blastocyst engraftment may be ameliorated through modification of PSCs using growth factors and small molecule inhibitors in cell culture medium during growth. Moreover, a landmark study from Cao et. al recently reported promising results from blastocyst complementation using ESCs under optimized cell culture conditions that produced a live birth of a chimeric monkey with high, 20–90%, donor cell contribution. These results have major implications for the future study of PSCs and optimization of corresponding cell culture conditions for blastocyst complementation.98
It has been previously shown that murine ESCs can exist in two pluripotent forms, ground form or naive form.99–101 Moreover, there are distinct murine ESCs derived from the post-implantation state known as epiblast-derived stem cells that are often found in a pre-pluripotent state. These epiblast-derived stem cells are notable because relative to human ESCs, they share a great deal of their gene expression and signaling patterns, making human iPSCs more similar to epiblast-derived stem cells than even murine ESCs.102 Human PSCs are primed for pluripotency and are often committed to lineage-specific development but remain epigenetically restricted. This is in direct contrast to naive PSCs which demonstrate a high degree of differentiation and can produce chimeric organisms via blastocyst complementation.103 As such, the utilization of naive human PSCs would be optimal for maximizing the likelihood of successful human-pig chimeric liver formation. Fortunately, the transformation of primed human PSCs into naive cells has been previously accomplished through altered cell culture conditions or exogenous expression of select transcription factors.94,104 Nevertheless, a well-characterized issue with this process lies in the fact that naive human PSCs possess a strong tendency to form karyotype abnormalities in prolonged cell culture conditions.91 A recent experiment involving direct isolation of naive human PSCs from an inner cell mass could be a solution to this problem.105
The evolutionary basis of interspecies cell competition has also been suggested as another contributing factor for low chimeric competence. Zheng et al conducted research on interspecies cell competition in tissue culture aggregates, investigating various paired combinations. They found that cells from distantly related species engaged in vigorous competition, resulting in the survival of cells from the victorious species while cells from the defeated species underwent apoptotic cell death.106 As such, when utilizing human PSCs for blastocyst complementation of pig embryos, this type of competition is a substantial barrier for ensuring a high degree of chimeric competence. If interspecies cell competition is based on evolutionary distinction, it has been proposed that embryonic stage-matching of the donor and recipient species may reduce the likelihood of competition.
Shetty et al conducted in silico stage-matching of different species to elucidate whether there were optimal embryonic stage matches to maximize the likelihood of success when generating chimeras via interspecies blastocyst complementation. Through their stage-matching analyses, the team identified the human blastocyst (E6/E7), the gastrulating mouse embryo (E6–E6.75), the marmoset late inner cell mass, and the pig late blastocyst as the four stages that displayed the closest similarities to each other. These stage-matching results have the potential to provide invaluable insights for both in vivo and in vitro interspecies chimerism and blastocyst complementation investigations. It is hoped that these results can be applied to pair donor cells with host embryos from different species, leading to improved chimerism for organogenesis enhancement.107 Moreover, another recent development that may assist in maximizing chimeric success is the emergence of improved methods for quantifying mixed cell types in chimeric and mosaic tissues. Suchy et. al created a droplet digital PCR assay that can discriminate single-nucleotide variations and is designed to detect chimerism among mouse strains. Relative to existing methods for their detection, this assay is unique in that it is compatible with crude lysates from all solid organs, simplifying the preparation process. It is adaptable to a range of chimeric species, and it can measure cell populations in chimeric and mosaic tissues with a high degree of accuracy. As such, with emergence of cutting-edge methods to better understand and quantify chimerism, the field can further optimize the creation of chimeric organs using blastocyst complementation.108
A possible solution for both the developmental arrest of human PSCs and interspecies cell competition was proposed by a recent study investigating the efficacy of eliminating insulin-like growth factor 1 (Igf1r) to facilitate the formation of a “cell-competitive niche” for interspecies blastocyst complementation.109 Through the deletion of Igf1r in mouse embryos, researchers were able to evade the deleterious developmental arrest in G1 and improve mouse-rat organ chimerism in mouse embryos, establishing the possibility of similar success with experiments involving naive human PSCs (Figure 5).110 Similarly, Yuri et. al reported success with a reverse-blastocyst complementation method in a Fgfr2b-deficient mouse model with the formation of rat-derived lungs. Their tetraploid-based organ complementation successfully generated functional lungs in a mouse model and more importantly, the rat epithelial lung cells exhibited no deficits in their developmental timing following injection.111 Moreover, a separate study recently reported novel human PSCs that possess broader differentiation and chimerism potential than naive cells. They had the ability to undergo differentiation into embryonic and extra-embryonic lineages in vivo and successfully form chimeras.112 These findings are instrumental in addressing the challenges and roadblocks surrounding pluripotency in the production of human-pig chimeric livers.
 |
Figure 5 Applications of Igf1r-null blastocyst complementation for rat-mouse and human-pig chimeras. |
Ethical Considerations
As interspecies blastocyst complementation has gained growing interest from the scientific community and even the public, there has been a parallel increase in ethical concerns surrounding its application for regenerative medicine. This is due, primarily, to its potential in generating human tissues and organs in animals, including brain tissue and germ cells.113,114 As previously discussed, Hhex is broadly involved in the formation of several organs, and this includes an integral role in the development of the forebrain.41,61,62 With this in mind, there is a reasonable possibility that interspecies blastocyst complementation with Hhex-expressing human PSCs could assist in neurogenesis alongside other organs that necessitate Hhex involvement during their development. Consequently, there is some basis for concerns regarding the production of humanoid brains through the Hhex model of interspecies blastocyst complementation. Nevertheless, a potential solution to this problem in the context of interspecies blastocyst complementation to generate human livers is direct differentiation of injected PSCs into hepatic lineage. This workaround is feasible, as previously demonstrated by Kobayashi et. al with the development of chimeric pancreases in mice via directed expression of Mix-like protein 1 to subsequently produce murine ESCs and endodermal organs following their injection into the blastocyst.115
One intriguing option is to extend the lifespan of ESCs to facilitate a greater degree of engraftment. Masaki et. al demonstrated the efficacy of this option by upregulating the expression of anti-apoptotic protein BCL2, which in turn augmented the survival of rat derived ESCs and facilitated their integration into pre-implantation blastocysts, alongside endothermal progenitor cells possessing Sox17. The increased BCL2 expression enabled the contribution of the Sox17 progenitor cells and rat ESCs within the mouse blastocyst to promote successful development of rat-mouse embryonic chimeras.116 Moreover, a recent study led by Hasimoto et. al illustrated a means by which chimeric organisms could be generated in the absence of any contribution to neural or germline cell development while still utilizing interspecies blastocyst complementation.26 This was first accomplished by creating mutant murine ESCs lacking the genes Prdm14 and Otx2, both of which are involved in the specification of the forebrain and primordial germ cells in mice.117,118 Following this, the mutant murine ESCs, which were also modified to express GFP, were microinjected into Pdx1 knockout murine blastocysts. Post-mortem tissue analysis of these chimeric mice revealed that the mutant ESCs did not participate in the development of neural or germline cells.26 An alternative solution for this problem would be to simply use liver-specific progenitor or stem cells in the place of PSCs, ensuring that only liver or liver-adjacent cells are formed following interspecies blastocyst complementation. For example, liver-derived progenitor cells could be implanted into a Hhex knockout blastocyst to only produce a chimeric liver from blastocyst complementation, while leaving the other organs untouched.119
Nevertheless, it is important to note that since 2015, the NIH has maintained a moratorium on funding for research involving human-animal chimeras generated through the insertion of human cells into early-stage, non-human vertebrate embryos.120 This moratorium was placed due to the sensitive nature of human-animal chimeras, but it has served as a major barrier for productive discussions surrounding the creation of regulations for the field. Furthermore, these restrictions also presently incentivize US based researchers to pursue research and product development related to the generation of human-animal chimeras to countries outside the US that possess fewer or no restrictions, in turn raising concerns for larger issues such as international regulations.121,122 As such, effective discussions centered upon regulation of research involving human-animal chimeras are necessary to allow for proper progression of the field, both at the national and international level.
Alternative Targets for Exclusive Formation of Chimeric Livers
In addition to the myriad of genes identified to ensure exclusive development of chimeric livers following interspecies blastocyst complementation, ubiquitin C (UbC) is also of interest as well as male abnormal 21-like protein 2 (Mab21L2) and transcription factors Tbx3, OC-1, and OC-2. UbC became a potential target following knockout experiments where mouse embryos would consistently perish in utero between E12.5 and E14.5. It was later revealed that this fatality was due to significant defects in hematopoietic stem cells as well as blunted proliferation of both bipotent liver epithelial progenitor cells and hepatocytes.123 When expression of UbC was returned in the mouse embryos, the previous deficits in hepatogenesis were completely reversed, thereby confirming a potentially vital role of UbC in liver development that has yet to be further elucidated.124 Mab21L2 gained attention when it was found to be expressed in the septum transversum mesenchyme and its absence during embryonic development in a mutant mouse model resulted in inadequate morphogenesis of the liver and diminished hepatoblast proliferation with demise occurring between E11.5 and E13.5.125 It has also been noted that the Mab21L2 protein is necessary for cardiac and ocular development as well. Functional analysis of Mab21L2 has shown that the protein interacts with the TGF-β pathway in addition to serving as an antagonist for BMP4 through its interactions with SMAD1.126
Research involving genetically modified mice has demonstrated that inhibiting notch signaling transcription factor Tbx3 can hinder the transformation of hepatoblasts into biliary epithelial cells, which ultimately form the biliary tree.127 OC-1 and OC-2, another set of transcription factors, are also possible targets secondary to the role they play in the production of ɑ2-macroglobulin and follistatin. These two molecules act as inhibitors of the TGF-β/activin pathway, which is crucial for the proper formation of the biliary tree. Additionally, during liver development, OC-1 and OC-2 also facilitate the migration of hepatoblasts within the septum transversum by promoting the degradation of the basal lamina surrounding the liver bud.8,128 Like Hhex, these handful of genes and transcription factors are of interest as potential targets for the exclusive formation of chimeric livers through interspecies blastocyst complementation in pigs. Finally, it is reasonable to predict that the complex ethical conundrum surrounding interspecies blastocyst complementation will ultimately be addressed to allow the development of a human-pig chimeric liver for transplantation.
Viral Transmission and Xenotransplantation
In addition to ethical considerations, there are also substantial safety concerns surrounding the xenotransplantation of chimeric livers into patients with ESLD given the elevated risk of associated zoonotic infections. Despite no reported cases of porcine xenografts causing infections, there is still the potential to transmit several viral infections including: porcine-derived lymphotropic herpesvirus, cytomegalovirus, and endogenous retroviruses in addition to hepatitis E genotypes 3 and 4, all of which can cause disease in humans if the xenografts are not robustly tested to ensure their safety.127–131 Furthermore, porcine influenza viruses, such as the infamous H1N1 virus that attacks airway epithelial cells in humans, present significant concerns for pig-derived xenografts.132 With this in mind, proper screening and surveillance of porcine xenografts will be of the utmost importance in order to ensure maximal safety and feasibility when utilizing xenotransplantation for the treatment of ESLD.133 Nevertheless, there are myriad means by which the field is attempting to mitigate as much of the viral transmission risk as possible for porcine xenografts. Yang et. al recently demonstrated the effectiveness of genome-wide inactivation of porcine endogenous retroviruses in preventing zoonotic infection in porcine renal epithelial cells. Through use of the CRISPR-cas9 system, researchers were able to remove 25 copies of porcine endogenous retroviruses from the genomes of pigs. These animals were then genetically engineered to highly express 9 human transgenes that augmented their blood-coagulation and immunological compatibility with humans.134 Furthermore, researchers removed three major xenoantigens from these pigs and in turn reduced the likelihood of xenograft rejection secondary to a host immune response.135 Host production of xenoantibodies is a daunting obstacle for safe and effective clinical xenotransplantation, but this study illustrated the feasibility of successful xenoantigen deletion in pigs.
Host Rejection of Xenografts
Perhaps the most formidable challenge for clinical xenotransplantation of chimeric livers is that of xenograft rejection. There are several potential causes of graft rejection, all of which serve as major obstacles for successful implementation of xenotransplantation. The first significant cause is that of hyperacute xenograft rejection, which occurs when antibodies from the graft recipient bind to xenoantigenic epitopes present on porcine-derived endothelial cells and activate the complement cascade, swiftly resulting in annihilation of the graft vasculature and rejection of the graft.136 Another significant cause of rejection is acute humoral xenograft rejection, which is initiated by a combination of the recipient’s cellular and humoral immune responses.137,138 The third major cause of rejection is acute cellular xenograft rejection, which is executed by the recipient’s immune cells following activation of their adaptive and innate immune systems.139,140 In order to overcome these critical challenges, efforts are directed towards the development of genetically modified pigs that lack primary components involved in the three major causes of graft rejection. Recently, to combat hyperacute xenograft rejection, α1,3-galactosyltransferase genetic knockout pigs were generated, and renal xenograft survival was drastically improved.141–143
Another pathway to address known difficulties surrounding hyperacute xenograft rejection is through modification of host endothelial cells, as they are involved in graft rejection for every organ. ETV2, a master regulator of hematoendothelial lineages, has become a major target for the generation of human-pig chimeras with human-derived endothelial cells to minimize the likelihood of hyperacute graft rejection.76 Due to its pivotal role in regulation and development of hematoendothelial lineages, deletion of ETV2 results in an absence of all blood and vasculature, thereby, ensuring the creation of an empty niche for interspecies blastocyst complementation.144 Das et. al successfully generated ETV2-null blastocysts and subsequently performed interspecies blastocyst complementation with human PSCs to create chimeric embryos from entirely human-derived endothelial cells.145,146 Moreover, a different group recently published promising results with efficient in vitro generation of ETV2-null pigs through the use of CRISPR-cas9 mediated genome modification.147 Alternatively, to combat immunological rejection of xenografts, genetically modified pigs have been engineered to express complement-regulatory proteins CD46, CD55, and CD59 in an attempt to repress any activation of the recipient’s complement cascade.148 Collectively, these findings have demonstrated novel avenues to effectively combat xenogeneic immunity following xenotransplantation and extend graft survival.
While substantial strides have been made to meet and overcome these barriers for feasible xenotransplantation of chimeric livers, pioneering cases have been unsuccessful. To date, the only xenotransplantation of a pig-derived liver into a human involved a 26-year-old patient with hepatitis. The xenograft was a heterotopic liver from a wild-type pig that was intended to serve as a bridge to allotransplantation. Unfortunately, the xenotransplantation failed and approximately 34 hours following the procedure, the patient died due to terminal brain damage.149 Although this is the sole known instance of pig to human xenotransplantation of a chimeric liver, similar results were observed following the first orthotopic xenotransplantation of livers derived from wild-type pigs into non-human primates. Under those circumstances, the primates exhibited a mean survival time of only 3.5 days following the procedures. Furthermore, a study involving xenotransplantation of livers derived from α1,3-galactosyltransferase genetic knockout pigs did not substantially improve the efficacy of the procedure with the recipient animals showing a mean survival time of 9 days.150 In comparison, relative success was achieved with an augmented mean survival time of 29 days in a study involving xenotransplantation from a pig to a non-human primate alongside administration of an anti-CD40 monoclonal antibody and continuous infusion of a human prothrombin complex in order to prevent any coagulation and permit the recovery of platelet counts.151 Nevertheless, lethal complications of interspecies incompatibility such as thrombotic microangiopathy, coagulopathy, and severe thrombocytopenia still serve as major barriers to safe and effective xenotransplantation of chimeric livers.152
While there are substantial shortcomings for procedural outcomes involving chimeric livers, fruitful results have been obtained for other chimeric organs in pig to primate xenotransplantation studies. As example, the mean survival time for chimeric hearts has been reported to be as long as 945 days, with 499 days for chimeric kidneys.152–154 Furthermore, there was extensive interest from the public and scientific community alike in January of 2022 when a 57-year-old patient with end-stage heart failure underwent the procedure and received a genetically engineered, pig-derived chimeric heart. Following the procedure, the patient exhibited no signs of acute xenograft rejection, and showed strong cardiac function. However, the patient developed sudden onset of heart failure about two months after the xenotransplantation and died shortly thereafter. While the sudden cause of death was not determined, it was discovered afterwards that the pig’s heart was infected with a porcine virus known as porcine cytomegalovirus. Nevertheless, within the scientific community, the patient’s brief survival following the procedure was considered a landmark achievement for the field of regenerative medicine.155
There is growing optimism surrounding the prospects of xenotransplantation for the treatment of ESLD, but additional studies are required for development of novel techniques to combat current pitfalls with xenograft rejection and complications. At present, a major avenue for additional study is centered around the generation of chimeric livers using ESCs from non-human primates to allow for more in-depth investigation of innovative strategies aimed at improving the safety and feasibility of hepatic xenografts. Alternatively, it is hoped that if a chimeric pig liver is not truly a sufficient alternative for the treatment of ESLD, chimeric livers may be able to serve as a “bridge” for OLT of a human liver. The average life expectancy of a patient with fulminant hepatic failure on a transplant waiting list is only 3 days. As such, even if a transplanted chimeric liver is only a suitable option for a few days or weeks, there is immense potential for improved mortality outcomes.156,157
Conclusions
The use of interspecies blastocyst complementation to generate and restore organs in humans within an extraordinarily short timeframe harbors immense potential for the future of regenerative medicine.158 When comparing contemporary techniques posed as solutions to the significant lack of available human livers for orthotopic transplantation in the treatment of ESLD, interspecies blastocyst complementation is an undoubtedly promising avenue. Additional studies are still required to ensure effective generation of human-pig chimeric livers and may even require successful xenotransplantation in large mammals, such as non-human primates, as proof-of-concept. It will be necessary to confirm optimal hepatic functions, such as detoxification and metabolism, and corresponding long-term survival prior to feasible production of chimeric livers using patient-derived PSCs and subsequent xenotransplantation. Regardless, it is clear that the production of human livers by means of interspecies blastocyst complementation demonstrates significant potential to pave a novel therapeutic avenue for ESLD and in turn, alleviate an abundance of human suffering.
When considering the urgent unmet medical need for an additional source or repository of transplantable livers, it is our belief and that of others that the American public is ready to accept human-animal chimera research.159,160 Recent polls in the United States have determined that the general public has an overall positive view towards xenotransplantation.161 The complex ethical and moral profile surrounding the technology remains an omnipresent concern. There is, however, a growing realization that xenotransplantation could provide critical help to those individuals with life-threatening conditions. This sentiment is also supported by the fact that even optimizing allotransplantation and primary prevention of organ failure will not alone alleviate the scarcity of organ donors.161–164 Thus, with further advancements in blastocyst complementation and xenotransplantation, it is critical that researchers and other stakeholders maintain the citizens’ trust through transparency, objectivity, and scientific integrity – only then will these 21st century technologies meet the needs of modern medicine and properly address unmet human suffering.
Acknowledgments
This work was supported, in part, by a grant from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (NIH/NIAID 1R01AI173804-01) to CJS. The authors would like to thank Dr. Andrew T. Crane for providing Figures 1 and 2.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sanchez W, Talwalkar JA. Palliative care for patients with end-stage liver disease ineligible for liver transplantation. Gastroenterol Clin North Am. 2006;35(1):201–219. doi:10.1016/j.gtc.2005.12.007
2. Cox-North P, Doorenbos A, Shannon SE, Scott J, Curtis JR. The transition to end-of-life care in end-stage liver disease. J Hospice Pall Nursing. 2013;15(4):209–215. doi:10.1097/NJH.0b013e318289f4b0
3. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. 2019;70(1):151–171. doi:10.1016/j.jhep.2018.09.014
4. Olson JC. Acute-on-chronic and decompensated chronic liver failure: definitions, epidemiology, and prognostication. Crit Care Clin. 2016;32(3):301–309. doi:10.1016/j.ccc.2016.02.001
5. Center for Disease Control. Saved request: underlying cause of death, 2018–2021, single race, D321F126. Available from: https://wonder.cdc.gov/controller/saved/D158/D321F126.
6. National data, 2021 - OPTN. Available from: https://optn.transplant.hrsa.gov/data/view-data-reports/national-data/#.
7. Founta KM, Papanayotou C. In vivo generation of organs by blastocyst complementation: advances and challenges. Int J Stem Cells. 2021;15(2):113–121. doi:10.15283/ijsc21122
8. Crane AT, Aravalli RN, Asakura A, et al. Interspecies organogenesis for human transplantation. Cell Transplant. 2019;28(9–10):1091–1105. doi:10.1177/0963689719845351
9. Aravalli RN, Belcher JD, Steer CJ. Liver-targeted gene therapy: approaches and challenges. Liver Transpl. 2015;21(6):718–737. doi:10.1002/lt.24122
10. Fitzpatrick E, Mitry RR, Dhawan A. Human hepatocyte transplantation: state of the art. J Intern Med. 2009;266(4):339–357. doi:10.1111/j.1365-2796.2009.02152.x
11. Yagi H, Fukumitsu K, Fukuda K, et al. Human-scale whole-organ bioengineering for liver transplantation: a regenerative medicine approach. Cell Transplant. 2013;22(2):231–242. doi:10.3727/096368912X654939
12. Hannoun Z, Steichen C, Dianat N, Weber A, Dubart-Kupperschmitt A. The potential of induced pluripotent stem cell derived hepatocytes. J Hepatol. 2016;65(1):182–199. doi:10.1016/j.jhep.2016.02.025
13. Choi HJ, Choi D. Successful mouse hepatocyte culture with sandwich collagen gel formation. J Korean Surg Soc. 2013;84(4):202. doi:10.4174/jkss.2013.84.4.202
14. Wang B, Jakus AE, Baptista PM, et al. Functional maturation of induced pluripotent stem cell hepatocytes in extracellular matrix–A comparative analysis of bioartificial liver microenvironments. Stem Cells Transl Med. 2016;5(9):1257–1267. doi:10.5966/sctm.2015-0235
15. Okuyama T, Yamazoe H, Mochizuki N, Khademhosseini A, Suzuki H, Fukuda J. Preparation of arrays of cell spheroids and spheroid-monolayer cocultures within a microfluidic device. J Biosci Bioeng. 2010;110(5):572–576. doi:10.1016/j.jbiosc.2010.05.013
16. Koide N, Sakaguchi K, Koide Y, et al. Formation of multicellular spheroids composed of adult rat hepatocytes in dishes with positively charged surfaces and under other nonadherent environments. Exp Cell Res. 1990;186(2):227–235. doi:10.1016/0014-4827(90)90300-y
17. Meier F, Freyer N, Brzeszczynska J, et al. Hepatic differentiation of human iPSCs in different 3D models: a comparative study. Int J Mol Med. 2017;40(6):1759–1771. doi:10.3892/ijmm.2017.3190
18. Ehrlich A, Duche D, Ouedraogo G, Nahmias Y. Challenges and opportunities in the design of liver-on-chip microdevices. Annu Rev Biomed Eng. 2019;21:219–239. doi:10.1146/annurev-bioeng-060418-052305
19. Mitry RR, Jitraruch S, Iansante V, Dhawan A. Alginate encapsulation of human hepatocytes and assessment of microbeads. Methods Mol Biol. 2017;1506:273–281. doi:10.1007/978-1-4939-6506-9_19
20. Li L, Zhang Y, Pan X. Preparation and characterization of alginate-chitosan microcapsule for hepatocyte culture. Methods Mol Biol. 2017;1479:199–206. doi:10.1007/978-1-4939-6364-5_15
21. Meier RPH, Montanari E, Morel P, et al. Microencapsulation of hepatocytes and mesenchymal stem cells for therapeutic applications. Methods Mol Biol. 2017;1506:259–271. doi:10.1007/978-1-4939-6506-9_18
22. Sauer V, Roy-Chowdhury N, Guha C, Roy-Chowdhury J. Induced pluripotent stem cells as a source of hepatocytes. Curr Pathobiol Rep. 2014;2(1):11–20. doi:10.1007/s40139-013-0039-2
23. Pareja E, Gómez-Lechón MJ, Tolosa L. Induced pluripotent stem cells for the treatment of liver diseases: challenges and perspectives from a clinical viewpoint. Ann Transl Med. 2020;8(8):566. doi:10.21037/atm.2020.02.164
24. Tricot T, Verfaillie CM, Kumar M. Current status and challenges of human induced pluripotent stem cell-derived liver models in drug discovery. Cells. 2022;11(3):442. doi:10.3390/cells11030442
25. Matsunari H, Watanabe M, Hasegawa K, et al. Compensation of disabled organogeneses in genetically modified pig fetuses by blastocyst complementation. Stem Cell Reports. 2019;14(1):21–33. doi:10.1016/j.stemcr.2019.11.008
26. Hashimoto H, Eto T, Yamamoto M, et al. Development of blastocyst complementation technology without contributions to gametes and the brain. Exp Anim. 2019;68(3):361–370. doi:10.1538/expanim.18-0173
27. Wu J, Platero Luengo A, Gil MA, et al. Generation of human organs in pigs via interspecies blastocyst complementation. Reprod Domest Anim. 2016;51(2):18–24. doi:10.1111/rda.1279
28. Ruiz-Estevez M, Crane AT, Rodriguez-Villamil P, et al. Liver development is restored by blastocyst complementation of HHEX knockout in mice and pigs. Stem Cell Res Ther. 2021;12(1):292. doi:10.1186/s13287-021-02348-z
29. Coppiello G, Barlabé P, Moya-Jódar M, et al. Generation of heart and vascular system in rodents by blastocyst complementation. Dev Cell. 2023;58(24):2881–2895.e7. doi:10.1016/j.devcel.2023.10.008
30. Miura A, Sarmah H, Tanaka J, et al. Conditional blastocyst complementation of a defective Foxa2 lineage efficiently promotes the generation of the whole lung. Elife. 2023;12:e86105. doi:10.7554/eLife.86105
31. Tanaka J, Miura A, Shimamura Y, et al. Generation of salivary glands derived from pluripotent stem cells via conditional blastocyst complementation. bioRxiv. 2023;2023:566845. doi:10.1101/2023.11.13.566845
32. Kano M, Mizuno N, Sato H, et al. Functional calcium-responsive parathyroid glands generated using single-step blastocyst complementation. Proc Natl Acad Sci USA. 2023;120(28):e2216564120. doi:10.1073/pnas.2216564120
33. Strell P, Johnson ST, Carchi C, Low WC. Neuronal transplantation for Alzheimer’s disease and prospects for generating exogenic neurons as a source of cells for implantation. Cell Transplant. 2023;32. doi:10.1177/09636897231164712
34. Nykonenko A, Vávra P, Zonča P. Anatomic peculiarities of pig and human liver. Exp Clin Transplant. 2017;15(1):21–26. doi:10.6002/ect.2016.0099
35. Wu J, Platero-Luengo A, Sakurai M, et al. Interspecies chimerism with mammalian pluripotent stem cells. Cell. 2017;168(3):473–486.e15. doi:10.1016/j.cell.2016.12.036
36. Aloia L. The influence of tissue spatial geometry and functional organisation on liver regeneration. Sem Cell Devel Biol. 2022;130:70–78. doi:10.1016/j.semcdb.2021.09.011
37. Ding C, Li Y, Guo F, et al. A cell-type-resolved liver proteome. Mol Cell Proteomics. 2016;15(10):3190–3202. doi:10.1074/mcp.M116.060145
38. Ishibashi H, Nakamura M, Komori A, Migita K, Shimoda S. Liver architecture, cell function, and disease. Semin Immunopathol. 2009;31(3):399–409. doi:10.1007/s00281-009-0155-6
39. Fu R, Yu D, Ren J, et al. Domesticated cynomolgus monkey embryonic stem cells allow the generation of neonatal interspecies chimeric pigs. Protein Cell. 2020;11(2):97–107. doi:10.1007/s13238-019-00676-8
40. Gordillo M, Evans T, Gouon-Evans V. Orchestrating liver development. Development. 2015;142(12):2094–2108. doi:10.1242/dev.114215
41. Tremblay KD, Zaret KS. Distinct populations of endoderm cells converge to generate the embryonic liver bud and ventral foregut tissues. Dev Biol. 2005;280(1):87–99. doi:10.1016/j.ydbio.2005.01.003
42. Zaret KS. From Endoderm to liver bud: paradigms of cell type specification and tissue morphogenesis. Curr Top Dev Biol. 2016;117:647–669. doi:10.1016/bs.ctdb.2015.12.015
43. Bort R, Signore M, Tremblay K, Martinez Barbera JP, Zaret KS. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev Biol. 2006;290(1):44–56. doi:10.1016/j.ydbio.2005.11.006
44. Rossi JM, Dunn NR, Hogan BL, Zaret KS. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001;15(15):1998–2009. doi:10.1101/gad.904601
45. Shiojiri N. Analysis of differentiation of hepatocytes and bile duct cells in developing mouse liver by albumin immunofluorescence. Dev Growth Differ. 1984;26(6):555–561. doi:10.1111/j.1440-169X.1984.00555.x
46. Watt AJ, Zhao R, Li J, Duncan SA. Development of the mammalian liver and ventral pancreas is dependent on GATA4. BMC Dev Biol. 2007;7(1):37. doi:10.1186/1471-213X-7-37
47. Haworth K, Samuel L, Black S, Kirilenko P, Latinkic B. Liver specification in the absence of cardiac differentiation revealed by differential sensitivity to Wnt/β catenin pathway activation. Front Physiol. 2019;10:155. doi:10.3389/fphys.2019.00155
48. Amaya E, Musci TJ, Kirschner MW. Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell. 1991;66(2):257–270. doi:10.1016/0092-8674(91)90616-7
49. McLin VA, Rankin SA, Zorn AM. Repression of Wnt/β-catenin signaling in the anterior endoderm is essential for liver and pancreas development. Development. 2007;134(12):2207–2217. doi:10.1242/dev.001230
50. Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–947. doi:10.1038/nature03649
51. Uemura M, Hara K, Shitara H, et al. Expression and function of mouse Sox17 gene in the specification of gallbladder/bile-duct progenitors during early foregut morphogenesis. Biochem Biophys Res Commun. 2010;391(1):357–363. doi:10.1016/j.bbrc.2009.11.063
52. Sosa-Pineda B, Wigle JT, Oliver G. Hepatocyte migration during liver development requires Prox1. Nat Genet. 2000;25(3):254–255. doi:10.1038/76996
53. Yatskievych TA, Pascoe S, Antin PB. Expression of the homeobox gene Hex during early stages of chick embryo development. Mech Dev. 1999;80(1):107–109. doi:10.1016/s0925-4773(98)00204-4
54. Crompton MR, Bartlett TJ, MacGregor AD, et al. Identification of a novel vertebrate homeobox gene expressed in haematopoietic cells. Nucleic Acids Res. 1992;20(21):5661–5667. doi:10.1093/nar/20.21.5661
55. Bedford FK, Ashworth A, Enver T, Wiedemann LM. HEX: a novel homeobox gene expressed during haematopoiesis and conserved between mouse and human. Nucleic Acids Res. 1993;21(5):1245–1249. doi:10.1093/nar/21.5.1245
56. Morgutti M, Demori E, Pecile V, Amoroso A, Rustighi A, Manfioletti G. Genomic organization and chromosome mapping of the human homeobox gene HHEX. Cytogen Cell Genet. 2001;94(1–2):30–32. doi:10.1159/000048778
57. Thomas PQ, Brown A, Beddington RS. Hex: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development. 1998;125(1):85–94. doi:10.1242/dev.125.1.85
58. Keng VW, Fujimori KE, Myint Z, Tamamaki N, Nojyo Y, Noguchi T. Expression of Hex mRNA in early murine postimplantation embryo development. FEBS Lett. 1998;426(2):183–186. doi:10.1016/s0014-5793(98)00342-1
59. Kubo A, Kim YH, Irion S, et al. The homeobox gene Hex regulates hepatocyte differentiation from embryonic stem cell-derived endoderm. Hepatology. 2010;51(2):633–641. doi:10.1002/hep.23293
60. Zhao R, Watt AJ, Li J, Luebke-Wheeler J, Morrisey EE, Duncan SA. GATA6 Is essential for embryonic development of the liver but dispensable for early heart formation. Mol Cell Biol. 2005;25(7):2622–2631. doi:10.1128/MCB.25.7.2622-2631.2005
61. Bogue CW, Ganea GR, Sturm E, Ianucci R, Jacobs HC. Hex expression suggests a role in the development and function of organs derived from foregut endoderm. Dev Dyn. 2000;219(1):84–89. doi:10.1002/1097-0177
62. Zhang W, Yatskievych TA, Baker RK, Antin PB. Regulation of Hex gene expression and initial stages of avian hepatogenesis by Bmp and Fgf signaling. Dev Biol. 2004;268(2):312–326. doi:10.1016/j.ydbio.2004.01.019
63. Denson LA, McClure MH, Bogue CW, Karpen SJ, Jacobs HC. HNF3β and GATA-4 transactivate the liver-enriched homeobox gene, Hex. Gene. 2000;246(1–2):311–320. doi:10.1016/s0378-1119(00)00082-2
64. Lu H, Ma J, Yang Y, Shi W, Luo L. EpCAM is an endoderm-specific Wnt derepressor that licenses hepatic development. Dev Cell. 2013;24(5):543–553. doi:10.1016/j.devcel.2013.01.021
65. Arterbery AS, Bogue CW. Hhex is necessary for the hepatic differentiation of mouse ES cells and acts via Vegf signaling. PLoS One. 2016;11(1):e0146806. doi:10.1371/journal.pone.0146806
66. Hunter MP, Wilson CM, Jiang X, et al. The homeobox gene Hhex is essential for proper hepatoblast differentiation and bile duct morphogenesis. Dev Biol. 2007;308(2):355–367. doi:10.1016/j.ydbio.2007.05.028
67. Goodings C, Smith E, Mathias E, et al. Hhex is required at multiple stages of adult hematopoietic stem and progenitor cell differentiation. Stem Cells. 2015;33(8):2628–2641. doi:10.1002/stem.2049
68. Hallaq H, Pinter E, Enciso J, et al. A null mutation of Hhex results in abnormal cardiac development, defective vasculogenesis and elevated Vegfa levels. Development. 2004;131(20):5197–5209. doi:10.1242/dev.01393
69. Villasenor A, Gauvrit S, Collins MM, Maischein HM, Stainier DYR. Hhex regulates the specification and growth of the hepatopancreatic ductal system. Dev Biol. 2020;458(2):228–236. doi:10.1016/j.ydbio.2019.10.021
70. Gao C, Huang W, Gao Y, et al. Zebrafish hhex-null mutant develops an intrahepatic intestinal tube due to de-repression of cdx1b and pdx1. J Mol Cell Biol. 2018;11(6):448–462. doi:10.1093/jmcb/mjy068
71. Brickman JM, Jones CM, Clements M, Smith JC, Beddington RS. Hex is a transcriptional repressor that contributes to anterior identity and suppresses Spemann organiser function. Development. 2000;127(11):2303–2315. doi:10.1242/dev.127.11.2303
72. Soufi A, Jayaraman PS. PRH/Hex: an oligomeric transcription factor and multifunctional regulator of cell fate. Biochem J. 2008;412(3):399–413. doi:10.1042/BJ20080035
73. Wu J, Izpisua Belmonte JC. Interspecies chimeric complementation for the generation of functional human tissues and organs in large animal hosts. Transgenic Res. 2016;25(3):375–384. doi:10.1007/s11248-016-9930-z
74. Oldani G, Peloso A, Lacotte S, Meier R, Toso C. Xenogeneic chimera–Generated by blastocyst complementation–As a potential unlimited source of recipient-tailored organs. Xenotransplantation. 2017;24(4). doi:10.1111/xen.12327
75. Liégeois NJ, Horner JW, DePinho RA. Lens complementation system for the genetic analysis of growth, differentiation, and apoptosis in vivo. Proc Natl Acad Sci U S A. 1996;93(3):1303–1307. doi:10.1073/pnas.93.3.1303
76. ho CY, Sorensen J, Garry DJ, Garry MG. Blastocyst complementation and interspecies chimeras in gene edited pigs. Front Cell Dev Biol. 2022;2022:10.
77. Sarmah H, Sawada A, Hwang Y, et al. Towards human organ generation using interspecies blastocyst complementation: challenges and perspectives for therapy. Front Cell Dev Biol. 2023;2023:11.
78. Zheng C, Ballard EB, Wu J. The road to generating transplantable organs: from blastocyst complementation to interspecies chimeras. Development. 2021;148(12):dev195792. doi:10.1242/dev.195792
79. Raju R, Chau D, Verfaille C, Hu WS. The road to regenerative liver therapies: the triumphs, trials and tribulations. Biotechnol Adv. 2013;31(7):1085–1093. doi:10.1016/j.biotechadv.2013.08.022
80. Freedman BS. Hopes and difficulties for blastocyst complementation. Nephron. 2018;138(1):42–47. doi:10.1159/000480370
81. Yamaguchi T. Hurdles to generating human islets in animals via blastocyst complementation. Curr Diab Rep. 2019;19(8):45. doi:10.1007/s11892-019-1167-9
82. Chen J, Lansford R, Stewart V, Young F, Alt FW. RAG-2-deficient blastocyst complementation: an assay of gene function in lymphocyte development. Proc Natl Acad Sci U S A. 1993;90(10):4528–4532. doi:10.1073/pnas.90.10.4528
83. Müller SM, Terszowski G, Blum C, et al. Gene targeting of VEGF-A in thymus epithelium disrupts thymus blood vessel architecture. Proc Natl Acad Sci U S A. 2005;102(30):10587–10592. doi:10.1073/pnas.0502752102
84. Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature. 2007;445:7130):886–891. doi:10.1038/nature05537
85. Ichi UJ, Kobayashi T, Yamaguchi T, Knisely AS, Nishinakamura R, Nakauchi H. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am J Pathol. 2012;180(6):2417–2426. doi:10.1016/j.ajpath.2012.03.007
86. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi:10.1038/nprot.2013.143
87. James D, Noggle SA, Swigut T, Brivanlou AH. Contribution of human embryonic stem cells to mouse blastocysts. Dev Biol. 2006;295(1):90–102. doi:10.1016/j.ydbio.2006.03.026
88. Kobayashi T, Yamaguchi T, Hamanaka S, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142(5):787–799. doi:10.1016/j.cell.2010.07.039
89. Isotani A, Hatayama H, Kaseda K, Ikawa M, Okabe M. Formation of a thymus from rat ES cells in xenogeneic nude mouse↔rat ES chimeras. Genes Cells. 2011;16(4):397–405. doi:10.1111/j.1365-2443.2011.01495.x
90. Oliviero C, Junnikkala S, Peltoniemi O. The challenge of large litters on the immune system of the sow and the piglets. Reprod Domest Anim. 2019;54(S3):12–21. doi:10.1111/rda.13463
91. Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG. The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat Immunol. 2020;21(9):1082–1093. doi:10.1038/s41590-020-0713-6
92. Liu Y, Kaneda R, Leja TW, et al. Hhex and Cer1 mediate the sox17 pathway for cardiac mesoderm formation in embryonic stem cells. Stem Cells. 2014;32(6):1515–1526. doi:10.1002/stem.1695
93. Gauvrit S, Villasenor A, Strilic B, et al. HHEX is a transcriptional regulator of the VEGFC/FLT4/PROX1 signaling axis during vascular development. Nat Commun. 2018;9(1):2704. doi:10.1038/s41467-018-05039-1
94. Takebe T, Sekine K, Enomura M, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:7459):481–484. doi:10.1038/nature12271
95. Theunissen TW, Friedli M, He Y, et al. Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell. 2016;19(4):502–515. doi:10.1016/j.stem.2016.06.011
96. Aksoy I, Rognard C, Moulin A, et al. Apoptosis, G1 phase stall, and premature differentiation account for low chimeric competence of human and Rhesus monkey naive pluripotent stem cells. Stem Cell Reports. 2021;16(1):56–74. doi:10.1016/j.stemcr.2020.12.004
97. Wu J, Okamura D, Li M, et al. An alternative pluripotent state confers interspecies chimaeric competency. Nature. 2015;521(7552):316–321. doi:10.1038/nature14413
98. Cao J, Li W, Li J, et al. Live birth of chimeric monkey with high contribution from embryonic stem cells. Cell. 2023;186(23):4996–5014.e24. doi:10.1016/j.cell.2023.10.005
99. Takahashi S, Kobayashi S, Hiratani I. Epigenetic differences between naïve and primed pluripotent stem cells. Cell Mol Life Sci. 2018;75(7):1191–1203. doi:10.1007/s00018-017-2703-x
100. Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–156. doi:10.1038/292154a0
101. Brons IGM, Smithers LE, Trotter MWB, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:7150):191–195. doi:10.1038/nature05950
102. Tesar PJ, Chenoweth JG, Brook FA, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:7150):196–199. doi:10.1038/nature05972
103. Nichols J, Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4(6):487–492. doi:10.1016/j.stem.2009.05.015
104. Takashima Y, Guo G, Loos R, et al. Resetting Transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158(6):1254–1269. doi:10.1016/j.cell.2014.08.029
105. Guo G, von Meyenn F, Santos F, et al. Naive pluripotent stem cells derived directly from isolated cells of the human inner cell mass. Stem Cell Reports. 2016;6(4):437–446. doi:10.1016/j.stemcr.2016.02.005
106. Zheng C, Hu Y, Sakurai M, et al. Cell competition constitutes a barrier for interspecies chimerism. Nature. 2021;592:272–276. doi:10.1038/s41586-021-03273-0
107. Shetty A, Lim S, Strell P, Steer CJ, Rivera-Mulia JC, Low WC. In silico stage-matching of human, marmoset, mouse, and pig embryos to enhance organ development through interspecies chimerism. Cell Transplant. 2023;32:9636897231158728. doi:10.1177/09636897231158728
108. Suchy FP, Nishimura T, Seki S, et al. Streamlined and quantitative detection of chimerism using digital PCR. Sci Rep. 2022;12(1):10223. doi:10.1038/s41598-022-14467-5
109. Nishimura T, Suchy FP, Bhadury J, Igarashi KJ, Charlesworth CT, Nakauchi H. Generation of functional organs using a cell-competitive niche in intra- and inter-species rodent chimeras. Cell Stem Cell. 2021;28(1):141–149.e3. doi:10.1016/j.stem.2020.11.019
110. Ballard EB, Wu J. Growth competition in interspecies chimeras: a new paradigm for blastocyst complementation. Cell Stem Cell. 2021;28(1):3–5. doi:10.1016/j.stem.2020.12.011
111. Yuri S, Murase Y, Isotani A. Generation of rat-derived lung epithelial cells in Fgfr2b-deficient mice retains species-specific development. Development. 2024;151(1):dev202081. doi:10.1242/dev.202081
112. Yang Y, Liu B, Xu J, et al. Derivation of pluripotent stem cells with in vivo embryonic and extraembryonic potency. Cell. 2017;169(2):243–257.e25. doi:10.1016/j.cell.2017.02.005
113. Koplin J, Wilkinson D. Moral uncertainty and the farming of human-pig chimeras. J Med Ethics. 2019;45(7):440–446. doi:10.1136/medethics-2018-105227
114. Koplin JJ. Human-animal chimeras: the moral insignificance of uniquely human capacities. Hastings Center Rep. 2019;49(5):23–32. doi:10.1002/hast.1051
115. Kobayashi T, Kato-Itoh M, Nakauchi H. Targeted organ generation using Mixl1-inducible mouse pluripotent stem cells in blastocyst complementation. Stem Cells Dev. 2015;24(2):182–189. doi:10.1089/scd.2014.0270
116. Masaki H, Kato-Itoh M, Takahashi Y, et al. Inhibition of apoptosis overcomes stage-related compatibility barriers to chimera formation in mouse embryos. Cell Stem Cell. 2016;19(5):587–592. doi:10.1016/j.stem.2016.10.013
117. Yamaji M, Seki Y, Kurimoto K, et al. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat Genet. 2008;40(8):1016–1022. doi:10.1038/ng.186
118. Fang F, Iaquinta PJ, Xia N, Liu L, Diao L, Reijo Pera RA. Transcriptional control of human gametogenesis. Hum Reprod Update. 2022;28(3):313–345. doi:10.1093/humupd/dmac002
119. Chen Y, Zhou H, Sarver AL, et al. Hepatic differentiation of liver-derived progenitor cells and their characterization by microRNA analysis. Liver Transpl. 2010;16(9):1086–1097. doi:10.1002/lt.22111
120. National Institutes of Health. NOT-OD-16-128: request for public comment on the proposed changes to the nih guidelines for human stem cell research and the proposed scope of an nih steering committees consideration of certain human-animal chimera research. Available from: https://grants.nih.gov/grants/guide/notice-files/NOT-OD-16-128.html.
121. The five: chimeras created by science | genetics | the Guardian. Available from: https://www.theguardian.com/technology/2019/aug/11/the-five-chimeras-human-monkey-hybrid-genetic.
122. Cyranoski D. Japan approves first human-animal embryo experiments. Nature. 2019. doi:10.1038/d41586-019-02275-3
123. Ryu KY, Maehr R, Gilchrist CA, et al. The mouse polyubiquitin gene UbC is essential for fetal liver development, cell-cycle progression and stress tolerance. EMBO J. 2007;26(11):2693–2706. doi:10.1038/sj.emboj.7601722
124. Park H, Yoon M-S, Ryu K-Y. Disruption of polyubiquitin gene Ubc leads to defective proliferation of hepatocytes and bipotent fetal liver epithelial progenitor cells. Biochem Biophys Res Commun. 2013;435(3):434–440. doi:10.1016/j.bbrc.2013.05.003
125. Saito Y, Kojima T, Takahashi N. Mab21l2 is essential for embryonic heart and liver development. PLoS One. 2012;7(3):e32991. doi:10.1371/journal.pone.0032991
126. Baldessari D, Badaloni A, Longhi R, Zappavigna V, Consalez GG. MAB21L2, a vertebrate member of the Male-abnormal 21 family, modulates BMP signaling and interacts with SMAD1. BMC Cell Biol. 2004;5(1):48. doi:10.1186/1471-2121-5-48
127. Suzuki A, Sekiya S, Büscher D, Izpisúa Belmonte JC, Taniguchi H. Tbx3 controls the fate of hepatic progenitor cells in liver development by suppressing p19ARF expression. Development. 2008;135(9):1589–1595. doi:10.1242/dev.016634
128. Margagliotti S, Clotman F, Pierreux CE, et al. The Onecut transcription factors HNF-6/OC-1 and OC-2 regulate early liver expansion by controlling hepatoblast migration. Dev Biol. 2007;311(2):579–589. doi:10.1016/j.ydbio.2007.09.013
129. Denner J. Porcine endogenous retroviruses and xenotransplantation, 2021. Viruses. 2021;13(11):2156. doi:10.3390/v13112156
130. Rodrigues Costa M, Fischer N, Gronewold A, Gulich B, Godehardt AW, Tönjes RR. Isolation of an ecotropic porcine endogenous retrovirus PERV-C from a Yucatan SLAD/D inbred miniature swine. J Virol. 2023;97(3):e00062–23. doi:10.1128/jvi.00062-23
131. Fishman JA. Infectious disease risks in xenotransplantation. Am J Transplant. 2018;18(8):1857–1864. doi:10.1111/ajt.14725
132. Sun H, Xiao Y, Liu J, et al. Prevalent Eurasian avian-like H1N1 swine influenza virus with 2009 pandemic viral genes facilitating human infection. Proc Natl Acad Sci U S A. 2020;117(29):17204–17210. doi:10.1073/pnas.1921186117
133. Mao H, Li J, Liao G, Gao M, Yang G, Bao J. The prevention strategies of swine viruses related to xenotransplantation. Virol J. 2023;20(1):121. doi:10.1186/s12985-023-02090-3
134. Yang L, Güell M, Niu D, et al. Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science. 2015;350(6264):1101–1104. doi:10.1126/science.aad1191
135. Yue Y, Xu W, Kan Y, et al. Extensive germline genome engineering in pigs. Nat Biomed Eng. 2021;5(2):134–143. doi:10.1038/s41551-020-00613-9
136. Yokoo T, Yamanaka S, Kobayashi E. Xeno-regenerative medicine: a novel concept for donor kidney fabrication. Xenotransplantation. 2020;27(5):e12622. doi:10.1111/xen.12622
137. Sykes M, Sachs DH. Transplanting organs from pigs to humans. Sci Immunol. 2019;4(41):eaau6298. doi:10.1126/sciimmunol.aau6298
138. Zhou Q, Li T, Wang K, et al. Current status of xenotransplantation research and the strategies for preventing xenograft rejection. Front Immunol. 2022;13:928173. doi:10.3389/fimmu.2022.928173
139. Deng S, Pan D, Dai Y, Wu J. Editorial: solutions for organ transplantation: xenotransplantation and interspecies chimaeras. Front Cell Dev Biol. 2023;11:1194654. doi:10.3389/fcell.2023.1194654
140. Arabi TZ, Sabbah BN, Lerman A, Zhu XY, Lerman LO. Xenotransplantation: current challenges and emerging solutions. Cell Transplant. 2023;32:09636897221148771. doi:10.1177/09636897221148771
141. Iwase H, Hara H, Ezzelarab M, et al. Immunological and physiological observations in baboons with life-supporting genetically engineered pig kidney grafts. Xenotransplantation. 2017;24(2). doi:10.1111/xen.12293
142. Rivard CJ, Tanabe T, Lanaspa MA, et al. Upregulation of CD80 on glomerular podocytes plays an important role in development of proteinuria following pig-to-baboon xeno-renal transplantation – an experimental study. Transpl Int. 2018;31(10):1164–1177. doi:10.1111/tri.13273
143. Yamada K, Bottino RE. Xenotransplantation for the therapy of diabetes: a new look. Front Endocrinol. 2023;14:1190442. doi:10.3389/fendo.2023.1190442
144. Gong W, Das S, Sierra-Pagan JE, et al. ETV2 functions as a pioneer factor to regulate and reprogram the endothelial lineage. Nat Cell Biol. 2022;24(5):672–684. doi:10.1038/s41556-022-00901-3
145. Das S, Koyano-Nakagawa N, Gafni O, et al. Generation of human endothelium in pig embryos deficient in ETV2. Nat Biotechnol. 2020;38(3):297–302. doi:10.1038/s41587-019-0373-y
146. Garry DJ, Weiner JI, Greising SM, Sachs DH, Garry MG. Xenotransplantation and exotransplantation: strategies to expand the number of donor organs. Xenotransplantation. 2023;30(1):e12786. doi:10.1111/xen.12786
147. Moya-Jódar M, Coppiello G, Rodríguez-Madoz JR, et al. One-step in vitro generation of ETV2-null pig embryos. Animals. 2022;12(14):1829. doi:10.3390/ani12141829
148. Klymiuk N, Aigner B, Brem G, Wolf E. Genetic modification of pigs as organ donors for xenotransplantation. Mol Reprod Dev. 2010;77(3):209–221. doi:10.1002/mrd.21127
149. Makowa L, Cramer DV, Hoffman A, et al. The use of a pig liver xenograft for temporary support of a patient with fulminant hepatic failure. Transplantation. 1995;59(12):1654–1659. doi:10.1097/00007890-199506270-00002
150. Kim K, Schuetz C, Elias N, et al. Up to 9-day survival and control of thrombocytopenia following alpha1,3-galactosyl transferase knockout swine liver xenotransplantation in baboons. Xenotransplantation. 2012;19(4):256–264. doi:10.1111/j.1399-3089.2012.00717.x
151. Shah JA, Patel MS, Elias N, et al. Prolonged survival following pig-to-primate liver xenotransplantation utilizing exogenous coagulation factors and costimulation blockade. Am J Transplant. 2017;17(8):2178–2185. doi:10.1111/ajt.14341
152. Mohiuddin MM, Singh AK, Corcoran PC, et al. Chimeric 2C10R4 anti-CD40 antibody therapy is critical for long-term survival of GTKO.hCD46.hTBM pig-to-primate cardiac xenograft. Nat Commun. 2016;7:11138. doi:10.1038/ncomms11138
153. Längin M, Mayr T, Reichart B, et al. Consistent success in life-supporting porcine cardiac xenotransplantation. Nature. 2018;564(7736):430–433. doi:10.1038/s41586-018-0765-z
154. Kim SC, Mathews DV, Breeden CP, et al. Long-term survival of pig-to-rhesus macaque renal xenografts is dependent on CD4 T cell depletion. Am J Transplant. 2019;19(8):2174–2185. doi:10.1111/ajt.15329
155. Mohiuddin MM, Singh AK, Scobie L, et al. Graft dysfunction in compassionate use of genetically engineered pig-to-human cardiac xenotransplantation: a case report. Lancet. 2023;402(10399):397–410. doi:10.1016/S0140-6736(23)00775-4
156. Patel MS, Louras N, Vagefi PA. Liver xenotransplantation. Curr Opin Organ Transplant. 2017;22(6):535–540. doi:10.1097/MOT.0000000000000459
157. Zhang X, Li X, Yang Z, et al. A review of pig liver xenotransplantation: current problems and recent progress. Xenotransplantation. 2019;26(3):e12497. doi:10.1111/xen.12497
158. Wang J, Xie W, Li N, et al. Generation of a humanized mesonephros in pigs from induced pluripotent stem cells via embryo complementation. Cell Stem Cell. 2023;30:1235–1245. doi:10.1016/j.stem.2023.08.003
159. Crane AT, Shen FX, Brown JL, et al. The American public is ready to accept human-animal chimera research. Stem Cell Reports. 2020;15(4):804–810. doi:10.1016/j.stemcr.2020.08.018
160. Brown JL, Voth JP, Person K, Low WC. A technological and regulatory review on human-animal chimera research: the current landscape of biology, law and public opinion. Cell Transplant. 2023;32:1–18. doi:10.1177/09636897231183112
161. Kögel J, Marckmann G. Xenotransplantation challenges us as a society. EMBO Rep. 2020;21(9):e50274. doi:10.15252/embr.202050274
162. Carrier AN, Verma A, Mohiuddin M, et al. Xenotransplantation: a new era. Front Immunol. 2022;13:900594. doi:10.3389/fimmu.2022.900594
163. Negri A, Wilson L. Future systems of xenotransplantation: melding historical and bioethical methodology. Cell Transplant. 2023;32:9636897231170510. doi:10.1177/09636897231170510
164. Lu T, Yang B, Wang R, Qin C. Xenotransplantation: current status in preclinical research. Front Immunol. 2019;10:3060. doi:10.3389/fimmu.2019.03060
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.