Back to Journals » International Medical Case Reports Journal » Volume 14
Childhood-Onset Leber Hereditary Optic Neuropathy: Particular Features
Authors Cunha AM ![]() , Vilares-Morgado R
, Vilares-Morgado R ![]() , Moleiro AF, Falcão-Reis F, Faria O
, Moleiro AF, Falcão-Reis F, Faria O
Received 24 January 2021
Accepted for publication 16 February 2021
Published 12 March 2021 Volume 2021:14 Pages 163—169
DOI https://doi.org/10.2147/IMCRJ.S303460
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Ana Maria Cunha,1 Rodrigo Vilares-Morgado,1 Ana Filipa Moleiro,1 Fernando Falcão-Reis,1,2 Olinda Faria1,2
1Department of Ophthalmology, Centro Hospitalar Universitário de São João, Porto, Portugal; 2Department of Surgery and Physiology, Faculty of Medicine, University of Porto, Porto, Portugal
Correspondence: Ana Maria Cunha
Department of Ophthalmology, Centro Hospitalar Universitário de São João, Avenida Prof. Hernâni Monteiro, Porto, 4202 – 451, Portugal
Tel +35 1225512100
Fax +35 1225513669
Email [email protected]
Background: Leber hereditary optic neuropathy (LHON) is an optic neuropathy of mitochondrial inheritance. Childhood-onset disease is relatively rare and there are limited data on this important patient subgroup.
Case Presentation: We present 3 particular presentations of LHON. Patient 1 was an 8-year-old boy admitted to the emergency department reporting a progressive bilateral visual loss and intermittent headaches. Neuro-ophthalmological examination revealed a bilateral pseudopapilledema. Lumbar puncture identified intracranial hypertension and the brain and orbits magnetic resonance imaging showed T2 hyperintensity in the posterior region of the left optic nerve and the optic chiasm. Patient 2 was a 12-year-old boy admitted to the emergency department reporting painless, progressive central vision loss in the right eye. Fundus examination revealed a hyperemic disc and vascular network papillary and peripapillary vascular microdilations. Three months later, the left eye presented visual loss. Patient 3 was a 6-year-old female child referred to the neuro-ophthalmology specialist due to painless central visual loss in both eyes. Her BCVA was 1/10 and counting fingers in right and left eye, respectively, and fundus examination revealed a pallor optic disc in the temporal sector.
Discussion: The phenotype of childhood-onset disease may present itself distinct from classical adult-onset LHON. The absence of classical clinical features could lead to initial misdiagnosis. There should exist a high index of suspicion in children presenting unexplained subnormal vision in order to avoid potential diagnostic delays.
Keywords: Leber hereditary optic neuropathy, childhood-onset disease, mitochondrial inheritance
Background
Leber hereditary optic neuropathy (LHON) is an optic neuropathy of mitochondrial inheritance.1 The three most common mutations representing 80 to 95% of LHON cases are m.11778G>A (p.R340H, mitochondrially encoded NADH dehydrogenase 4, ND4), m.3460G>A (p.A52T, mitochondrially encoded NADH dehydrogenase 1, ND1), and m.14484T>C (p.M64V, mitochondrially encoded NADH dehydrogenase 6, ND6), all involving encoding complex I subunits of mitochondrial respiratory chain.2
The pathophysiology involves a primary point mtDNA mutation causing failure of the electron transport oxidative phosphorylation pathway, with the end result being selective retinal ganglion cells (RGC) apoptosis.3,4 The presence of specific mitochondrial point mutation does not guarantee the development of symptomatic LHON in individuals harbouring the mutation. Incomplete penetrance is shown in around 50% of males and 10% of females, with the mutation later causing visual loss to develop.3–5
Clinically, the symptomatic phase of LHON is frequently characterized by an acute or subacute bilateral painless central visual loss associated with disc hyperemia and telangiectatic vessels in the peripapillary region, without leakage on fluorescein angiography5,6 Although disease conversion can occur anywhere from the first to the eighth decade of life, the peak age of onset of visual loss among LHON carriers is between 15 and 30 years.7 Childhood-onset disease is relatively rare and less than 10% of patients were 12 years old or younger at the time of diagnosis.7–9 Although there are limited data on this important patient subgroup, the phenotype seems distinct from classical adult-onset LHON, with atypical patterns of vision loss.9,10
Case Presentation
Patient 1
An 8-year-old Caucasian boy was admitted to the emergency department reporting a progressive bilateral visual loss. One month before, the patient had started to exhibit a decrease in vision initially in the left eye and latterly in the right eye. He also complained of intermittent headaches, which varied in severity, taking 8 months to evolve, eventually occurring as frequently as on a daily basis, and most frequently in the month prior to admittance. The patient had a normal Body Mass Index. There was no family history of visual loss nor substance abuse.
Neuro-ophthalmological examination revealed best correct visual acuity (BCVA) of 1/10 in the right eye and counting fingers in the left eye, pupils with few reactions to light but with relative afferent pupillary defect in the left eye. Slit-lamp examination and intraocular pressure were normal. Fundus examination demonstrated bilateral moderate optic disc elevation, suggesting papilledema (Figure 1A and B). Lumbar puncture was performed and identified intracranial hypertension with opening pressure of 320 mm H2O and normal cell count, protein, and glucose levels.
 |
Figure 1 (A and B) Fundus photograph of patient 1 with a bilateral moderate optic disc elevation. (C and D) Fundus fluorescein angiography revealed bilateral peripapillary microangiopathy and no optic nerve leakage. (E and F) Campimetric study showed cecocentral scotoma. |
Fundus fluorescein angiography (FA) revealed a bilateral peripapillary microangiopathy and no optic nerve leakage, which indicated a pseudopapilledema (Figure 1C and D). Brain and orbits Magnetic Resonance Imaging (MRI) showed T2 hyperintensity in the posterior region of the left optic nerve and the optic chiasm (Figure 2). The result of an extensive workup for inflammatory, infectious and auto-immune conditions was negative. Campimetric study showed cecocentral scotoma in both eyes (Figure 1E and F).
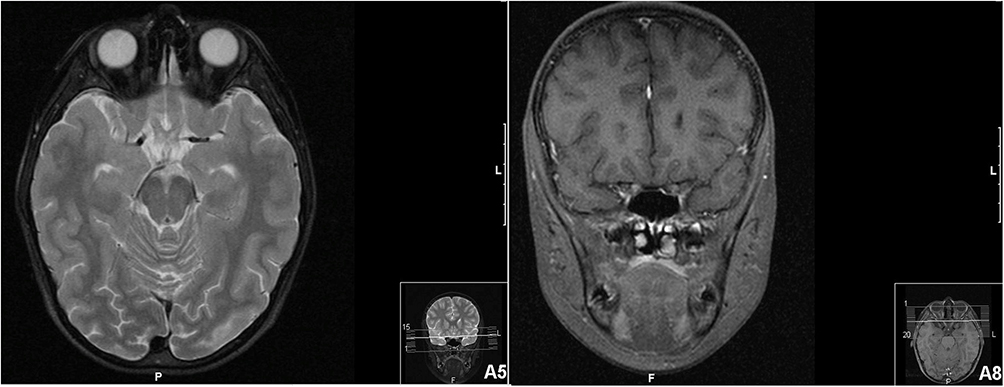 |
Figure 2 Brain and orbits magnetic resonance imaging (MIR) of patient 1. T2 hyperintensity in the posterior region of the left optic nerve and the optic chiasm. |
Due to the idiopathic intracranial hypertension, treatment with acetazolamide was initiated. The headaches improved but the loss of visual acuity remained. Targeted mutation analysis for LHON was performed, yielding homoplasmy for MTND4* LHON11778A mutation. After 1 year of clinical follow-up, his best visual acuity was counting fingers and presented pallor optic discs in both eyes. Currently, he is 17 years old and shows the same visual outcome.
Patient 2
A 12-year-old Caucasian boy was admitted to the emergency department reporting painless, progressive central vision loss in the right eye. On ophthalmological examination, BCVA was 2/10 in the right eye and 10/10 in the left eye. A slight relative afferent pupillary defect in the right eye was identified. Biomicroscopy of the anterior segment and intraocular pressure was unremarkable. The fundus examination revealed a hyperemic disc and vascular bilateral dilatation, without macular changes. SD-OCT imaging documented peripapillary retinal nerve fiber layer (pRNFL) thickening in superior and inferior sectors (Figure 3). FA revealed vascular network papillary and peripapillary vascular microdilations, with no diffusion in the late stages, without inflammatory features (Figure 4). Electrophysiology studies revealed visual evoked potential suggestive of optic neuropathy. Kinetic study assessment revealed a cecocentral scotoma in the right eye. The Brain and orbits MRI imaging was normal. Three months later, his BCVA was 1/10 in the right eye and 6/10 in the left eye with an enlarged central scotoma in the right eye and developed a cecocentral scotoma in the left eye.
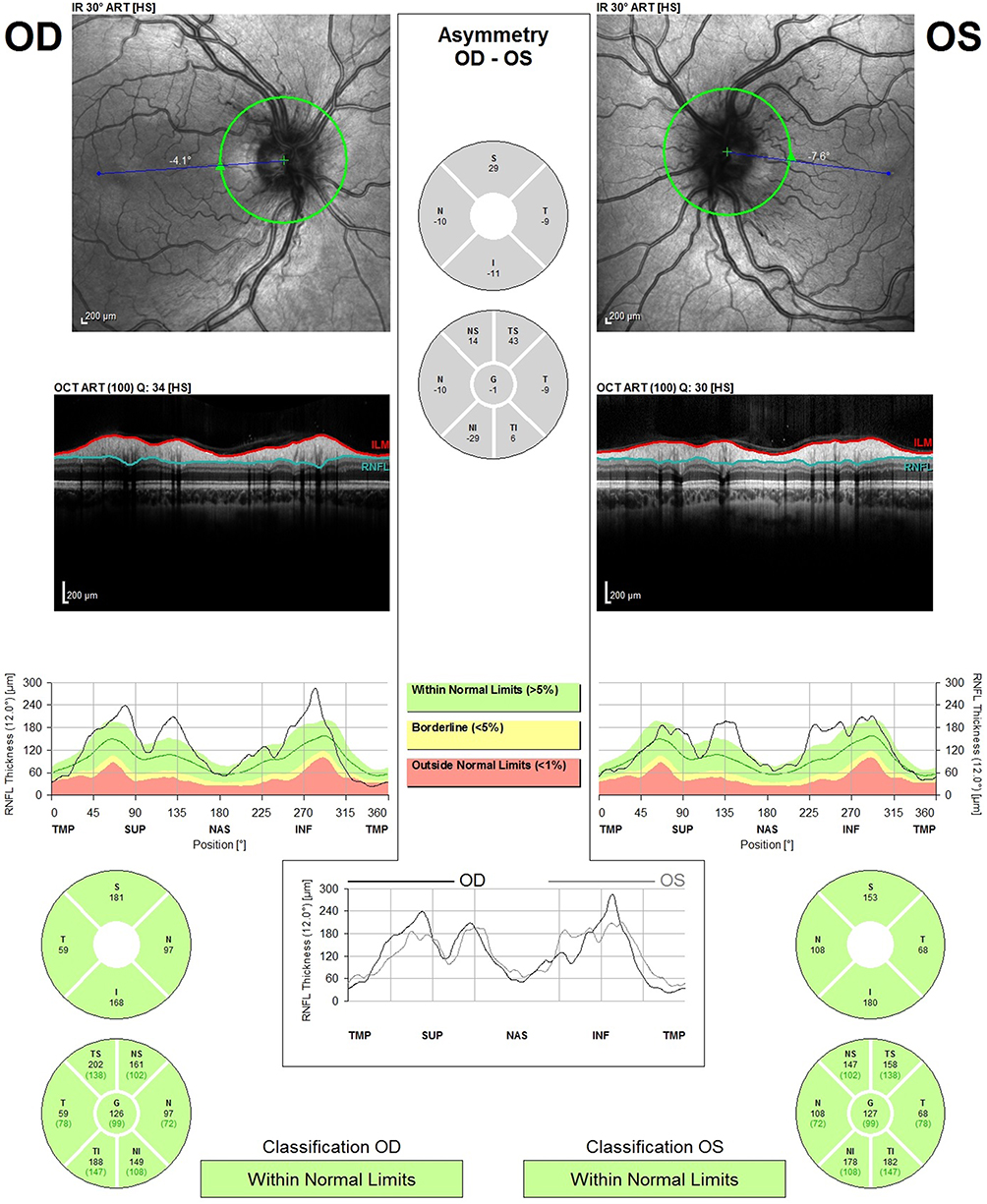 |
Figure 3 Spectral-domain optical coherence tomography (SD-OCT Spectralis, Heidelberg) analysis of patient 2. SD-OCT imaging documented peripapillary retinal nerve fiber layer (pRNFL) thickening in superior and inferior sectors. |
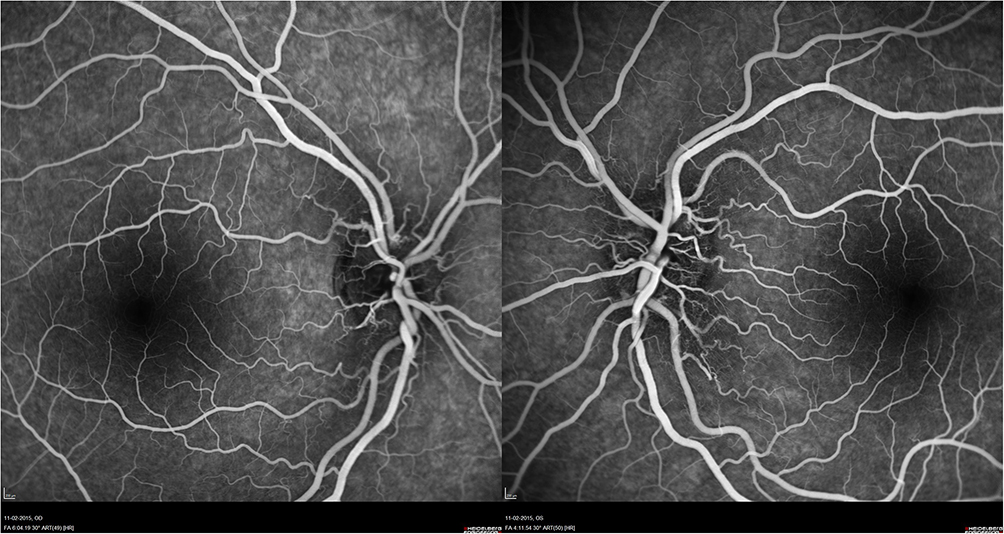 |
Figure 4 FA of patient 2 revealed vascular network papillary and peripapillary vascular microdilations, without diffusion in late stages. |
The patient had a family history of LHON. Targeted mutation analysis for LHON was performed and identified a homoplasmic mutation in MTND4*LHON11778A mutation. He underwent treatment with idebenone. Two months later, the BCVA was 0.05 in the right eye and 0.1 in the left eye. Presently, he is 19 years-old and his BCVA is counting fingers in both eyes, with temporal optic disc pallor and an RNFL thinning in all sectors.
Patient 3
A 6-year-old Caucasian female child was referred to the neuro-ophthalmology specialist due to painless central visual loss in both eyes. On ophthalmological examination, BCVA was 1/10 in the RE and counting fingers in LE, without relative afferent pupillary defect. The anterior segment slit-lamp examination and intraocular pressure were normal. Fundus examination revealed a pallor optic disc in the temporal sector. Electrophysiological exams revealed in visual evoked potential a P100 decreased in 50% and a latency increased in 20 ms. Brain MRI was normal.
The patient had a family history of LHON. Targeted mutation analysis for LHON was performed, with identification of a homoplasmic mutation in MTND4*LHON11778A mutation.
Currently, at 15 years old, her BCVA is counting fingers in both eyes, with an optic disc pallor in the temporal sector and SD-OCT revealed pRNFL thinning in all sectors.
Discussion
LHON is a mitochondrial disorder widely considered in adult or juvenile age, but the disease conversion to a symptomatic phase can occur during childhood.10 Due to its relative rarity, there are limited data on the clinical features and visual prognosis of childhood LHON. However, childhood-onset disease is an important patient subgroup, and the phenotype seems distinct from classical adult-onset LHON with atypical patterns of vision loss. Children with LHON are often described as experiencing a milder form of the disease than adults-onset. The subclassification of the mode of onset and progression of visual loss of these patients is usually classified as acute, if visual acuity has deteriorated rapidly, reaching its nadir within 6 months of onset of the disease; slowly progressive, if visual deterioration has occurred over a period exceeding 6 months; and insidious or subclinical, if the patient was clinically asymptomatic at the time that a diagnosis of optic atrophy or subnormal vision was made, and there was no change in visual acuity during subsequent follow-ups.7,9,10
Patient 1 had an atypical presentation with an elevation of the optic disc, an idiopathic intracranial hypertension and optic nerve and optic chiasm hyperintensity in the MR imaging. Such atypical features during the initial presentation might give an impression of other optic neuropathies as the aetiology for visual loss. However, the absence of leakage on fluorescein angiography was fundamental to the differentiation of pseudopapilledema from papilledema.3,6,11 Moreover, usually LHON had normal findings on MRI. Few studies, however, reported hyperintensity and enhancement of the visual pathway involved in LHON, as described in this case.12–14
The onset of LHON visual loss could be triggered by many aetiologies. It had been reported that the symptomatic phase of LHON was elicited by exposures to certain agents such as smoking, alcohol and certain medications. However, the environmental factors are unlikely to be aetiologically important in young children. Other factors could include optic nerve insults due to other optic neuropathies or even trauma.12,15,16 In the present case, intracranial hypertension could act as the triggering factor of LHON. Lamirel et al12 reported one case of 11778 mtDNA LHON-specific mutations with confirmed diagnosis in a young adult male with idiopathic intracranial hypertension with papilledema. However, to our knowledge, there has not been reported any onset in children with idiopathic intracranial hypertension at presentation. It was hypothesized that idiopathic intracranial hypertension could cause a disturbance to pre-existing sub-normal axoplasmic transport in LHON, possibly responsible for additional imbalance of oxidative regulation in retinal ganglion cells.11
The second case was admitted with a slowly progressive visual deterioration and a subclinical presentation of the right eye. According to the previous studies, although classical acute patterns of visual loss are the most common presentation in childhood LHON, over a third of the patients were suffering from slow progression or subclinical disease evolution, and this rate is higher than the adult onset.10 Moreover, the slow progression subgroup of patients usually had a better prognosis due to the slow rate and asynchronous loss of nerve fiber with a higher rate of spontaneous improvement in visual acuity.9 However, despite the visual deterioration occurring over a period exceeding 6 months in our case, after a follow-up of seven years, results show a poor outcome, with BCVA of counting fingers in both eyes and a temporal optic disc pallor with an RNFL thinning in all sectors.
In contrast with the first two cases, the child in the third case had an acute presentation, with a rapid visual deterioration. The acute childhood LHON presentation usually had the worse prognosis and mimicked adult-onset LHON. There is a rapid early loss of the papillomacular bundle followed by complete optic atrophy after disease onset, usually without the possibility of some degree of visual recovery.
Diagnosis of LHON can usually be made based on patient and family clinical history as well as baseline investigations including a formal neuroophthalmological examination and mtDNA genetic testing.
In conclusion, we described three peculiar presentations of childhood LHON. Although mitochondrial turnover is implicated in the pathogenesis of LHON, age-related factors could be implicated in the distinct phenotypical subgroup of patients.17,18 The absence of classical clinical features could lead to initial misdiagnosis. There should exist a high index of suspicion in children presenting unexplained subnormal vision in order to avoid potential diagnostic delays. Diagnostic confirmation is crucial, especially in LHON because of the clinical course, prognosis and hereditary pattern of the disease.
Ethics Approval and Consent to Participate
Written informed consent was obtained from all legal guardian, for this study and for allowing the use of clinical images and information for the purposes of publication. This study of de-identified case report was exempt from ethical approval by the Ethics Committee of Centro Hospitalar Universitário de São João.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Newman NJ, Biousse V. Hereditary optic neuropathies. Eye. 2004;18:1144–1160. doi:10.1038/sj.eye.6701591
2. Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55:299e334.
3. Meyerson C, Van Stavern G, McClelland C. Leber hereditary optic neuropathy: current perspectives. Clin Ophthalmol. 2015;9:1165–1176. doi:10.2147/OPTH.S62021
4. Ueda K, Morizane Y, Shiraga F, et al. Nationwide epidemiological survey of leber hereditary optic neuropathy in japan. J Epidemiol. 2017;27(9):447–450. doi:10.1016/j.je.2017.02.001
5. Yu-Wai-Man P, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet. 2002;39:162–169. doi:10.1136/jmg.39.3.162
6. Piotrowska A, Korwin M, Bartnik E, Tonska K. Leber hereditary optic neuropathy - historical report in comparison with the current knowledge. Gene. 2015;555(1):41–49.
7. Nikoskelainen EK, Huoponen K, Juvonen V, et al. Ophthalmologic findings in Leber Hereditary Optic Neuropathy, with special reference to mtDNA mutations. Ophthalmology. 1996;103:504–514. doi:10.1016/S0161-6420(96)30665-9
8. Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol. 1991;111:750–762. doi:10.1016/S0002-9394(14)76784-4
9. Barboni P, Savini G, Valentino ML, et al. Leber’s hereditary optic neuropathy with childhood onset. Invest Ophthalmol Vis Sci. 2006;47:5303–5309. doi:10.1167/iovs.06-0520
10. Majander A, Bowman R, Poulton J. Childhood-onset Leber hereditary optic neuropathy.Br J Ophthalmol. 2017:1–5.
11. Arianti A, Chuman H, Kajihara N, Sakamoto N, Nao N. Atypical clinical and neuroimaging findings in Leber’s hereditary optic neuropathy: a case report. JOJ Ophthalmol. 2018;6(5):555698. doi:10.19080/JOJO.2018.06.555698
12. Lamirel C, Cassereau J, Cochereau I, et al. Papilloedema and MRI enhancement of the prechiasmal optic nerve at the acute stage of leber hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 2010;81(5):578–580. doi:10.1136/jnnp.2009.174953
13. Ong E, Biotti D, Abouaf L, et al. Teaching neuroimages: chiasmal enlargement and enhancement in leber hereditary optic neuropathy. Neurology. 2013;7:e126–e127. doi:10.1212/WNL.0b013e3182a95698
14. Van Westen D, Hammar B, Bynke G. Magnetic resonance findings in the pregeniculate visual pathways in leber hereditary optic neuropathy. J Neuroophthalmol. 2013;31(1):48–51. doi:10.1097/WNO.0b013e3181f3f203
15. Riordan-Eva P, Sanders MD, Govan GG, et al. The clinical features of leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118(Pt 2):319–337. doi:10.1093/brain/118.2.319
16. Apinyawasisuk S, Chan JW, Arnold AC. Trauma-associated leber hereditary optic neuropathy. Neuro-Ophthalmology. 2016;40(4):192–196. doi:10.1080/01658107.2016.1183682
17. Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial bi006Fgenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain. 2014;137:335–353. doi:10.1093/brain/awt343
18. Dombi E, Diot A, Morten K, et al. The m.13051G>A mitochondrial DNA mutation results in variable neurology and activated mitophagy. Neurology. 2016;86:1921–1923. doi:10.1212/WNL.0000000000002688
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.