Back to Journals » Cancer Management and Research » Volume 6
Chemotherapy-induced peripheral neuropathy in adults: a comprehensive update of the literature
Authors Argyriou A, Kyritsis A, Makatsoris T, Kalofonos H
Received 31 December 2013
Accepted for publication 9 February 2014
Published 19 March 2014 Volume 2014:6 Pages 135—147
DOI https://doi.org/10.2147/CMAR.S44261
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Andreas A Argyriou,1,3 Athanasios P Kyritsis,2 Thomas Makatsoris,3 Haralabos P Kalofonos3
1Department of Neurology, "Saint Andrew's" General Hospital of Patras, Greece; 2Department of Neurology, University Hospital of Ioannina, Greece; 3Department of Medicine-Division of Oncology, University of Patras Medical School, Rion-Patras, Greece
Abstract: Commonly used chemotherapeutic agents in oncology/hematology practice, causing toxic peripheral neuropathy, include taxanes, platinum compounds, vinca alkaloids, proteasome inhibitors, and antiangiogenic/immunomodulatory agents. This review paper intends to put together and discuss the spectrum of chemotherapy-induced peripheral neuropathy (CIPN) characteristics so as to highlight areas of future research to pursue on the topic. Current knowledge shows that the pathogenesis of CIPN still remains elusive, mostly because there are several sites of involvement in the peripheral nervous system. In any case, it is acknowledged that the dorsal root ganglia of the primary sensory neurons are the most common neural targets of CIPN. Both the incidence and severity of CIPN are clinically under- and misreported, and it has been demonstrated that scoring CIPN with common toxicity scales is associated with significant inter-observer variability. Only a proportion of chemotherapy-treated patients develop treatment-emergent and persistent CIPN, and to date it has been impossible to predict high- and low-risk subjects even within groups who receive the same drug regimen. This issue has recently been investigated in the context of pharmacogenetic analyses, but these studies have not implemented a proper methodological approach and their results are inconsistent and not really clinically relevant. As such, a stringent approach has to be implemented to validate that information. Another open issue is that, at present, there is insufficient evidence to support the use of any of the already tested chemoprotective agents to prevent or limit CIPN. The results of comprehensive interventions, including clinical, neurophysiological, and pharmacogenetic approaches, are expected to produce a consistent advantage for both doctors and patients and thus allow the registration and analysis of reliable data on the true characteristics of CIPN, eventually leading to potential preventive and therapeutic interventions.
Keywords: neurotoxicity, incidence, diagnosis, treatment
Introduction
Chemotherapy drugs used to treat cancer can be neurotoxic by either exerting a direct noxious effect on the brain or the peripheral nerves.1,2 Nonetheless, chemotherapy-induced peripheral neurotoxicity (CIPN) is considered to be among the most common non-hematological adverse effects of a number of effective chemotherapeutic agents. Depending on its severity, CIPN can be dose limiting and may also significantly diminish the quality of life (QOL) of patients, because it can persist or even intensify long after the completion of chemotherapy.2 Moreover, the economic cost of neurotoxicity secondary to antineoplastic agents on health systems is significant, as cancer patients with CIPN have significant excess health care costs and resource use.3
CIPN can usually affect the dorsal root ganglia (DRG) of the primary sensory neurons, but other sites, ie, the nerve terminals (distal terminations of the branches of an axon), may also be involved. Its clinical features vary depending on the type of the offending agent involved and the site of action, ranging from pure sensory or sensory–motor peripheral nerve damage of large myelinated or small unmyelinated fibers. Damage to peripheral nerve systems from chemotherapy can present with or without autonomic impairment. Rarely, cranial nerve involvement occurs.4
The diagnosis of CIPN usually relies on traditional clinical grading scales, such as the World Health Organization (WHO), Ajani, and Eastern Cooperative Oncology Group (ECOG) scales.2,4 Nevertheless, the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) for sensory and motor neuropathy are considered to be the standard method for assessing CIPN. NCI-CTCAE version 3 grades its severity from 1) loss of deep tendon reflexes or paresthesia (including tingling) but not interfering with function/subjective weakness with no objective findings, to 4) permanent sensory loss that interferes with function/paralysis. Grade 5 is assigned to death from neurotoxicity. In summary, NCI-CTCAEv3 has been constructed to grade the severity of neurosensory and neuromotor symptoms with particular relevance to their interference with function. However, clinical experience shows that the use of NCI-CTCAEv3 is associated with underestimation of CIPN prevalence and severity.5 The newer NCI-CTCAEv4 was released in May 2009, and its most important difference when compared with version 3 is that NCI-CTCAEv4 harmonizes with the lowest level terms from the Medical Dictionary for Regulatory Activities (MedDRA), which are listed based on their MedDRA primary system organ class, eg, immune system disorders or nervous system disorders.6 One would expect that the changes in NCI-CTCAEv4 would impact the prevalence estimates of CIPN. However, this does not seem to be the case, because again, no formal neurological examination is required and grading of CIPN severity remains subjective, solely relying on patients’ reported symptoms. Moreover, the use of the same tool of peripheral sensory neuropathy as a separate item and addition of definitions, ie, dysesthesia, paresthesia, or neuralgia as distinct items seems to confuse rather than offer clarity.7
Alternatively to NCI-CTCAE, the 11-item neurotoxicity subscale (FACT/GOG-Ntx [Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity]) developed by the Gynecologic Oncology Group has also been used.8 However, significant inter-observer disagreement occurs in scoring CIPN with these scales and as such, initiatives have been launched to define the optimal outcome measures of CIPN assessment.9,10
Recently, the CI-PeriNoms (chemotherapy-induced peripheral neuropathy) study group reported initial validity and reliability findings for grading scales, such as the NCI-CTCAE, the Total Neuropathy Score clinical version (TNSc©), the modified Inflammatory Neuropathy Cause and Treatment (INCAT) scale, and the group sensory sum score (mISS).11 The TNSc, a shorter and more easily applied clinical version of the formal TNS, is a composite measure that includes symptoms, signs, and ability aspects, and its use is proposed because it appears superior to NCI-CTCAE in terms of responsiveness.11–15
Furthermore, patients have reported a validated scale that has been used to assess CIPN in a more comprehensive and accurate manner: the Rasch-built Overall Disability Scale (CIPN-R-ODS), consisting of 28 items, was able to detect disability in CIPN patients with proper validity and reliability, and to bypass the difficulties with ordinal-based measures.16 In any case, it seems that clinical and patients’ reported outcome measures should always be combined to achieve a comprehensive knowledge of CIPN, including a reliable assessment of both the severity and the quality of CIPN-related sensory impairment.17
Commonly used chemotherapeutic agents in oncology/hematology practice causing peripheral neuropathy are taxanes, platinum compounds, vinca alkaloids, proteasome inhibitors, and antiangiogenic/immunomodulatory agents. We will herein review and discuss the spectrum of CIPN characteristics resulting from the administration of chemotherapeutic agents. Tables summarizing the common sites of involvement (Table 1), risk factors of CIPN (Table 2), as well as the type of neuropathy and clinical pattern of CIPN (Table 3) by neurotoxic drug classification are also presented. Additionally, we will highlight areas for future research to pursue.
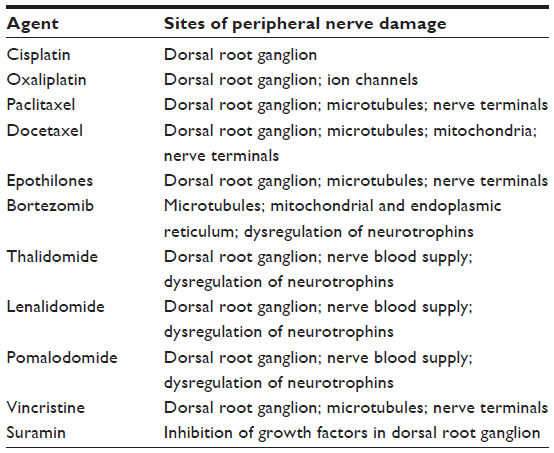 | Table 1 Common sites of involvement by neurotoxic drug classification |
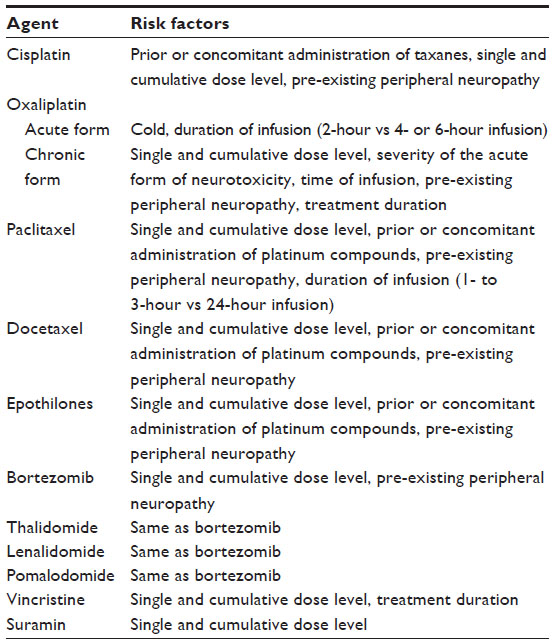 | Table 2 Risk factors of CIPN by neurotoxic drug classification |
 | Table 3 Type of neuropathy and clinical pattern of CIPN by neurotoxic drug classification |
Taxanes (paclitaxel/docetaxel)
Pathogenesis
Conventionally, the mechanisms underlying the pathogenesis of taxane-induced peripheral neuropathy (TIPN) include interference with microtubule-based axonal transport, macrophage activation in both the DRG and peripheral nerve, as well as microglial activation within the spinal cord.18 As a result of the problematic signal transduction, there is evidence of a “dying back” process starting from the distal nerve endings followed by effects on Schwann cells and neuronal bodies, or disturbed axonal transport changes in the affected neurons.19,20
Recent evidence shows that the activation of spinal astrocytes and the inhibition of microtubule-based fast axonal transport may al so be significant contributors to TIPN.21 The structure of internodal myelin in peripheral nerves remains unaffected in TIPN.22
Incidence, severity, and risk factors
Paclitaxel appears to be more neurotoxic than docetaxel with an overall incidence of about 60% and 15% for each agent, respectively.23,24 Current evidence shows that the most important triggering factor of TIPN is the accumulation of doses over the course of chemotherapy with a neurotoxic threshold of 1,000 mg/m2 for paclitaxel and 400 mg/m2 for docetaxel.25 Grade 3–4 sensory neuropathy is much more common with paclitaxel than with docetaxel.2,26 The nanoparticle albumin-bound (Nab) form of paclitaxel was formulated to enable lower doses and reduce toxicity, but clinical experience shows that grade ≥2 peripheral neuropathy still remains a significant treatment-limiting toxicity.27,28
Other risk factors include prior or concomitant administration of platinum compounds, pre-existing peripheral neuropathy due to various medical conditions, and duration of infusion (1- to 3-hour infusion vs 24-hour infusion).29 The issue relating to the risk of neurotoxicity after the administration of weekly versus every 3 weeks paclitaxel treatment schedules has been conflictingly addressed. There was evidence to suggest that the risk is lower with the weekly paclitaxel schedule, while the opposite was demonstrated in other studies.30,31 The risk appears to be unrelated to advanced age.32
Clinical and electrophysiological characteristics
Usually, patients affected by TIPN complain of paresthesia, numbness, and/or neuropathic pain in a stocking-and-glove distribution. Clinical examination documents loss of proprioception and suppression or loss of deep tendon reflexes (DTRs).23 Nerve conduction studies reveal the decrease or abolishment of sensory responses in keeping with an axonal sensory neuropathy as a result of axonal loss from sensory nerves.33 The sural nerve is particularly affected.34 Motor involvement with reduction of compound muscle action potential responses and myopathy with proximal weakness is less frequently seen.35 The significance of sympathetic skin response to provide electrophysiological evidence of small fiber neuropathy in taxane-treated patients merits further study.36
Course of neurotoxicity
Symptoms usually improve or resolve within 3 months after the discontinuation of treatment, whereas severe symptoms may persist for a longer period.23,33
Options for treatment or prevention
Previous evidence in relation to the symptomatic management of painful TIPN shows that amitriptyline, glutamine, low-dose of oral prednisone, and gabapentin may alleviate the patients’ pain.4 Newer drugs, such as duloxetine alone or in combination with pregabalin have provided additional measures of success in reducing pain, myalgia, and arthralgia.37,38
Although several neuroprotective agents, including amifostine, glutamine, acetyl-l-carnitine, and vitamin E hold promise as possible neuroprotective factors, clinical data are still controversial and their routine use is currently not recommended in everyday clinical practice.39–44
Epothilones (ixabepilone/sagopilone)
Pathogenesis
Epothilones are able to evoke peripheral neuropathy by inducing tubulin polymerization into microtubules, by interfering with the normal process of anterograde and retrograde axonal transport.45,46 Additionally, it seems that epothilones are able to induce damage to the ganglion soma cells and peripheral neuroaxons through the disruption of microtubules of the mitotic spindle and by interfering with axonal transport and cytoplasmic flow in the affected neurons.47 Besides axonopathy, DRG changes have been demonstrated in epothilone-treated animal models.48
Incidence, severity, and risk factors
The neurotoxicity threshold for ixabepilone is reached at a dose of 40 mg/m2.49 Above that dose, a significant percentage of patients exposed to ixabepilone treatment, varying from 40%–88%, exhibit sensory peripheral neuropathy.50,51 The incidence of treatment-related advanced sensory or motor neurotoxicity can affect up to 24% and 5% of patients, respectively.50–52
ZK-EPO (sagopilone), a third-generation epothilone B derivative, has been clinically tested in metastatic breast or platinum-resistant ovarian cancer patients, and safety data show that the incidence and severity of neurotoxicity was comparable to that of taxanes and ixabepilone.53 Sagopilone-induced peripheral neuropathy occurs in up to 81.5% of patients, usually in the form of a dose-related sensory neuropathy.54,55 The rates of treatment-emergent neurotoxicity are significant, as up to 16% of patients can experience grade 3 peripheral neuropathy.55
Clinical and electrophysiological characteristics
Both ixabepilone and sagopilone have been reported to produce a clinical and electrophysiological spectrum of neurotoxicity, similar to taxanes.49
Course of neurotoxicity
Current knowledge shows that recovery from neurotoxicity symptoms is relatively faster than the period required for the recovery from symptoms of taxane-related neuropathy; it usually takes only 4–6 weeks for grade 3–4 symptoms to improve by at least one NCI-CTCAE grade after the finalization of treatment with either ixabepilone or sagopilone.54,56
Options for treatment or prevention
To date, the literature has provided only weak evidence to support the use of any prophylactic treatment against ixabepilone-induced neurotoxicity.49 A quite recent Phase II European multicenter clinical trial failed to support the efficacy of acetyl-L-carnitine against the neurotoxicity induced by sagopilone, an analog of ixabepilone.57 Therefore, adherence to dose modification guidelines is clearly warranted.54,58
Platinum compounds (cisplatin/carboplatin/oxaliplatin)
Pathogenesis
DRG represent the main structure that is affected by the deposition of platinum compounds, thereby generating neurotoxicity.59 Two putative mechanisms are primarily involved in platinum-induced neurotoxicity, and DRG neuron apoptosis is the common cornerstone.60,61
Firstly, they are able to alter the tertiary structure of DNA by forming intrastrand adducts and interstrand crosslinks.62 Moreover, it has been documented that the neuronal apoptosis on DRG could be triggered by oxidative stress, mitochondrial dysfunction with a release of the cytochrome-c pathway, independence of Fas receptor activation, or by increased activity of p53, p38, and ERK1/2.63–66
The pathogenesis of oxaliplatin-induced neurotoxicity does not share the same characteristics, as this platinum compound induces two clinically distinct forms of neurotoxicity, namely acute and chronic. The chronic, sensory form is considered to be induced by the morphologic and functional changes in the DRG cells, resulting from the local deposition and accumulation of oxaliplatin. On the other hand, the acute form is thought to be caused by a dysfunction of nodal axonal voltage-gated Na+ channels, likely resulting from the oxalate chelating effect on both Ca2+ and Mg2+.67
Incidence, severity, and risk factors
For cisplatin, evidence of peripheral nerve damage of any grade has been reported in about 60% of patients receiving a total cumulative drug dose ranging from 225–500 mg/m2.2 However, only 10% of them experience treatment-emergent grade 3–4 neurotoxicity.68,69 The combination of cisplatin/paclitaxel exerts additive effects in producing neuropathy at higher rates than cisplatin monotherapy.33,70
Available data show that carboplatin is almost unrelated to peripheral neuropathy when given as monotherapy at an area under the curve of 6 (AUC6), while its administration at an AUC12 is not associated with the occurrence of grade 3–4 neurotoxicity.71,72 As such, carboplatin is definitely much less neurotoxic than cisplatin or oxaliplatin.2
As to the acute form of neurotoxicity induced by oxaliplatin, it is generally acknowledged that the vast majority of patients treated with various oxaliplatin-based regimens at a dose ranging from 85–130 mg/m2 experience some grade of neurotoxicity.4,73,74 Severe acute OXLIPN (oxaliplatin-induced peripheral neuropathy) that requires prolongation of oxaliplatin infusion or treatment discontinuation may occur in up to 22% of treated patients.75 Cold temperatures and the time of oxaliplatin infusion are the main risk factors of acute OXLIPN.76
On the other hand, the overall rate of neurosensory symptoms in the context of chronic oxaliplatin-induced neurotoxicity (OXAIPN) can range from 60%–75% in patients assigned to be treated with oxaliplatin-based regiments, including FOLFOX4, FOLFOX6, or XELOX.77,78 Data from large studies show that treatment-emergent grade 3–4 neurotoxicity can occur in up to 20% of oxaliplatin-treated patients, but it can be predicted by clinical and neurophysiological information obtained at mid-treatment.77,79
The cumulative oxaliplatin dose, time of infusion, and the existence of peripheral neuropathy prior to the initiation of chemotherapy rank among the most important triggers of chronic OXAIPN genesis.80 In addition to these well-known risk factors, recent evidence from a large homogeneous series of colorectal cancer patients showed that patients who have a more complex combination of acute phenomena related to axonal hyperexcitability are those who eventually develop more severe chronic neurotoxicity.75 Furthermore, it seems that the chemotherapy regimen may also represent a risk factor of OXAIPN. This view was supported by a recently published study, which documented that XELOX may be the preferable regimen to avoid the more severe neurotoxicity associated with FOLFOX, despite comparable oxaliplatin cumulative dose.81 Advanced age does not seem to represent a significant risk factor of OXAIPN in patients without any other significant comorbidity.82
Clinical and electrophysiological characteristics
The clinical spectrum of cisplatin-induced peripheral neuropathy is comprised of sensory symptoms in a stocking-and-glove distribution, decreased vibration, and proprioception and suppression or loss of DTRs. Neurophysiology is in keeping with an axonal sensory peripheral neuropathy with a decrease or abolishment of sensory action potentials and normal sensory conduction velocities.4,33
Signs and symptoms of acute OXLIPN may begin during the infusion or within 1–2 days of oxaliplatin administration and mostly include distal and perioral cold-induced paresthesias and dysesthesias. However, other uncommon symptoms, such as shortness of breath, jaw spasm, fasciculations, cramps, and difficulty swallowing may also be present at significant rates. Voice and visual changes, ptosis, and pseudo-laryngospasm rarely occur.83 Recording of repetitive compound action potentials, high-frequency discharges of motor unit multiplets, and bursts of muscle fiber action potentials are evident during nerve conduction study and needle electromyography examination. This pattern is in keeping with neuromyotonia as a result of excessive nerve excitability, distally attenuated.84 The clinical and neurophysiological characteristics of chronic OXLIPN are generally similar to those of cisplatin.2
Long-term outcome
Although there are few specifically designed studies to assess the long-term course of platinum-induced neurotoxicity, it is expected to improve or completely resolve within 1 year after the discontinuation of treatment. However, there have been cases in which CIPN remained persistent or at best, partially reversible, because of the “coasting” phenomenon resulting from the capacity of platinum compounds to accumulate in DRG for a long time.25
Options for treatment or prevention
Based on results from randomized controlled trials (RCTs), there are no effective symptomatic treatments, and only duloxetine at a dose of 60 mg per day has been shown in a well-designed RCT to be effective in alleviating oxaliplatin-associated neuropathic pain.37 As to prophylaxis, there have been insufficient data thus far, to support the use of any candidate chemoprotective agents, such as acetylcysteine, amifostine, calcium and magnesium, diethyldithiocarbamate, glutathione, Org 2766, oxcarbazepine, or vitamin E to prevent or limit the neurotoxicity of platinum compounds.85 As such, adherence to the non-pharmacological stop-and-go approach (ie, intermittent oxaliplatin dosing) may be warranted to prevent platinum compound-induced peripheral neuropathy, particularly OXAIPN.86
Vinca alkaloids (vincristine)
Pathogenesis
Vincristine detrimentally affects both fast- and slow-conducting peripheral nerve fibers by interfering with axonal transport at the level of the cell body and alterations in the cellular microtubuli structure.87
Incidence, severity, and risk factors
Vincristine-induced peripheral neuropathy is dose dependent, as up to 60% of patients may develop a clinically significant (grade 1–2) primarily sensory or sensorimotor neuropathy at vincristine cumulative doses between 30–50 mg.88
Clinical and electrophysiological characteristics
At the initial stage, the clinical manifestations of vincristine- induced peripheral neuropathy include bilateral and symmetrical painful paresthesia and hyperesthesia, distally attenuated. Muscle cramps and mild distal weakness are frequently seen. Neurological examination reveals proprioception and DTR abnormalities. Autonomic dysfunction is frequently seen in vincristine-treated patients, with evidence of orthostatic hypotension, constipation, and erectile impotence. Few cases of cranial nerve palsies have been reported while patients were being treated with chemotherapy using vincristine.89 Nerve conduction abnormalities are in keeping with a length-dependent axonal sensory or sensorimotor peripheral neuropathy.90
Long-term outcome
Neurotoxic symptoms of vincristine are reversible after discontinuation of treatment.88 However, off-therapy worsening of neurotoxic symptoms and signs might unexpectedly occur.91
Options for treatment or prevention
To date, there has been insufficient evidence to recommend the use of any neuroprotectant against vincristine-induced peripheral neuropathy in clinical practice.2
Binding of growth factor inhibitors (suramin)
Pathogenesis
The cardinal pathogenetic hallmark of suramin neurotoxicity is the axonal degeneration in DRG and the accumulation of glycolipid lysosomal inclusions, probably because of the competition between suramin and nerve growth factor (NGF) at the high-affinity NGF receptor.92
Incidence, severity, and risk factors
Suramin-induced peripheral neuropathy is dose dependent, as up to 60% of patients may develop a clinically significant (grade 1–2) primarily sensory or sensorimotor neuropathy at suramin plasma peak levels higher than 350 μg/mL.93
Clinical and electrophysiological characteristics
At the initial stage, the clinical manifestations of suramin-induced peripheral neuropathy include bilateral and symmetrical painful paresthesia and hyperesthesia, distally attenuated. Neurological examination reveals proprioception and DTR abnormalities. Nerve conduction abnormalities are in keeping with a length-dependent axonal sensory or sensorimotor peripheral neuropathy.93 Single case reports of subacute demyelinating and inflammatory polyneuropathy secondary to suramin therapy have also been occasionally published.94
Long-term outcome
Neurotoxic symptoms of suramin are usually reversible after discontinuation of treatment.93
Options for treatment or prevention
To date, none of the candidate neuroprotective agents have been proven effective to treat suramin-induced peripheral neuropathy.2
Proteasome inhibitors (bortezomib)
Pathogenesis
The pathogenetic hallmark of bortezomib-induced peripheral neuropathy (BIPN) consists of morphological alterations in the spinal cord, DRG, and peripheral nerves with specific functional alterations in Aδ and C peripheral nerve fibers.95,96 In addition, proteasome inhibition, increased a-tubulin polymerization, mitochondrial and endoplasmic reticulum damage, and dysregulation of neurotrophins through inhibition of NFkB activation may also significantly contribute to BIPN genesis.97–99
Incidence, severity, and risk factors
The incidence of clinically significant, ie, grade 1–2 BIPN, can occur in up to 75% patients with multiple myeloma who have relapsed after or were refractory to frontline therapy, while treatment emergent grade 3–4 neurotoxicity may appear in 12% of bortezomib-treated patients.100,101 BIPN is usually exacerbated in patients with pre-existing neuropathy and comorbidities associated with peripheral nerve damage.98,102 However, the cumulative dose effect of bortezomib remains the main triggering factor of BIPN, although the severity of neurotoxicity is escalated until the completion of the first five cycles of bortezomib administration and thereafter remains stable.103
Bortezomib administered subcutaneously rather than intravenously has an improved safety profile and appears to be ideal for patients with pre-existing neuropathy or at a high risk of developing neurotoxicity. In a large randomized, Phase III, non-inferiority study enrolling 222 patients with relapsed multiple myeloma (148 on subcutaneous versus (vs) 74 patients on intravenous bortezomib), it was documented that peripheral neuropathy of any grade (38% vs 53%; P=0.044), grade ≥2 (24% vs 41%; P=0.012), and grade ≥3 (6% vs 16%; P=0.026) was significantly less common with subcutaneous than with intravenous administration.104
Clinical and electrophysiological characteristics
The cardinal symptom of BIPN is neuropathic pain and paresthesias in distal extremities of limbs, in keeping with a painful neuropathy due to dysfunction in all three major fiber (Aβ, Aδ, and C) types of sensory nerves, as demonstrated in both clinical and animal models.95,105 Neurological examination reveals distal sensory loss to all modalities and changes in proprioception, while DTRs are either suppressed or absent. Nerve conduction study usually reveals typical findings of CIPN consistent with a distal, sensory, axonal neuronopathy. Motor involvement is occasionally present. Reversal of BIPN usually occurs after a median interval of 3 months following the discontinuation of bortezomib treatment, but it may persist for up to 2 years or remain indefinitely in some cases.98,103
Options for treatment or prevention
Lafutidine, a H2-blocker with gastroprotective activity, may be able to prevent or improve BIPN, based on the results of a recently published small case series of eight patients.106 However, the protective activity of lafutidine against BIPN needs to be further demonstrated in large RCTs. As such, there is no proven effective prophylactic treatment to prevent the development of BIPN, and medication towards this aspect is merely symptomatic.98 Therefore, likewise to the case of EIPN, adherence to the dose-modification guidelines is advised.
Antiangiogenic/immunomodulatory agents (thalidomide/lenalidomide/pomalidomide)
Pathogenesis
Neurotoxicity is considered to be generated by both thalidomide and lenalidomide as either the result of their antiangiogenic properties with a reduction of nerve blood supply or because of functional and metabolic changes in the DRG. Dysregulation of neurotrophin activity may also play a significant role in the pathogenesis of neurotoxicity.107
Incidence, severity, and risk factors
Quoting the results of an analysis of clinical trials assessing the efficacy and safety of thalidomide monotherapy in patients with relapsed or refractory multiple myeloma, the overall incidence of PN can range up to 44%, with a rate of early treatment discontinuation of about 15%.108 Moreover, another meta-analysis of safety data after thalidomide monotherapy in multiple myeloma patients revealed that the rate of treatment-emergent neurotoxicity (grade 3–4) was 6%, whereas according to the same report, there is no well-established factor to increase the risk of thalidomide-induced neurotoxicity.109
Lenalidomide appears to be less neurotoxic and better tolerated than thalidomide at a dose of 30 mg/day in relapsed/refractory multiple myeloma patients, with only 3% of significant grade 3 neurotoxicity after the completion of chemotherapy.110,111
Orally administered pomalidomide was recently approved by the US Food and Drug Administration for use in multiple myeloma patients who have received at least two prior therapies, including lenalidomide and bortezomib, and whose disease did not respond to treatment and progressed within 60 days of the last treatment. Grade 1–2 peripheral neurotoxicity can occasionally (up to 9% of patients) be seen but no grade 3–4 peripheral neuropathy was reported in large RCTs testing the efficacy and safety of this agent.112,113
Clinical and electrophysiological characteristics
The clinical and neurophysiological characteristics of neurotoxicity secondary to thalidomide, lenalidomide, and pomalidomide are generally similar to those of bortezomib.107
Options for treatment or prevention
The symptomatic treatment of painful peripheral neuropathy secondary to these agents does not differ from that of BIPN.107
Other drugs less commonly associated with CIPN
Fluorouracil or 5-FU is a pyrimidine analog, and peripheral neuropathy associated with its administration is unusual. Anyhow, the literature contains a small series of two patients experiencing neurotoxicity while they were receiving chemotherapy with 5-FU.114 Gemcitabine may occasionally evoke peripheral neuropathy with paresthesias and myalgias, whereas neurotoxicity rarely occurs while patients are treated with methotrexate, cytosine arabinoside (Ara-C), or topoisomerase inhibitors, such as irinotecan or topotecan.25
Rehabilitation and complementary/alternative therapies against CIPN
CIPN can significantly undermine the daily living activities and QOL of patients. Sensory ataxia is a common clinical phenomenon in the context of neuronopathies and it is associated with unsteady gait and impaired balance and coordination, as well as poor mobility. As a consequence, falls may occur in a significant proportion of cancer survivors. An increased risk of falls was associated with the degree of CIPN, particularly with evidence of sensory ataxia and severe muscle weakness.115
A variety of rehabilitative methods, such as balance and gait retraining as well as the use of gait aids and orthotics, have been tested with positive effects to prevent falls in patients with peripheral neuropathies and to assist them in adapting their activities and their environment.116 Both aerobic and resistance exercise, mindfulness, occupational therapy, and environmental planning are also proposed among effective self-management strategies in reducing the impact of CIPN symptoms.117
Acupuncture is the most widely used complementary intervention in CIPN subjects. However, a recently published meta-analysis of clinical studies found that there is no evidence to support the use of acupuncture for treating CIPN, and further studies with robust methodology are needed before one can conclude with confidence about its true usefulness.118 Supplementation with single medical herbs or herbal combinations might hold promise for its ability to exert neuroprotection or neuroregeneration in CIPN. However, it is similar to acupuncture in that the level of evidence is at present too low to establish a standard practice.119
CIPN in the era of pharmacogenetics
Although candidate gene approaches have been launched during the last decade to extract results from single nuclear polymorphisms (SNPs) in genes involved in the pharmacokinetic and pharmacodynamic properties of neurotoxic drugs, no reliable biomarker has thus far been identified to detect patients at high risk of developing CIPN. In the past, several SNPs have been reported to be associated with CIPN. For instance, SNPs in genes involved in the pharmacokinetic, transport, and pharmacodynamic properties of taxanes have been shown to be relevant for TIPN.120,121 Likewise, increased susceptibility to peripheral neurotoxicity after exposure to oxaliplatin and other platinum compounds has been associated with pharmacogenetic variations in genes encoding for drug transporters, detoxification enzymes, genes involved in DNA repair mechanisms, and integrin B3 Leu33Pro polymorphism.122–124
However, the results of most previous pharmacogenetic studies focused on oxaliplatin were limited, and with several methodological flaws, including small sample size, retrospective study design, and the implementation of a post-hoc analysis of oncology-based databases of different, not pre-planned sizes. Other major limitations of these studies include the lack of a pre-study hypothesis based on the known role of the investigated targets in the peripheral nervous system and the inappropriate outcome measures for neurological impairment.122,123,125
A recently published collaborative international study attempted to overcome all of those limitations, thoroughly investigating a series of SNPs in genes coding for neurologically relevant targets in an adequately powered, prospective cohort of well-characterized patients, such as the voltage-gated sodium channels (SCNA). The results of this study provided evidence to support a causal relationship between SCN4A and SCN10A polymorphisms and increased incidence and/or severity of oxaliplatin-induced peripheral neuropathy.78 Further SCNA SNPs, such as the SCN2A R19K polymorphism, have been previously investigated with negative results.126
Other polymorphisms in genes involved with immune function, reflexive coupling within Schwann cells, drug binding, neuron function, and steroid hormone biosynthesis have been associated with BIPN.127,128 Table 4 summarizes genetic biomarkers that have been linked with liability to CIPN, by neurotoxic drug classification.
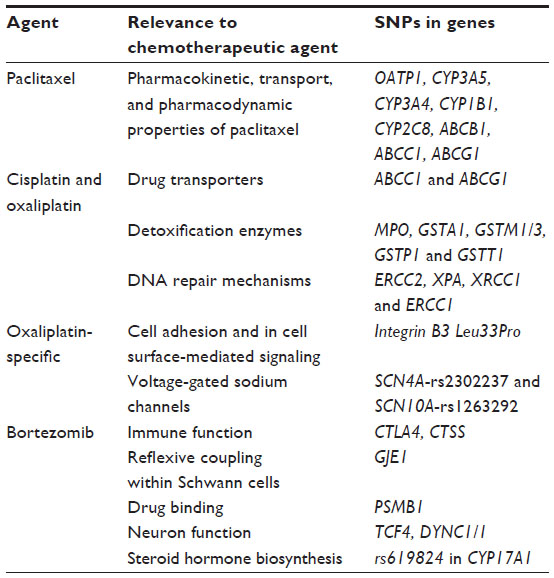 | Table 4 Genetic biomarkers linked to CIPN by neurotoxic drug classification |
Conclusion and future perspectives for research
CIPN is one of the most severe adverse effects of treatment, with a significant impact on the QOL of affected patients, mostly because the long-term effects of the persistence of symptoms/signs cannot be estimated. In the framework of promoting cancer therapies with fewer adverse effects, there are several open issues to be addressed in the future.
Important clinically relevant questions include: how to measure the incidence of neurotoxicity; how to grade the severity of the peripheral neuropathy; how to estimate its long-term course after the discontinuation of chemotherapy; and how to utilize this information clinically. An additional important issue is to determine reliable biomarkers to allow prompt identification of patients at high risk to develop CIPN. To address these gaps in knowledge, further large systematic prospective collection of data on CIPN is needed, comprising a comprehensive set of reliable clinical assessments and patient-reported outcomes, with the support of focused neurophysiological examinations, skin biopsies, and DNA analysis. Of note, skin biopsy may be a useful tool to examine the clinical applicability and correlation of intraepidermal nerve fiber density in CIPN with other clinical outcome measures, eventually leading to both possible preventive and therapeutic intervention.129
The results of such interventions would significantly contribute to improved comfort and QOL of cancer survivors. Proper and well-evaluated approaches would also produce a consistent advantage for both doctors and patients to allow the registration and analysis of reliable data on the incidence, prevalence, risk factors, and long-term impact of CIPN, eventually leading to both potential preventive and therapeutic multidimensional interventions.
Author contributions
Each author jointly contributed in the preparation of this review paper.
Disclosure
The authors report no conflicts of interest in this work. No funding source had a role in the preparation of this paper or in the decision to submit it for publication.
References
Argyriou AA, Assimakopoulos K, Iconomou G, Giannakopoulou F, Kalofonos HP. Either called “Chemobrain” or “Chemofog,” the long-term chemotherapy-induced cognitive decline in cancer survivors is real. J Pain Symptom Manage. 2011;41(1):126–139. | |
Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol. 2012;82(1):51–77. | |
Pike CT, Birnbaum HG, Muehlenbein CE, Pohl GM, Natale RB. Healthcare costs and workloss burden of patients with chemotherapy-associated peripheral neuropathy in breast, ovarian, head and neck, and non-small cell lung cancer. Chemother Res Pract. 2012;2012: 913848. | |
Argyriou AA, Zolota V, Kyriakopoulou O, Kalofonos HP. Toxic peripheral neuropathy associated with commonly used chemotherapeutic agents. J BUON. 2010;15(3):435–446. | |
Cavaletti G, Frigeni B, Lanzani F, et al. Chemotherapy-induced peripheral neurotoxicity assessment: a critical revision of the currently available tools. Eur J Cancer. 2010;46(3):479–494. | |
National Cancer Institute Common Terminology Criteria for Adverse Events v4.0 NCI, NIH, DHHS. May 29, 2009; NIH publication # 09-7473. | |
Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol. 2010;6(12):657–666. | |
Calhoun EA, Welshman EE, Chang CH, et al. Psychometric evaluation of the Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity (Fact/GOG-Ntx) questionnaire for patients receiving systemic chemotherapy. Int J Gynecol Cancer. 2003;13(6):741–748. | |
Griffith KA, Merkies IS, Hill EE, Cornblath DR. Measures of chemotherapy-induced peripheral neuropathy: a systematic review of psychometric properties. J Peripher Nerv Syst. 2010;15(4):314–325. | |
Frigeni B, Piatti M, Lanzani F, et al. Chemotherapy-induced peripheral neurotoxicity can be misdiagnosed by the National Cancer Institute Common Toxicity scale. J Peripher Nerv Syst. 2011;16(3):228–236. | |
Cavaletti G, Cornblath DR, Merkies IS, et al; CI-PeriNomS Group. The chemotherapy-induced peripheral neuropathy outcome measures standardization study: from consensus to the first validity and reliability findings. Ann Oncol. 2013;24(2):454–462. | |
Cornblath DR, Chaudhry V, Carter K, et al. Total neuropathy score: validation and reliability study. Neurology. 1999;53(8):1660–1664. | |
Cavaletti G, Bogliun G, Marzorati L, et al. Grading of chemotherapy-induced peripheral neurotoxicity using the total neuropathy scale. Neurology. 2003;61:1297–1300. | |
Cavaletti G, Jann S, Pace A, et al; Italian NETox Group. Multi-center assessment of the total neuropathy score for chemotherapy-induced peripheral neurotoxicity. J Periph Nerv Syst. 2006;11:135–141. | |
Cavaletti G, Frigeni B, Lanzani F, et al; Italian NETox Group. The Total Neuropathy Score as an assessment tool for grading the course of chemotherapy-induced peripheral neurotoxicity: comparison with the National Cancer Institute-Common Toxicity Scale. J Peripher Nerv Syst. 2007;12(3):210–215. | |
Binda D, Vanhoutte EK, Cavaletti G, et al; CI-PeriNomS study group. Rasch-built Overall Disability Scale for patients with chemotherapy-induced peripheral neuropathy (CIPN-R-ODS). Eur J Cancer. 2013;49(13):2910–2918. | |
Alberti P, Rossi E, Cornblath DR, et al; CI-PeriNomS Group. Physician-assessed and patient-reported outcome measures in chemotherapy-induced sensory peripheral neurotoxicity: two sides of the same coin. Ann Oncol. 2014;25(1):257–264. | |
Argyriou AA, Koltzenburg M, Polychronopoulos P, Papapetropoulos S, Kalofonos HP. Peripheral nerve damage associated with administration of taxanes in patients with cancer. Crit Rev Oncol Hematol. 2008;66(3):218–228. | |
Rowinsky EK, Donehower RC. Paclitaxel (Taxol). N Engl J Med. 1995;332:1004–1014. | |
Peters CM, Jimenez-Andrade JM, Jonas BM, et al. Intravenous paclitaxel administration in the rat induced a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp Neurol. 2007;203(1):42–54. | |
LaPointe NE, Morfini G, Brady ST, Feinstein SC, Wilson L, Jordan MA. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology. 2013;37:231–239. | |
Gilardini A, Avila RL, Oggioni N, et al. Myelin structure is unaltered in chemotherapy-induced peripheral neuropathy. Neurotoxicology. 2012;33(1):1–7. | |
Argyriou AA, Polychronopoulos P, Iconomou G, Koutras A, Kalofonos HP, Chroni E. Paclitaxel plus carboplatin-induced peripheral neuropathy. A prospective clinical and electrophysiological study in patients suffering from solid malignancies. J Neurol. 2005;252(12):1459–1464. | |
Krzakowski M, Ramlau R, Jassem J, et al. Phase III trial comparing vinflunine with docetaxel in second-line advanced non-small-cell lung cancer previously treated with platinum-containing chemotherapy. J Clin Oncol. 2010;28(13):2167–2173. | |
Grisold W, Cavaletti G, Windebank AJ. Peripheral neuropathies from chemotherapeutics and targeted agents: diagnosis, treatment, and prevention. Neuro Oncol. 2012;14 Suppl 4:iv45–iv54. | |
Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol. 2006;24(10):1633–1642. | |
Ma P, Mumper RJ. Paclitaxel nano-delivery systems: a comprehensive review. J Nanomed Nanotechnol. 2013;4(2):1000164. | |
Seidman AD, Conlin AK, Bach A, et al. Randomized phase II trial of weekly vs every 2 weeks vs every 3 weeks nanoparticle albumin-bound paclitaxel with bevacizumab as first-line chemotherapy for metastatic breast cancer. Clin Breast Cancer. 2013;13(4):239–246. e1. | |
Sioka C, Kyritsis AP. Central and peripheral nervous system toxicity of common chemotherapeutic agents. Cancer Chemother Pharmacol. 2009;63(5):761–767. | |
Mauri D, Kamposioras K, Tsali L, et al. Overall survival benefit for weekly vs three-weekly taxanes regimens in advanced breast cancer: a meta-analysis. Cancer Treat Rev. 2010;36(1):69–74. | |
Fountzilas G, Dafni U, Dimopoulos MA, et al. A randomized phase III study comparing three anthracycline-free taxane-based regimens, as first line chemotherapy, in metastatic breast cancer: a Hellenic Cooperative Oncology Group study. Breast Cancer Res Treat. 2009;115(1):87–99. | |
Argyriou AA, Polychronopoulos P, Koutras A, et al. Is advanced age associated with increased incidence and severity of chemotherapy-induced peripheral neuropathy? Support Care Cancer. 2006;14(3):223–229. | |
Argyriou AA, Polychronopoulos P, Koutras A, et al. Clinical and electrophysiological features of peripheral neuropathy induced by administration of cisplatin plus paclitaxel-based chemotherapy. Eur J Cancer Care (Engl). 2007;16(3):231–237. | |
Argyriou AA, Polychronopoulos P, Koutras A, et al. Peripheral neuropathy induced by administration of cisplatin and paclitaxel-based chemotherapy. Could it be predicted? Support Care Cancer. 2005;13(8):647–651. | |
Behin A, Psimaras D, Hoang-Xuan K, Leger JM. Neuropathies in the context of malignancies. Curr Opin Neurol. 2008;21(5):534–539. | |
Argyriou AA, Koutras A, Polychronopoulos P, et al. The impact of paclitaxel or cisplatin-based chemotherapy on sympathetic skin response: a prospective study. Eur J Neurol. 2005;12(11):858–861. | |
Smith EM, Pang H, Cirrincione C, et al; Alliance for Clinical Trials in Oncology. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA. 2013;309(13):1359–1367. | |
Takenaka M, Iida H, Matsumoto S, Yamaguchi S, Yoshimura N, Miyamoto M. Successful treatment by adding duloxetine to pregabalin for peripheral neuropathy induced by paclitaxel. Am J Hosp Palliat Care. 2013;30(7):734–736. | |
Moore DH, Donnelly J, McGuire WP, et al; Gynecologic Oncology Group. Limited access trial using amifostine for protection against cisplatin- and three-hour paclitaxel-induced neurotoxicity: a phase II study of the Gynecologic Oncology Group. J Clin Oncol. 2003;21(22):4207–4213. | |
Davis ID, Kiers L, MacGregor L, et al. A randomized, double-blinded, placebo-controlled phase II trial of recombinant human leukemia inhibitory factor (rhuLIF, emfilermin, AM424) to prevent chemotherapy-induced peripheral neuropathy. Clin Cancer Res. 2005;11(5):1890–1898. | |
Stubblefield MD, Vahdat LT, Balmaceda CM, Troxel AB, Hesdorffer CS, Gooch CL. Glutamine as a neuroprotective agent in high-dose paclitaxel-induced peripheral neuropathy: a clinical and electrophysiologic study. Clin Oncol (R Coll Radiol). 2005;17(4):271–276. | |
Maestri A, De Pasquale Ceratti A, et al. A pilot study on the effect of acetyl-L-carnitine in paclitaxel- and cisplatin-induced peripheral neuropathy. Tumori. 2005;91(2):135–138. | |
Argyriou AA, Chroni E, Koutras A, et al. Vitamin E for prophylaxis against chemotherapy-induced neuropathy: a randomized controlled trial. Neurology. 2005;64(1):26–31. | |
Argyriou AA, Chroni E, Koutras A, et al. Preventing paclitaxel-induced peripheral neuropathy: a phase II trial of vitamin E supplementation. J Pain Symptom Manage. 2006;32(3):237–244. | |
Nakata T, Yorifuji H. Morphological evidence of the inhibitory effect of taxol on the fast axonal transport. Neurosci Res. 1999;35(2):113–122. | |
Theiss C, Meller K. Taxol impairs anterograde axonal transport of microinjected horseradish peroxidase in dorsal root ganglia neurons in vitro. Cell Tissue Res. 2000;299(2):213–224. | |
Park SB, Krishnan AV, Lin CS, Goldstein D, Friedlander M, Kiernan MC. Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Curr Med Chem. 2008;15(29):3081–3094. | |
Chiorazzi A, Nicolini G, Canta A, et al. Experimental epothilone B neurotoxicity: results of in vitro and in vivo studies. Neurobiol Dis. 2009;35(2):270–277. | |
Argyriou AA, Marmiroli P, Cavaletti G, Kalofonos HP. Epothilone-induced peripheral neuropathy: a review of current knowledge. J Pain Symptom Manage. 2011;42(6):931–940. | |
Vansteenkiste J, Lara PN Jr, Le Chevalier T, et al. Phase II clinical trial of the epothilone B analog, ixabepilone, in patients with non small-cell lung cancer whose tumors have failed first-line platinum-based chemotherapy. J Clin Oncol. 2007;25(23):3448–3455. | |
Huang H, Menefee M, Edgerly M, et al. A phase II clinical trial of ixabepilone (Ixempra; BMS-247550; NSC 710428), an epothilone B analog, in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2010;16(5):1634–1641. | |
Sparano JA, Vrdoljak E, Rixe O, et al. Randomized phase III trial of ixabepilone plus capecitabine versus capecitabine in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2010;28(20):3256–3263. | |
Dorff TB, Gross ME. The epothilones: new therapeutic agents for castration-resistant prostate cancer. Oncologist. 2011;16(10):1349–1358. | |
Morrow PK, Divers S, Provencher L, et al. Phase II study evaluating the efficacy and safety of sagopilone (ZK-EPO) in patients with metastatic breast cancer that has progressed following chemotherapy. Breast Cancer Res Treat. 2010;123(3):837–842. | |
Rustin G, Reed N, Jayson GC, et al. A phase II trial evaluating two schedules of sagopilone (ZK-EPO), a novel epothilone, in patients with platinum- resistant ovarian cancer. Ann Oncol. 2011;22(11):2411–2426. | |
Thomas ES, Gomez HL, Li RK, et al. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J Clin Oncol. 2007;25(33):5210–5227. | |
Campone M, Berton-Rigaud D, Joly-Lobbedez F, et al. A double-blind, randomized phase II study to evaluate the safety and efficacy of acetyl-L-carnitine in the prevention of sagopilone-induced peripheral neuropathy. Oncologist. 2013;18(11):1190–1191. | |
IXEMPRA™ kit (ixabepilone) for injection [prescribing information]. Princeton: Bristol-Myers Squibb Company; 2009. | |
Krarup-Hansen A, Rietz B, Krarup C, Heydorn K, Rorth M, Schmalbruch H. Histology and platinum content of sensory ganglia and sural nerves in patients treated with cisplatin and carboplatin: an autopsy study. Neuropathol Appl Neurobiol. 1999;25:29–40. | |
Ta LE, Espeset L, Podratz J, Windebank AJ. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. Neurotoxicology. 2006;27:992–1002. | |
Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101:2842–2850. | |
McDonald ES, Randon KR, Knight A, Windebank AJ. Cisplatin preferentially binds to DNA in dorsal root ganglion neurons in vitro and in vivo: a potential mechanism for neurotoxicity. Neurobiol Dis. 2005;18:305–313. | |
Zhang L, Cooper AJ, Krasnikov BF, et al. Cisplatin-induced toxicity is associated with platinum deposition in mouse kidney mitochondria in vivo and with selective inactivation of the alpha-ketoglutarate dehydrogenase complex in LLC-PK1 cells. Biochemistry. 2006;45: 8959–8971. | |
Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signalling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68:6425–6434. | |
McDonald ES, Windebank AJ. Cisplatin-induced apoptosis of DRG neurons involves bax redistribution and cytochrome c release but not fas receptor signalling. Neurobiol Dis. 2002;9:220–233. | |
Scuteri A, Galimberti A, Maggioni D, et al. Role of MAPKs in platinum-induced neuronal apoptosis. Neurotoxicology. 2009;30(2):312–319. | |
Argyriou AA, Polychronopoulos P, Iconomou G, Chroni E, Kalofonos HP. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat Rev. 2008;34:368–377. | |
Sutton G, Brunetto VL, Kilgore L, et al. A phase III trial of ifosfamide with or without cisplatin in carcinosarcoma of the uterus: a Gynecologic Oncology Group Study. Gynecol Oncol. 2000;79(2):147–153. | |
Gatzemeier U, von Pawel J, Gottfried M, et al. Phase III comparative study of high-dose cisplatin versus a combination of paclitaxel and cisplatin in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2000;18(19):3390–3399. | |
Berger T, Malayeri R, Doppelbauer A, et al. Neurological monitoring of neurotoxicity induced by paclitaxel/cisplatin chemotherapy. Eur J Cancer. 1997;33(9):1393–1399. | |
Gore M, Mainwaring P, A’Hern R, et al. Randomized trial of dose-intensity with single-agent carboplatin in patients with epithelial ovarian cancer. London Gynaecological Oncology Group. J Clin Oncol. 1998;16(7):2426–2434. | |
Thigpen JT. Dose-intensity in ovarian carcinoma: hold, enough? J Clin Oncol. 1997;15(4):1291–1293. | |
de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938–2947. | |
Kemeny N, Garay CA, Gurtler J, et al. Randomized multicenter phase II trial of bolus plus infusional fluorouracil/leucovorin compared with fluorouracil/leucovorin plus oxaliplatin as third-line treatment of patients with advanced colorectal cancer. J Clin Oncol. 2004;22(23):4753–4761. | |
Argyriou AA, Cavaletti G, Briani C, et al. Clinical pattern and associations of oxaliplatin acute neurotoxicity: a prospective study in 170 patients with colorectal cancer. Cancer. 2013;119(2):438–444. | |
Gamelin E, Gamelin L, Bossi L, Quasthoff S. Clinical aspects and molecular basis of oxaliplatin neurotoxicity: current management and development of preventive measures. Semin Oncol. 2002;29(5 Suppl 15):21–33. | |
Argyriou AA, Polychronopoulos P, Iconomou G, et al. Incidence and characteristics of peripheral neuropathy during oxaliplatin-based chemotherapy for metastatic colon cancer. Acta Oncol. 2007;46(8):1131–1137. | |
Argyriou AA, Cavaletti G, Antonacopoulou A, et al. Voltage-gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin-induced peripheral neurotoxicity: results from a prospective multicenter study. Cancer. 2013;119(19):3570–3577. | |
Velasco R, Bruna J, Briani C, et al. Early predictors of oxaliplatin-induced cumulative neuropathy in colorectal cancer patients. J Neurol Neurosurg Psychiatry. Epub June 29, 2013. | |
Grothey A. Clinical management of oxaliplatin-associated neurotoxicity. Clin Colorectal Cancer. 2005;5(Suppl 1):S38–S46. | |
Argyriou AA, Velasco R, Briani C, et al. Peripheral neurotoxicity of oxaliplatin in combination with 5-fluorouracil (FOLFOX) or capecitabine (XELOX): a prospective evaluation of 150 colorectal cancer patients. Ann Oncol. 2012;23(12):3116–3122. | |
Argyriou AA, Briani C, Cavaletti G, et al. Advanced age and liability to oxaliplatin-induced peripheral neuropathy: post hoc analysis of a prospective study. Eur J Neurol. 2013;20(5):788–794. | |
Lucchetta M, Lonardi S, Bergamo F, et al. Incidence of atypical acute nerve hyperexcitability symptoms in oxaliplatin-treated patients with colorectal cancer. Cancer Chemother Pharmacol. 2012;70(6):899–902. | |
Wilson RH, Lehky T, Thomas RR, Quinn MG, Floeter MK, Grem JL. Acute oxaliplatin-induced peripheral nerve hyperexcitability. J Clin Oncol. 2002;20(7):1767–1774. | |
Albers JW, Chaudhry V, Cavaletti G, Donehower RC. Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database Syst Rev. 2011;(2):CD005228. | |
Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer-a GERCOR study. J Clin Oncol. 2006;24(3):394–400. | |
Pan YA, Misgeld T, Lichtman JW, Sanes JR. Effects of neurotoxic and neuroprotective agents on peripheral nerve regeneration assayed by time-lapse imaging in vivo. J Neurosci. 2003;23:11479–11488. | |
Postma TJ, Benard BA, Huijgens PC, Ossenkoppele GJ, Heimans JJ. Long-term effects of vincristine on the peripheral nervous system. J Neurooncol. 1993;15:23–27. | |
Dixit G, Dhingra A, Kaushal D. Vincristine induced cranial neuropathy. J Assoc Physicians India. 2012;60:56–58. | |
Pal PK. Clinical and electrophysiological studies in vincristine induced neuropathy. Electromyogr Clin Neurophysiol. 1999;39(6):323–330. | |
Verstappen CC, Koeppen S, Heimans JJ, et al. Dose-related vincristine-induced peripheral neuropathy with unexpected off-therapy worsening. Neurology. 2005;64(6):1076–1077. | |
Russell JW, Windebank AJ, Podratz JL. Role of nerve growth factor in suramin neurotoxicity studied in vitro. Ann Neurol. 1994;36:221–228. | |
Chaudhry V, Eisenberger MA, Sinibaldi VL, Sheikh K, Griffin JW, Cornblath DR. A prospective study of suramin-induced peripheral neuropathy. Brain. 1996;119:101–114. | |
Bachmann T, Koetter KP, Mühler J, Fuhrmeister U, Seidel G. Guillain-Barré syndrome after simultaneous therapy with suramin and interferon-alpha. Eur J Neurol. 2003;10(5):599. | |
Carozzi VA, Renn CL, Bardini M, et al. Bortezomib-induced painful peripheral neuropathy: an electrophysiological, behavioral, morphological and mechanistic study in the mouse. PLoS One. 2013;8(9):e72995. | |
Staff NP, Podratz JL, Grassner L, et al. Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. Neurotoxicology. 2013;39:124–131. | |
Meregalli C, Chiorazzi A, Carozzi VA, et al. Evaluation of tubulin polymerization and chronic inhibition of proteasome as citotoxicity mechanisms in bortezomib-induced peripheral neuropathy. Cell Cycle. Epub December 12, 2013. | |
Argyriou AA, Iconomou G, Kalofonos HP. Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood. 2008;112(5):1593–1599. | |
Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res. 2005;65(9):3828–3836. | |
Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–2617. | |
Jagannath S, Barlogie B, Berenson J, et al. A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol. 2004;127(2):165–172. | |
Lanzani F, Mattavelli L, Frigeni B, et al. Role of a pre-existing neuropathy on the course of bortezomib-induced peripheral neurotoxicity. J Peripher Nerv Syst. 2008;13(4):267–274. | |
Cavaletti G, Jakubowiak AJ. Peripheral neuropathy during bortezomib treatment of multiple myeloma: a review of recent studies. Leuk Lymphoma. 2010;51(7):1178–1187. | |
Moreau P, Pylypenko H, Grosicki S, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011;12(5):431–440. | |
Cata JP, Weng HR, Burton AW, Villareal H, Giralt S, Dougherty PM. Quantitative sensory findings in patients with bortezomib-induced pain. J Pain. 2007;8(4):296–306. | |
Tsukaguchi M, Shibano M, Matsuura A, Mukai S. The protective effects of lafutidine for bortezomib induced peripheral neuropathy. J Blood Med. 2013;4:81–85. | |
Mohty B, El-Cheikh J, Yakoub-Agha I, Moreau P, Harousseau JL, Mohty M. Peripheral neuropathy and new treatments for multiple myeloma: background and practical recommendations. Haematologica. 2010;95(2):311–319. | |
Prince HM, Schenkel B, Mileshkin L. An analysis of clinical trials assessing the efficacy and safety of single-agent thalidomide in patients with relapsed or refractory multiple myeloma. Leuk Lymphoma. 2007;48(1):46–55. | |
Glasmacher A, Hahn C, Hoffmann F, et al. A systematic review of phase-II trials of thalidomide monotherapy in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2006;132(5):584–593. | |
Richardson PG, Blood E, Mitsiades CS, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108(10):3458–3464. | |
Briani C, Torre CD, Campagnolo M, et al. Lenalidomide in patients with chemotherapy-induced polyneuropathy and relapsed or refractory multiple myeloma: results from a single-centre prospective study. J Peripher Nerv Syst. 2013;18(1):19–24. | |
Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. Epub January 13, 2014. | |
Miguel JS, Weisel K, Moreau P, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–1066. | |
Stein ME, Drumea K, Yarnitsky D, Benny A, Tzuk-Shina T. A rare event of 5-fluorouracil-associated peripheral neuropathy: a report of two patients. Am J Clin Oncol. 1998;21(3):248–249. | |
Tofthagen C, Overcash J, Kip K. Falls in persons with chemotherapy-induced peripheral neuropathy. Support Care Cancer. 2012;20(3):583–589. | |
Stubblefield MD, McNeely ML, Alfano CM, Mayer DK. A prospective surveillance model for physical rehabilitation of women with breast cancer: chemotherapy-induced peripheral neuropathy. Cancer. 2012;118(Suppl 8):2250–2260. | |
Speck RM, DeMichele A, Farrar JT, et al. Scope of symptoms and self-management strategies for chemotherapy-induced peripheral neuropathy in breast cancer patients. Support Care Cancer. 2012;20(10):2433–2439. | |
Franconi G, Manni L, Schröder S, Marchetti P, Robinson N. A systematic review of experimental and clinical acupuncture in chemotherapy-induced peripheral neuropathy. Evid Based Complement Alternat Med. 2013;2013:516916. | |
Schröder S, Beckmann K, Franconi G, et al. Can medical herbs stimulate regeneration or neuroprotection and treat neuropathic pain in chemotherapy-induced peripheral neuropathy? Evid Based Complement Alternat Med. 2013;2013:423713. | |
Sissung TM, Mross K, Steinberg SM, et al. Association of ABCB1 genotypes with paclitaxel-mediated peripheral neuropathy and neutropenia. Eur J Cancer. 2006;42:2893–2896. | |
Sissung TM, Baum CE, Deeken J, et al. ABCB1 genetic variation influences the toxicity and clinical outcome of patients with androgen-independent prostate cancer treated with docetaxel. Clin Cancer Res. 2008;14:4543–4549. | |
Cavaletti G, Alberti P, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity in the era of pharmacogenomics. Lancet Oncol. 2011;12(12):1551–1561. | |
Cecchin E, D’Andrea M, Lonardi S, et al. A prospective validation pharmacogenomic study in the adjuvant setting of colorectal cancer patients treated with the 5-fluorouracil/leucovorin/oxaliplatin (FOLFOX4) regimen. Pharmacogenomics J. 2013;13(5):403–409. | |
Antonacopoulou AG, Argyriou AA, Scopa CD, et al. Integrin beta-3 L33P: a new insight into the pathogenesis of chronic oxaliplatin-induced peripheral neuropathy? Eur J Neurol. 2010;17(7):963–968. | |
Sittl R, Lampert A, Huth T, et al. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc Natl Acad Sci U S A. 2012;109(17):6704–6709. | |
Argyriou AA, Antonacopoulou AG, Scopa CD, et al. Liability of the voltage-gated sodium channel gene SCN2A R19K polymorphism to oxaliplatin-induced peripheral neuropathy. Oncology. 2009;77(3–4):254–256. | |
Favis R, Sun Y, van de Velde H, et al. Genetic variation associated with bortezomib-induced peripheral neuropathy. Pharmacogenet Genomics. 2011;21(3):121–129. | |
Corthals SL, Kuiper R, Johnson DC, et al. Genetic factors underlying the risk of bortezomib induced peripheral neuropathy in multiple myeloma patients. Haematologica. 2011;96(11):1728–1732. | |
Lauria G, Cornblath DR, Johansson O, et al; European Federation of Neurological Societies. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur J Neurol. 2005;12(10):747–758. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.