Back to Journals » International Journal of Nanomedicine » Volume 11
Chemically cross-linked silk fibroin hydrogel with enhanced elastic properties, biodegradability, and biocompatibility
Received 15 February 2016
Accepted for publication 14 April 2016
Published 23 June 2016 Volume 2016:11 Pages 2967—2978
DOI https://doi.org/10.2147/IJN.S106467
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Min Hee Kim, Won Ho Park
Department of Advanced Organic Materials and Textile Engineering System, Chungnam National University, Daejeon, Korea
Abstract: In this study, the synthesis of silk fibroin (SF) hydrogel via chemical cross-linking reactions of SF due to gamma-ray (γ-ray) irradiation was investigated, as were the resultant hydrogel’s properties. Two different hydrogels were investigated: physically cross-linked SF hydrogel and chemically cross-linked SF hydrogel irradiated at different doses of γ-rays. The effects of the irradiation dose and SF concentration on the hydrogelation of SF were examined. The chemically cross-linked SF hydrogel was compared with the physically cross-linked one with regard to secondary structure and gel strength. Furthermore, the swelling behavior, crystallinity, and biodegradation of the SF hydrogels were characterized. To assay cell proliferation, the cell viability of human mesenchymal stem cells on the lyophilized SF hydrogel scaffolds was evaluated, and no significant cytotoxicity against human mesenchymal stem cells was observed.
Keywords: silk fibroin, hydrogels, biodegradation rate, gamma irradiation, cross-linking
Introduction
Regenerated Bombyx mori (B. mori) silk fibroin (SF) has excellent biological and mechanical properties, including biocompatibility, programmable biodegradability, and remarkable strength and toughness.1 Due to its superior properties, SF has excellent potential for biomedical materials and has been investigated with regard to applications for tissue-engineered blood vessel, skin, bone, and cartilage. Fibroin is the structural protein of silk fibers, and sericin is the water-soluble glue that binds the fibroin fibers together.1 The SF molecule consists of heavy and light chain polypeptides of ~350 and ~25 kDa, respectively, connected by a disulfide link.2 Silk-based system can be prepared using a water-based solution under mild manufacturing conditions, such as room temperature with a neutral pH and without application of strong shear forces, because it is water soluble in its α-helix and random coil forms.3 Adaptable and tunable properties of SF are possible through structural variations under different processing conditions.4 One of the important physical forms for biomaterials is the formation of hydrogels, which has been extensively studied for a variety of polymers such as alginates, chitosan, and collagen.2 In order to improve the strength of SF, it has been cross-linked via a number of methods in recent years. Physically cross-linked SF hydrogels (SF P-gel) have been produced through the induction of beta sheet (β-sheet) structure in solution.5 The sol–gel transition is dependent on the concentration of SF, temperature, and pH. In the SF hydrogel, structural transitions from random coil to β-sheet are noted during the process of hydrogelation. The formation of thermodynamically stable β-sheet structures serves as physical cross-links, stabilizing the SF molecules. Sufficient energy to overcome the energy barrier for β-sheet crystallization, by heating, vortex shearing, ultrasonication, or electric current, accelerates the hydrogelation of SF. Due to the β-sheet formation, SF exhibits relatively slow degradation in vitro and in vivo.4,6–9 In chemical cross-linking, SF is cross-linked using chemical cross-linking agents such as genipin and glutaraldehyde. The advantage of using a natural cross-linker is reduced cytotoxicity when compared to a synthetic one. However, the use of a natural cross-linker is time-consuming and it reacts mainly with lysine and arginine in the protein structure.10 These amino acids make up a low ratio in the total SF composition with ~0.6 mole% for each. The enzymatic cross-linking of SF using tyrosinase was also reported.11 This reaction is also time-consuming. Ruthenium-mediated photo cross-linking of SF was reported by Whittaker et al.5 The mechanism is based on tyrosine and is rapid, but additive electron acceptors such as ammonium persulfate have some cytotoxicity. Therefore, it would be extremely important and useful to find a rapid and simple cross-linking method to develop chemically cross-linked SF hydrogels (SF C-gel).
The radiation technique seems to be an excellent pathway for the preparation of polymer hydrogels because polymer solutions easily undergo chemical cross-linking and graft copolymerization upon irradiation to yield a hydrogel. Simple procedural control, no additives such as initiators and cross-linkers, no waste, and relatively low operating costs make the irradiation technique a suitable choice for the synthesis of hydrogels.12 The gamma-ray (γ-ray) irradiation has been used to induce the cross-linking or chain scission for SF gels and fibers. In previous studies, Borisut et al prepared SF/polyvinyl alcohol (PVA) blend hydrogel for dermal scaffolds using γ-ray irradiation.13 The secondary structural change of blended hydrogel also occurred by PVA during gelation, and thus higher PVA content shortened the gelation time.13,14 Also, Amornthep et al attempted to reduce the biodegradation period of SF fibers through high γ-ray dose. Radiation directly decreased the tensile strength of SF fibers, but the relative content of the crystalline and amorphous phases in the SF fibers was not changed with the variation of radiation dose.15
In this study, the synthesis of SF hydrogel via a chemical cross-linking reaction using γ-ray irradiation was investigated and the resultant hydrogel was characterized. Two different hydrogels were investigated: SF P-gel induced by β-sheet structure and chemically cross-linked hydrogel prepared by γ-ray irradiation. The secondary structures and strengths of the gels were compared. Furthermore, the swelling behavior, biodegradation, and cell viability of human mesenchymal stem cells (hMSCs) on the SF hydrogel were evaluated.
Experimental
Materials
Degummed B. mori silkworm silk fiber was supplied by BJ Silk Co. Ltd (Jinju-si, Korea). Calcium chloride dihydrate (71.0%–77.5%) and ethanol (94.5%) were purchased from Samchun Chemical (Pyeongtaek, Korea). Dialysis cellulose membranes were purchased from Sigma-Aldrich Co. (cutoff 12,000–13,000 Da) and Membrane Filtration Products Inc. (Cellu-Sep 5030-43, cutoff 3,500 Da) and presoaked in distilled water for 10 minutes before use. Polyethylene glycol was purchased from KPX Green Chemical Co. Ltd (Seoul, Korea). Phosphate buffer solution (1.0 M, pH 7.4), protease from Streptomyces griseus, rhodamine B (Rho), and fluorescein (Flu) were purchased from Sigma-Aldrich Co. All the chemical agents were used without further purification.
Preparation of aqueous SF solutions
Degummed B. mori silk fiber was dissolved in a ternary solvent system composed of calcium chloride, ethanol, and water (1:2:8 molar ratio) at 85°C for 4 hours. This solution was dialyzed against distilled water using dialysis with cellulose tubular membranes (250-7; Sigma-Aldrich Co.) for 72 hours to remove the salt. The solution was optically clear after dialysis and was centrifuged at 3,000 rpm for 10 minutes at room temperature to remove silk aggregates formed during the process. The final concentration of the resultant aqueous SF solution was ~2.3 wt%, determined by weighing the remaining solid after drying. A higher concentration SF solution was prepared by dialysis against 25 wt% polyethylene glycol (20,000 g/mol) at room temperature. The SF concentration after dialysis was ~7.9 wt%. The regenerated SF solution was stored at 4oC for further use.
Preparation of SF P-gel formation
SF P-gel was prepared as described previously.16 Briefly, a prepared SF solution was kept at 37°C. Structural transitions from random coils to β-sheets are involved during the process of gelation. The formation of the thermodynamically stable β-sheet structure serves as physical cross-links, stabilizing SF molecules. The gelation time was determined when the sample did not flow from an inverted vial within 30 seconds. The gelation was typically completed after 3–5 days.
Preparation of SF C-gel formation
SF C-gel was prepared as described in Figure 1. Freshly regenerated SF solutions of various concentrations (2.3%–7.9%) were poured into a petri dish and irradiated with γ-rays from a Co-60 source. The radiation dose varied from 15–60 kGy and the dose rate was 15 kGy/h, at room temperature. The irradiated samples were cut into small pieces to compare with SF P-gel with regard to various properties.
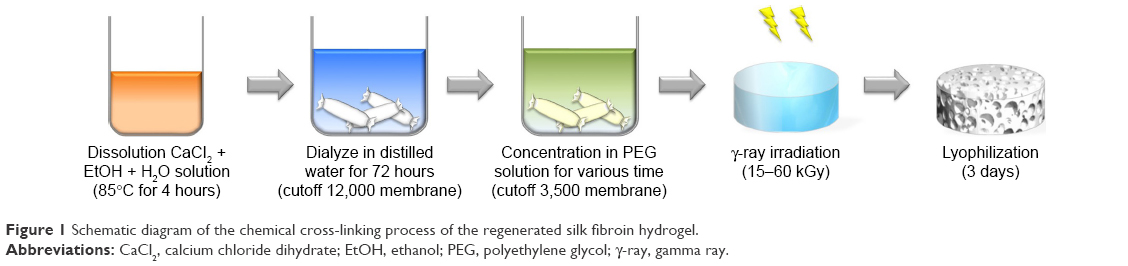 | Figure 1 Schematic diagram of the chemical cross-linking process of the regenerated silk fibroin hydrogel. |
Characterization
The hydrolysate SF solution (hydrolyzed in 6 M HCl at 120°C for 24 hours) was subjected to amino acid analysis (L-8900; Hitachi Ltd.) to examine the change in the amino acid composition of irradiated SF hydrogels.17 The physical and chemical adsorption between SF hydrogels and mercury, the specific surface area, pore volume, pore size distribution, and porosity were measured using surface area and pore characterization system (ASAP 2020; Micromeritics).
The samples were freeze-dried after irradiation (WA) and then soaked in formic acid for 30 minutes in order to remove the soluble fraction. The gels were then dried in air (WB). The gel fraction was calculated by the following equation:
|
|
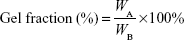
Infrared (IR) spectroscopy in attenuated total internal reflection mode (ATR) was performed using attenuated total reflectance infrared spectroscopy (ATR-IR; ALPHA-P; Bruker Optik GmbH). The secondary structure content of SF was calculated from the areas of the individual assigned bands and their fraction of the total area in the amide I region. 13C solid-state cross-polarization, magic-angle spinning (CP/MAS) nuclear magnetic resonance (NMR) spectra of the SF hydrogels were obtained on a DSX 400 NMR spectrometer (Bruker Optik GmbH), using a CP pulse sequence and MAS at 6.5 kHz. The morphology of the hydrogels was investigated with a scanning electron microscope (SEM; S-235; Hitachi Ltd.) in low vacuum mode. Compressive strength was measured at room temperature on cube-shaped SF hydrogels (10×10×10 mm) using an Instron 5848 mechanical tester machine with a crosshead speed of 5 mm/min. Recovery ability of the hydrogels was measured following multiple compression cycles. Swelling studies were conducted on the SF hydrogels as a function of time. Dried hydrogel samples (Wd) were immersed in water and PBS at pH 7.4 and 37°C for different time intervals up to 120 hours. After each time interval, excess water was removed and the samples weighed (Ws). The degree of swelling was calculated according to the following equation:18
|
|
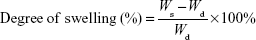
Enzymatic biodegradation behavior
The two types of freeze-dried SF hydrogels were cut into small pieces (Wd). SF samples were immersed in the appropriate 1.5 mg/mL protease type XIV (isolated from S. griseus) enzyme solution (3.5 U/mL) with PBS (pH 7.4) at 37°C. Solution was changed daily. After 1, 3, 5, 7, 14, 21, 28, 35, and 42 days, samples were rinsed in H2O and lyophilized and weighed. For a control, a porous SF sample was immersed in PBS (pH 7.4) without the enzyme.19
|
|
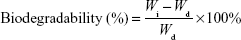
Measurements were performed on n=3 samples for each sample type, and results were expressed as the mean ± standard deviation.
Drug release behavior
For the investigation of drug release properties of SF hydrogels, Rho and Flu were used as model drugs (MDs). SF samples were loaded with 2 mg/mL Rho and Flu by immersion in an aqueous solution for 3 hours. The release of the MDs from the hydrogels was measured by placing the gels in a vessel containing 10 mL of different buffer solutions at a constant shaking rate at 37°C. At each time interval, aliquots of 3 mL were drawn from the medium following the release of the MDs. MD release was determined by a spectrophotometric method using a UV-2450 (Shimadzu) UV–vis spectrophotometer at λmax =544 nm of Rho and λmax =512 nm of Flu, respectively.19
Cell culture condition and cell viability assessment (MTS assay)
To obtain hMSCs, bone marrow was aspirated from the alveolar bone of patients during oral surgery. This study, the procedure, and all patient participation was approved by the Institute Research Review Board in the Department of Periodontology at Wonkwang University Dental Hospital, and all patients provided written informed consent. The properties of the hMSCs were discussed in our previous study.20 The cells were cultured in α-modified Eagle’s medium (Gibco™ Cell Culture; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum and 1% antibiotics (penicillin G 10,000 units/mL, amphotericin B 25 μg/mL; Gibco-BRL), incubated at 37°C, in a 5% CO2 environment. In this study, cells passaged for 3–6 times were used, and the culture medium was changed every 2 days. Various pre-freezed SF C-gels and SF P-gels were cut into small rounds of 5 mm in diameter, and these specimens were transferred to 96-well, 24-well, and 6-well tissue culture dishes. All specimens were sterilized with ethylene oxide gas. In this study, the 5 mm rounds were used for the 3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay, and the 12 mm rounds were used for live/dead staining.19 Proliferation of the cells attached to and subsequently grown on the scaffold was assessed using CellTiter96® Aqueous One Solution (Promega Corporation, Madison, WI, USA). Briefly, hMSCs were seeded on sterilized specimens with a diameter of 5 mm in a 96-well culture dish. For measurement at the indicated intervals, 25 μL of MTS reagent was added to each well and incubated for 3 days. Absorbance was measured at 490 nm on an ELISA reader (SpectraMAX M3; Molecular Devices LLC, Sunnyvale, CA, USA).21
Results and discussion
Gelation behavior
The radiation technique seems to be an excellent pathway for the preparation of hydrogels because polymer solutions undergo chemical cross-linking on irradiation to yield a hydrogel. By contrast, most chemical cross-linking reactions of SF can induce a modification of proteins, resulting in structural destabilization and inflammation by exposure to toxic organic solvents and cross-linking agents.22 In this study, the SF C-gel was prepared by γ-ray irradiation, which induced intermolecular cross-linking reactions. In order to investigate the effect of irradiation on the gelation of the SF solution, the solutions were γ-ray irradiated at various doses of 15, 30, 45, 60, 75, and 90 kGy. Regardless of the SF concentration and irradiation doses, SF hydrogel was formed immediately after irradiation, as shown in Figure 2A. However, as shown in Figure 2B, cracks occurred in the hydrogel at doses higher than 60 kGy. A suitable absorption dose for an SF solution was 60 kGy. Therefore, the experiment was performed using the dose of 60 kGy. From the result of the amino acid analysis, no significant changes in amino acid composition were observed after irradiation, as described in Figure 2C, and the value of the gel fraction was almost the same irrespective of the SF concentration, as shown in Figure 2D. The gel fraction of the γ-ray irradiated hydrogels was determined using Equation (1).
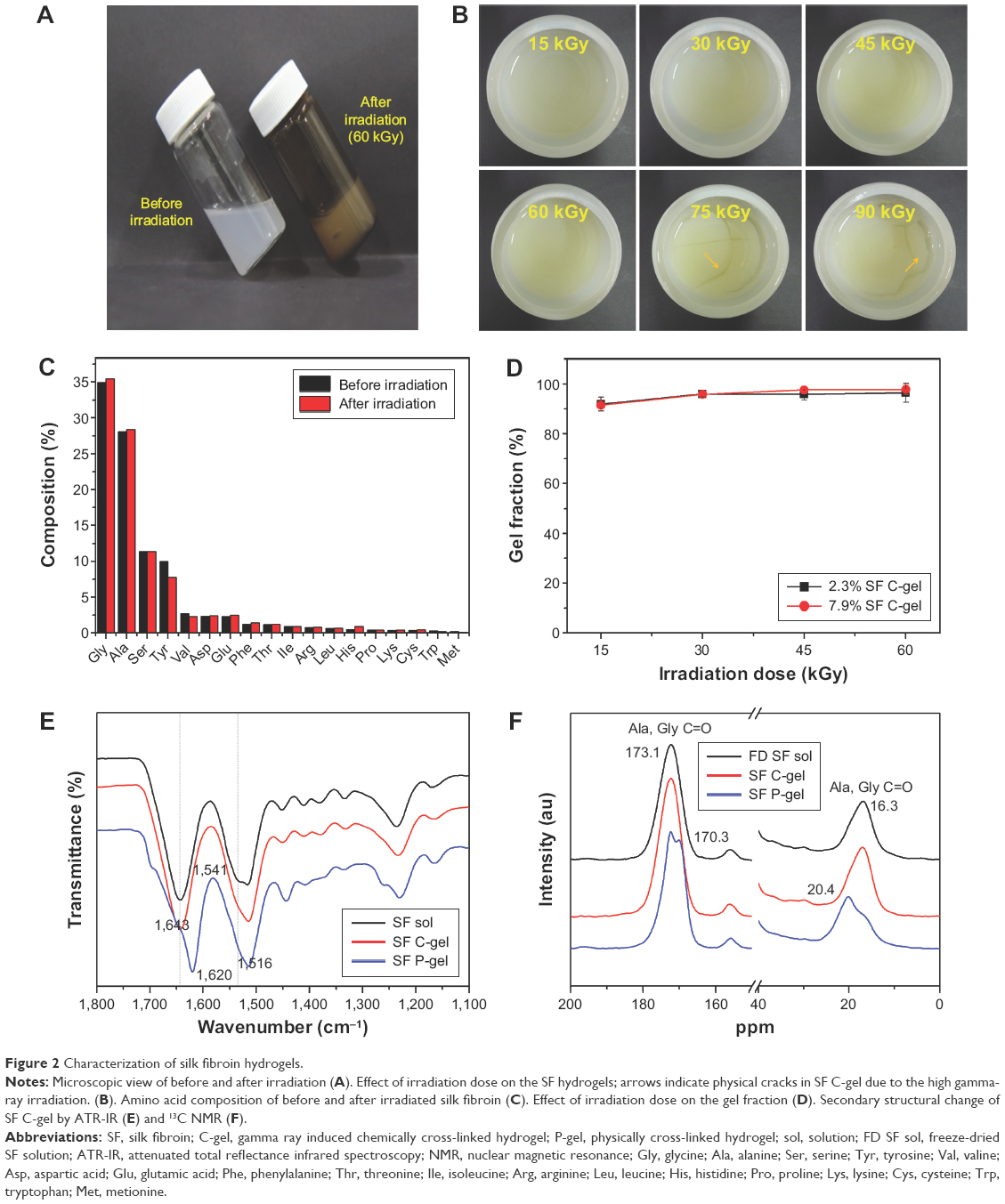 | Figure 2 Characterization of silk fibroin hydrogels. |
To better understand structural differences between SF C-gels and SF P-gels, ATR-IR and 13C CP/MAS NMR analysis were carried out to assess secondary structural changes of the SF during the sol–gel transition. The infrared (IR) region of 1,700–1,500 cm−1 corresponds to peptide backbone absorption for amide I (1,700–1,600 cm−1) and amide II (1,600–1,500 cm−1) bands. The absorbance spectrum in this region is used for determining the secondary structural change of various proteins, including SF. Figure 2E shows a typical amide I and amide II absorbance spectrum for SF solution, SF C-gel, and SF P-gel. The peaks at 1,643 and 1,541 cm−1 correspond to random coil structure (dashed line), and the peaks at 1,620 and 1,516 cm−1 correspond to β-sheet structure.4 Table S1 shows vibrational band assignment of IR peak in the amide I region for B. mori SF. Regenerated SF solution showed typical random coil conformations and α-helix structures (1,541, 1,643 cm−1), and amide I and II bands shifted from 1,541 to 1,643 cm−1 after physical cross-link.23,24 Interestingly, no significant peak shift in γ-ray-induced chemically cross-linked hydrogel (C-gel) was observed, indicating that the C-gel still had a random coil conformation. To further analyze the structure of SF hydrogel, 13C CP/MAS NMR spectra of SF solution, SF C-gel, and SF P-gel were taken and are shown in Figure 2F. In the 13C NMR spectrum of pure SF solution, peaks at 173.1 and 16.3 ppm were attributed to carbonyl carbons of SF, Cα and Cβ of Ala, and Cα of Gly.25 The NMR spectrum of SF P-gel showed characteristic chemical shifts. However, no significant peak shift was observed in the C-gel samples. From the results, no change in the secondary structure of SF C-gel formed by γ-ray irradiation was observed, indicating that the gelation of SF occurred via chemical cross-linking reactions without secondary structural change.
Morphology
The porous structure of freeze-dried SF hydrogel was characterized by SEM images. Three-dimensional SF scaffolds were fabricated using 2.3%–7.9% SF solution with pre-freezing temperatures of −20°C, −50°C, −80°C, and −196°C in molds, followed by freeze-drying. The porous structure of hydrogel was closely associated with SF concentration, irradiation dose, and pre-freezing temperature. Both SF hydrogels (C-gel and P-gel) had uniform pore sizes and an interconnected porous network structure, as shown in Figure 3A, but larger pore sizes were observed with the SF C-gel. When SF C-gels were fabricated using a 2.3% SF solution, larger pores were observed with an irradiation dosage of 15 kGy but the pore size was reduced with irradiation dosages of 30, 45, and 60 kGy due to the increased chemical cross-linking reactions (Figure 3B). At constant 60 kGy irradiation dosage, the pore sizes of the SF C-gel were decreased with increasing SF concentration (from 81.8 μm at 2.3% SF concentration to 12.3 μm at 7.9% SF concentration, as observed in Figure 3C and Table 1). The glass transition temperature of SF was reported to occur between −20°C and −30°C, which immensely affects the pore size within the SF matrix.26 The higher the pre-freezing temperature above the glass transition, the longer it takes for the ice to form and grow in size.27 With a 2.3% SF C-gel, a larger pore size was observed with a pre-freezing temperature of −20°C, and the pore size decreased as the pre-freezing temperature decreased to −80°C and −196°C, as shown in Figure 3D, due to lower sub-zero temperatures and faster freezing rate. The ice crystals formed at −196°C gave rise to smaller and highly interconnected pores upon freeze-drying due to rapid pre-freezing.
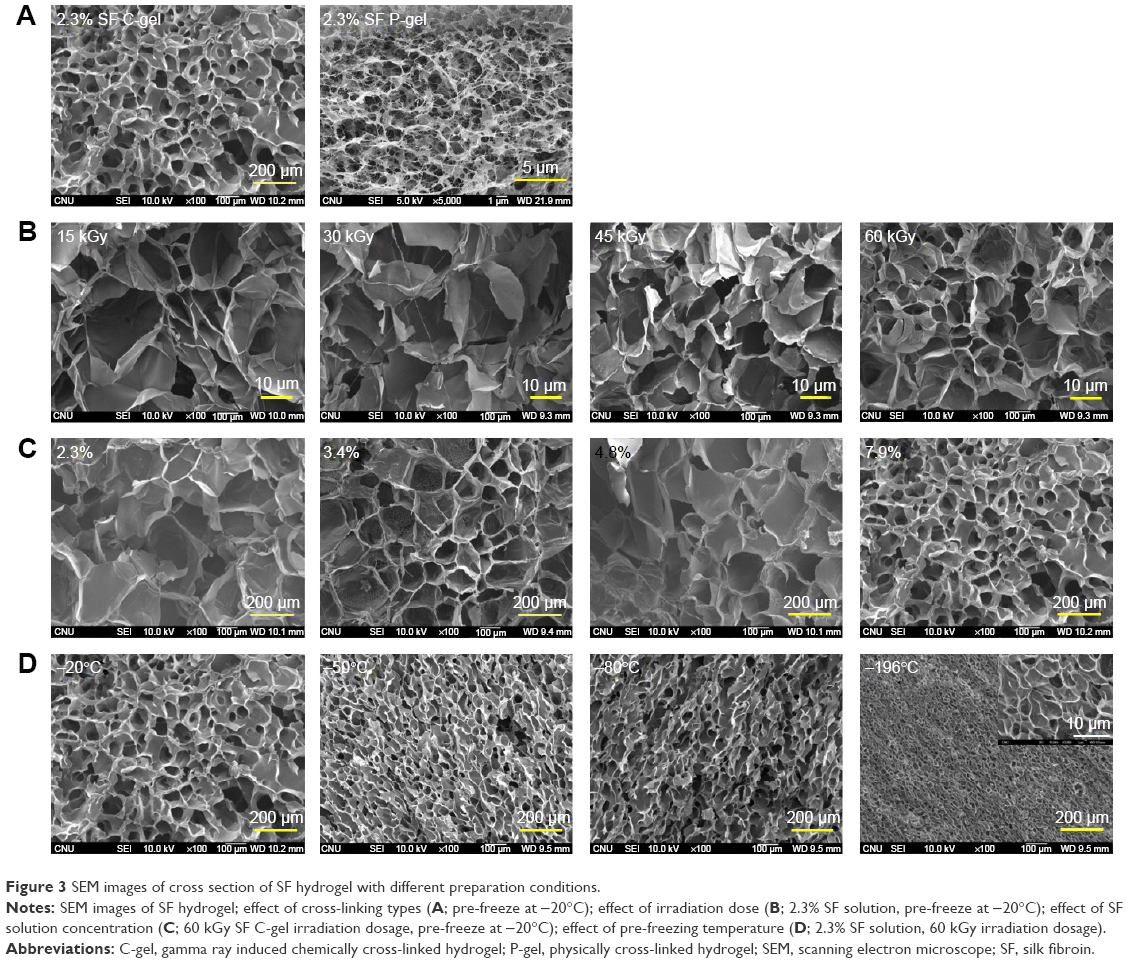 | Figure 3 SEM images of cross section of SF hydrogel with different preparation conditions. |
 | Table 1 Results of the porosity analysis of SF C-gel and SF P-gel depend on pore size, SF concentration, and cross-linking types |
Table 1 shows surface area and pore dimension data. The SF C-gel fabricated using γ-ray irradiation gave higher porosity, compared to the SF P-gel. The SF C-gel fabricated at a pre-freezing temperature of −196°C gave the highest porosity, followed by pre-freezing temperatures of −80°C and −20°C. Fast freezing at −196°C imparted a short time interval for ice formation and growth, and ice crystals remained small in size, giving rise to small pores but high porosity. Furthermore, an SF C-gel with up to 90.90% porosity was formed on pre-freezing at −80°C using 2.3% concentration. Even at 7.9% SF C-gel, a minimum porosity of 87.06% was achieved at −80°C.
Mechanical properties
Compression studies were carried out to determine the compressive properties of the SF P-gels and SF C-gels. The second cycle was recorded, and the stress–strain curve was calculated. On the first cycle, the maximum compressive strength of the SF P-gel was higher than that of the SF C-gel due to the β-sheet formation. Also, the SF P-gel did not show a compression recovery, because it had a low elasticity and the water in the SF P-gel was released. The compressive behavior of SF P-gel is shown in Figure 4A and B. Compared with the SF P-gel, the SF C-gel showed recovery from compressive strain, and had lower maximum compressive strength and higher elasticity due to low crystallinity and intermolecular cross-linking reaction. The compressive behavior of the SF C-gel is shown in Figure 4C and D. Also, the stiffness of the SF C-gel increased with the SF concentration (data not shown).
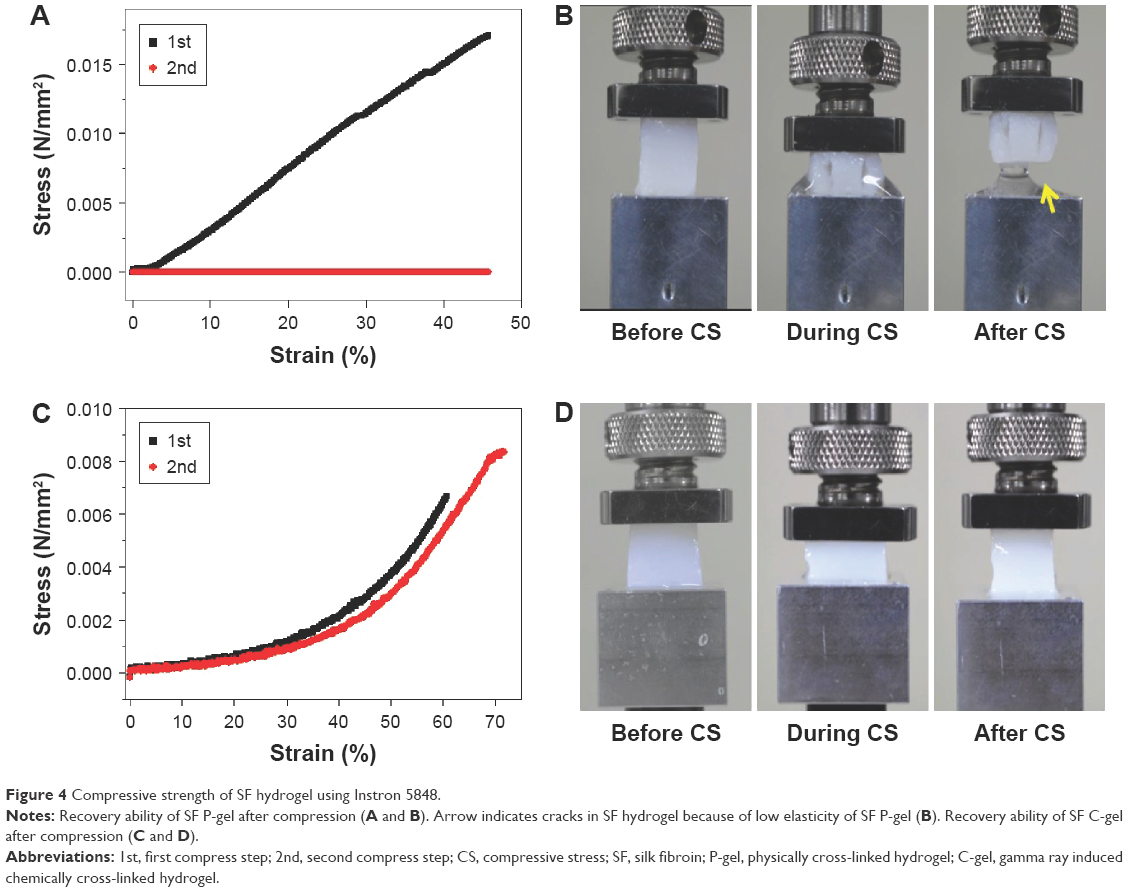 | Figure 4 Compressive strength of SF hydrogel using Instron 5848. |
Swelling behavior
Swelling properties are very important in hydrogels. To determine the swelling properties of SF hydrogels, gels were immersed in two kinds of media for an extended period of time until equilibrium was achieved. The swelling ratios of SF hydrogels at 37°C were determined by using Equation (2) and are plotted as a function of concentration, as shown in Figure 5. In a deionized (DI) water medium, comparing the SF C-gel with the SF P-gel, it was found that the latter had a lower swelling ratio, as shown in Figure 5A, and had lost its stability rapidly leading to below 0% swelling ratio due to the physical cross-linking. With the SF C-gel, the DI water uptake increased with time until it attained equilibrium. However, crystallinity increased because of tension generated by swelling, and the swelling ratio decreased (Figure S1). The swelling behavior of the SF P-gel in PBS solution was similar to that in DI water. But the swelling ratio of the SF C-gel decreased in PBS solution, as shown in Figure 5B. This de-swelling in the PBS solution was likely due to electrostatic shielding and osmotic pressure, resulting in different SF concentrations.28 Therefore, these swelling properties of the SF C-gel sensitive to aqueous medium were more relevant to its use in drug delivery system.
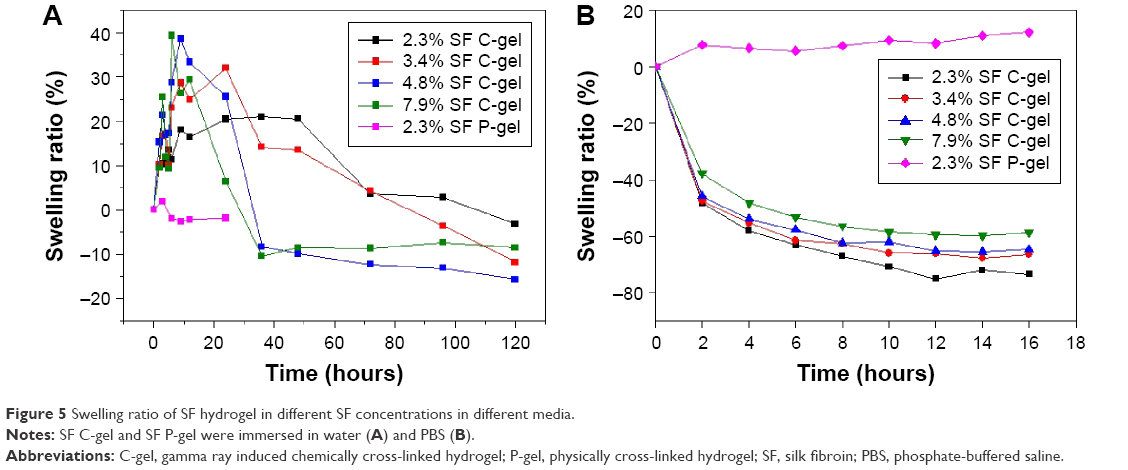 | Figure 5 Swelling ratio of SF hydrogel in different SF concentrations in different media. |
Biodegradation properties
SF is promising for use as a porous tissue matrix, a carrier for controlled-release medicine, and other uses. But the biodegradation behavior of this material needs to be studied. To assess its biodegradation, the freeze-dried SF gels were immersed in protease XIV enzymatic solution. The biodegradation rates of the SF C-gel and SF P-gel were significantly different. The gel weight immersed in the enzymatic solution decreased as the degradation time increased. In particular, due to low crystallinity, the degradation rate of the SF C-gel was faster than that of the SF P-gel, and furthermore the SF C-gel was completely degraded in 2 weeks when exposed to 1.0 U/mL protease XIV solution at 37°C, as shown in Figure 6A, whereas the SF P-gel was degraded slowly. The significantly different rates of enzymatic degradation of the SF C-gel and P-gel were, in part, due to differences in crystallinity and porosity: the lower crystallinity and higher porosity of the SF C-gel would accelerate the degradation by protease due to increased penetration of the enzyme. When the pore size was controlled by varying pre-freezing temperature, the degradation rate of the SF C-gel was almost the same, as observed in Figure 6B. However, the degradation of the SF C-gel was dependent on the SF concentration. When the concentration was increased from 2.3% to 7.9% (w/v), the degradation time to reach 50% mass loss in the experiment was increased from 4 to 15 days, as shown in Figure 6C. The control sample SF C-gels incubated in PBS solution without protease showed a slow degradation throughout the incubation period.29 The fast degradation of SF C-gel due to proteolytic processes may be suitable for various applications, such as in tissue engineering or rapid drug delivery systems. The structural changes in the degradation process were further investigated using SEM. As shown in Figure 6D, for the degraded 7.9% SF C-gel sample, the pore structures were maintained until degradation for 28 days, and after a certain period of time, cracks in the gel occurred and the pore shape collapsed.
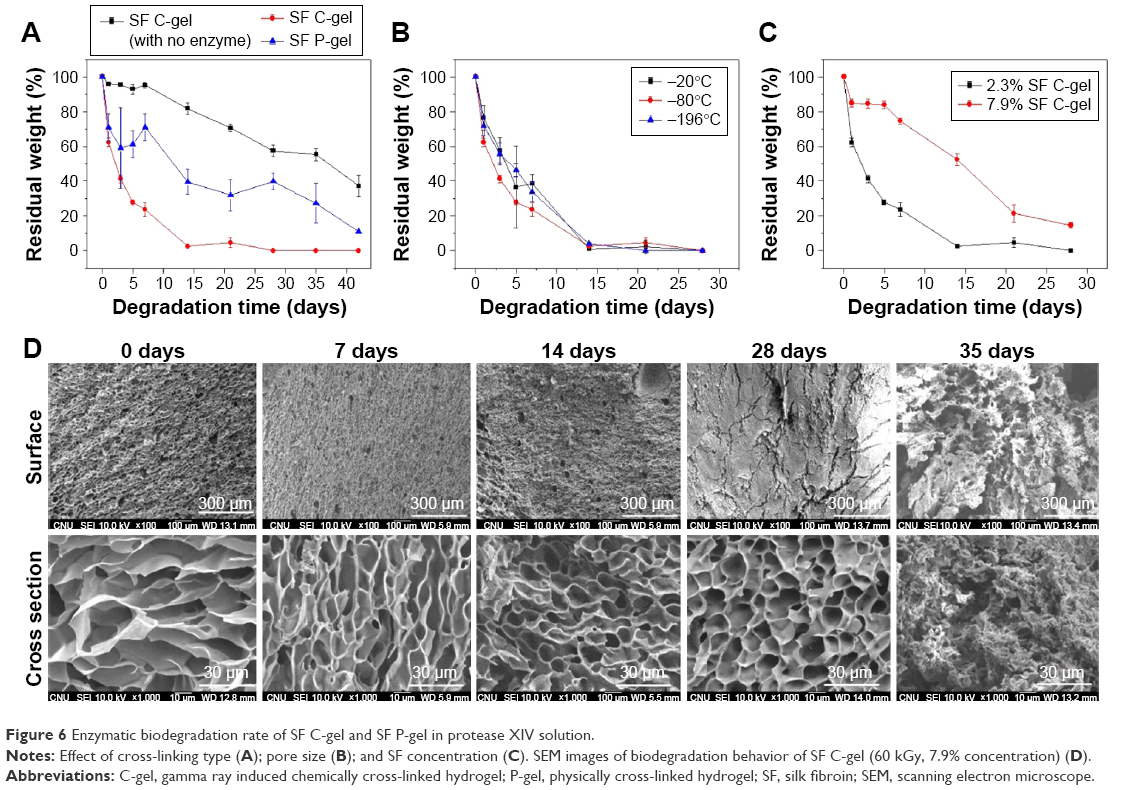 | Figure 6 Enzymatic biodegradation rate of SF C-gel and SF P-gel in protease XIV solution. |
Drug release properties
In order to investigate the applicability of SF hydrogels as a drug delivery system, two MDs (Rho an Flu) were loaded on SF hydrogels. Upon incubation of hydrogels in the drug solution, drug molecules adhered to the surface followed by diffusion into the SF matrix. Table S2 shows the loading efficiency of MDs in the SF hydrogel. The loading was studied with respect to the molar ratio of MD to SF. The loading efficiency was slightly different depending on the type of MD.30 The in vitro release behavior of MD from the SF hydrogels is shown in Figure 7. Positively charged molecules (Rho) were released in a more prolonged or sustained fashion, and the release rate of the drugs was associated with the SF concentration. The influence of the secondary structure of the SF hydrogels on their release behavior was studied. The loading efficiency of negatively charged molecules (Flu) was higher for the SF C-gel, and showed an increased release rate over the whole period of time, compared to the SF P-gel.
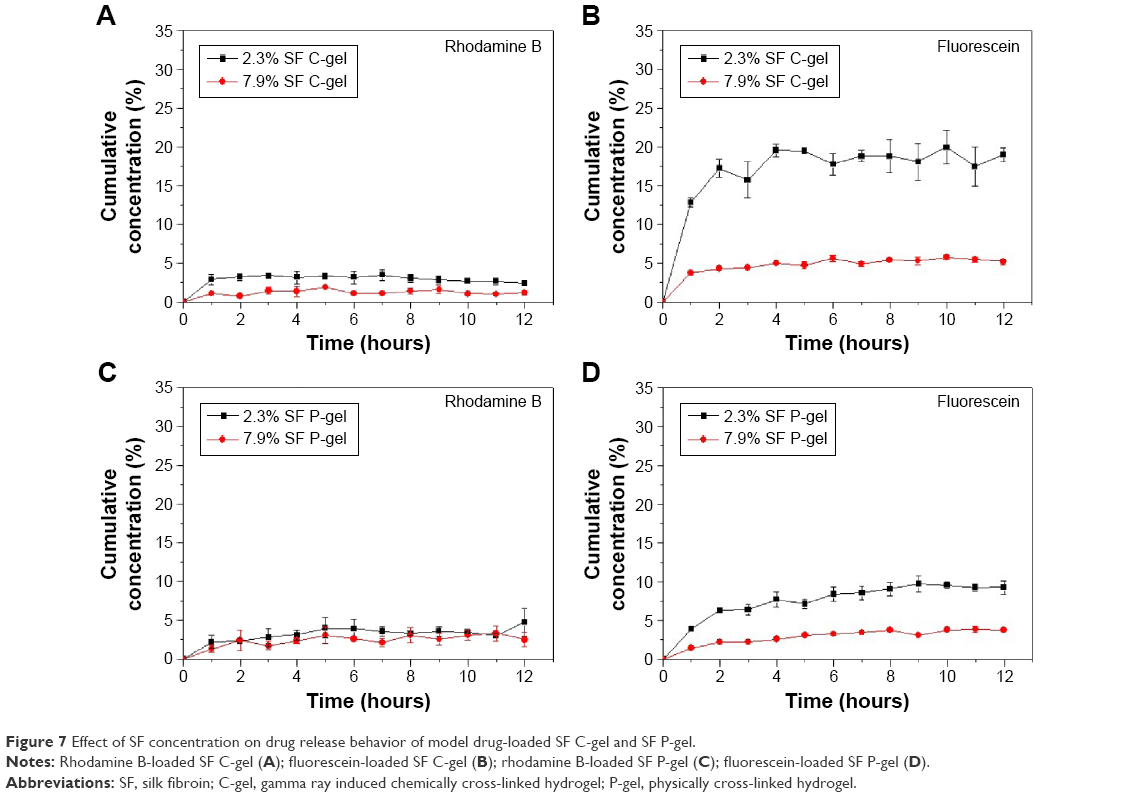 | Figure 7 Effect of SF concentration on drug release behavior of model drug-loaded SF C-gel and SF P-gel. |
Cell viability
The cytotoxicity of the SF hydrogels was determined using the standard MTS assay with hMSCs to evaluate the potential of these materials as practical biomaterials. At a high concentration (7.9%), the SF C-gel showed no cytotoxicity with respect to hMSCs, whereas at a lower concentration, lower cell viability was observed, as shown in Figure 8A. Furthermore, the cell viability of the SF C-gel was higher than that of the SF P-gel, and there was no significant effect on the pore size. Figure 8B shows SEM images of the hMSCs on the gel after 14 days of culturing. In all experimental groups, hMSCs adhered well and formed cell clusters. Especially, the SF C-gel at a higher concentration showed higher elongation and spread on the scaffolds.21 Cells were stained with a Live-Dead™ kit after 7 days of culture and then observed with confocal microscopy. Green color represents live cells, while red color represents dead cells and a stained SF matrix.31 After 3 days in culture, most cells presented green fluorescence indicating no significant cell death inside all types of SF gels under culture, as shown in Figure 8C. As the present irradiation-induced SF C-gel samples demonstrated no significant cytotoxicity against hMSCs, they have noteworthy potential as practical biomaterials.
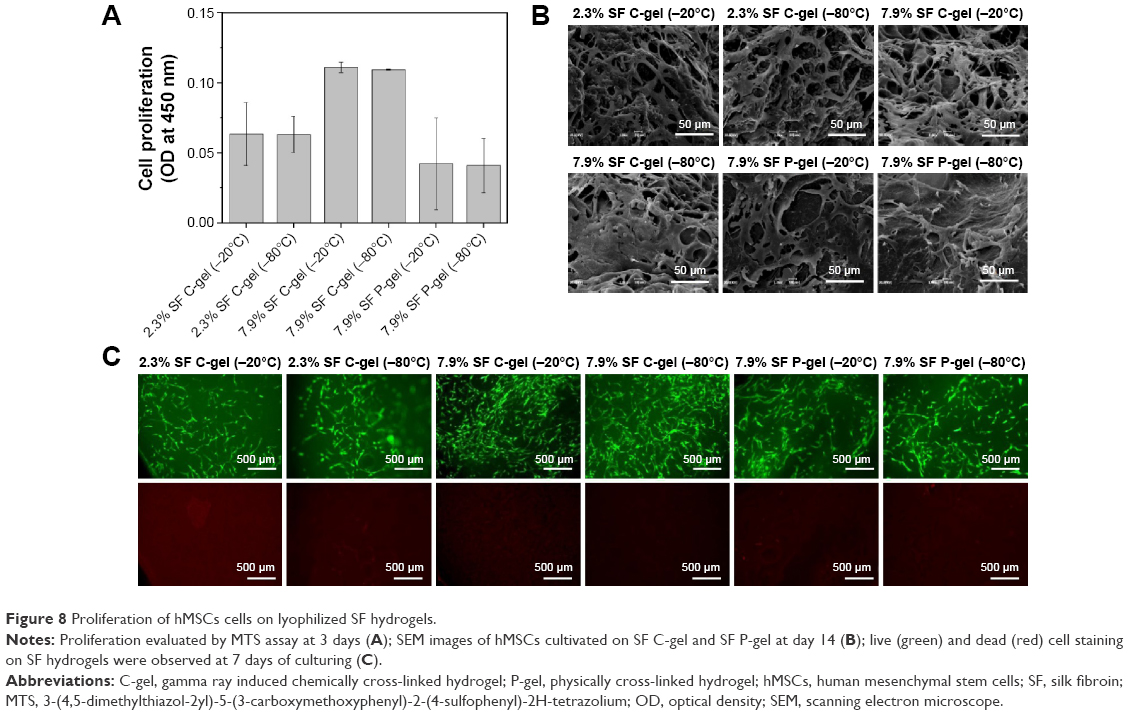 | Figure 8 Proliferation of hMSCs cells on lyophilized SF hydrogels. |
Conclusion
In this study, SF C-gel was prepared by γ-ray irradiation, which induced intermolecular cross-linking reactions. The change in the secondary structure of SF C-gel was investigated by ATR-IR. From the results, no change in the secondary structure of SF C-gel was observed, indicating that the gelation of SF occurred via chemical cross-linking reactions. The porous structure of lyophilized SF C-gel was characterized by SEM images. The pore size of SF C-gel decreased with increasing SF concentration, γ-ray irradiation dose, and decreasing pre-freezing temperature. Compared with SF P-gel, SF C-gel had higher elasticity due to intermolecular cross-linking. The biodegradation rates between SF C-gel and SF P-gel were significantly different. The degradation rate of SF hydrogels was dependent on their crystallinity and porosity. The drug loading efficiency of C-gel and P-gel was significantly different. The drug loading efficiency of SF hydrogels was dependent on the crystallinity and affinity of MD for the gel. The MD release from SF hydrogel increased with decreasing SF concentration and crystallinity. hMSCs cultured in SF scaffolds could attach, spread, and proliferate well in these hydrogels, confirming the applicability for cell culture. These findings demonstrate that SF C-gel without additive or cross-linker are biocompatible, and can be used for various tissue engineering and regenerative medicine applications.
Acknowledgment
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (2015M2A2A6A03044942).
Disclosure
The authors report no conflicts of interest in this work.
References
Kim UJ, Park J, Kim HJ, Wada M, Kaplan DL. Three-dimensional aqueous-derived biomaterial scaffolds from silk fibroin. Biomaterials. 2005;26(15):2775–2785. | ||
Kim UJ, Park J, Li C, Jin HJ, Valluzzi R, Kaplan DL. Structure and properties of silk hydrogels. Biomacromolecules. 2004;5(3): 786–792. | ||
Kundu B, Rajkhowa R, Kundu SC, Wang X. Silk fibroin biomaterials for tissue regenerations. Adv Drug Deliv Rev. 2013;65(4):457–470. | ||
Lin Y, Xia X, Shang K, et al. Tuning chemical and physical cross-links in silk electrogels for morphological analysis and mechanical reinforcement. Biomacromolecules. 2013;14(8):2629–2635. | ||
Whittaker JL, Choudhury NR, Dutta NK, Zannettino A. Facile and rapid ruthenium mediated photo-crosslinking of Bombyx mori silk fibroin. J Mater Chem B. 2014;2(37):6259–6270. | ||
Matsumoto A, Chen J, Collette AL, et al. Mechanisms of silk fibroin sol-gel transitions. J Phys Chem B. 2006;110(43):21630–21638. | ||
Yucel T, Cebe P, Kaplan DL. Vortex-induced injectable silk fibroin hydrogels. Biophys J. 2009;97(7):2044–2050. | ||
Xiao W, Liu W, Sun J, Dan X, Wei D, Fan H. Ultrasonication and genipin cross-linking to prepare novel silk fibroin–gelatin composite hydrogel. J Bioact Compat Polym. 2012:0883911512448692. | ||
Gil ES, Spontak RJ, Hudson SM. Effect of β-sheet crystals on the thermal and rheological behavior of protein-based hydrogels derived from gelatin and silk fibroin. Macromol Biosci. 2005;5(8):702–709. | ||
Silva SS, Maniglio D, Motta A, Mano JF, Reis RL, Migliaresi C. Genipin-modified silk-fibroin nanometric nets. Macromol Biosci. 2008;8(8):766–774. | ||
Kang GD, Lee KH, Ki CS, Park YH. Crosslinking reaction of phenolic side chains in silk fibroin by tyrosinase. Fiber Polym. 2004;5(3):234–238. | ||
Pourjavadi A, Soleyman R, Bardajee GR, Seidi F. γ-Irradiation synthesis of a smart hydrogel: optimization using Taguchi method and investigation of its swelling behavior. Scientia Iranica C. 2010;17(1):15–23. | ||
Borisut SH, Bovornlak O, Vichai S, Vivat V, Prasert S. De novo method of developing silk fibroin hydrogel aim for using as a dermal scaffold by gamma irradiation. Inter J Appl Biomed Eng. 2011;4:14–18. | ||
Peawpun I, Bovornlak O. A study of irradiated silk fibroin-poly vinyl alcohol hydrogel for artificial skin substitutes. J Metals Mater Minerals. 2010;20(3):119–122. | ||
Amornthep K, Prateep M, Boonya S, Lertyot T, Rachanee U, Bovornlak O. Effect of gamma radiation on biodegradation of Bombyx mori silk fibroin, Inter Biodeter Biodegrad. 2008;62:487–490. | ||
Park CH, Jeong L, Cho D, Kwon OH, Park WH. Effect of methylcellulose on the formation and drug release behavior of silk fibroin hydrogel. Carbohyd Polym. 2013;98(1):1179–1185. | ||
Min-Hui W, Jing-Xia Y, Yu-Qing Z. Ultrafiltration recovery of sericin from the alkaline waste of silk floss processing and controlled enzymatic hydrolysis. J Clean Prod. 2014;76:154–160. | ||
El-Din HMN, Alla SGA, El-Naggar AWM. Swelling and drug release properties of acrylamide/carboxymethyl cellulose networks formed by gamma irradiation. Radiat Phys Chem. 2010;79(6):725–730. | ||
Li M, Ogiso M, Minoura N. Enzymatic degradation behavior of porous silk fibroin sheets. Biomaterials. 2003;24(2):357–365. | ||
Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25(11):2739–2749. | ||
Kim BS, Park KE, Kim MH, You HK, Lee J, Park WH. Effect of nanofiber content on bone regeneration of silk fibroin/poly (ε-caprolactone) nano/microfibrous composite scaffolds. Int J Nanomed. 2015;10:485–502. | ||
Couet F, Rajan N, Mantovani D. Macromolecular biomaterials for scaffold-based vascular tissue engineering. Macromol Biosci. 2007;7(5):701–718. | ||
Hui L, Gopakumar S, Armando GM. Lignin valorization by forming thermally stimulated shape memory copolymeric elastomers-partially crystalline hyperbranched polymer as crosslinks. J Appl Polym Sci. 2014;131:41103–41112. | ||
Hui L, Gopakumar S, Armando GM. Highly biobased thermally-stimulated shape memory copolymeric elastomers derived from lignin and glycerol-adipic acid based hyperbranched prepolymer. Ind Crops Prod. 2015;67:143–154. | ||
Yoshimizu H. The structure of Bombyx mori silk fibroin membrane swollen by water studied with ESR, 13C-NMR, and FT-IR spectroscopies. J Appl Polym Sci. 1990;40(9–10):1745–1756. | ||
Mandal BB, Kundu SC. Non-bioengineered silk fibroin protein 3D scaffolds for potential biotechnological and tissue engineering applications. Macromol Biosci. 2008(9);8:807–818. | ||
Nazarov R, Jin HJ, Kaplan DL. Porous 3-D scaffolds from regenerated silk fibroin. Biomacromolecules. 2004;5(3):718–726. | ||
Partlow BP, Hanna CW, Rnjak-Kovacina J, et al. Highly tunable elastomeric silk biomaterials. Adv Funct Mater. 2014;24(29):4615–4624. | ||
Wang X, Kluge JA, Leisk GG, Kaplan DL. Sonication-induced gelation of silk fibroin for cell encapsulation. Biomaterials. 2008;29(8):1054–1064. | ||
Lammel AS, Hu X, Park SH, Kaplan DL, Scheibel TR. Controlling silk fibroin particle features for drug delivery. Biomaterials. 2010;31(16):4583–4591. | ||
Wang Y, Bella E, Lee CS, et al. The synergistic effects of 3-D porous silk fibroin matrix scaffold properties and hydrodynamic environment in cartilage tissue regeneration. Biomaterials. 2010;31(17):4672–4681. |
Supplementary materials
 | Table S1 Vibrational band assignments in the amide I region for B. mori SF |
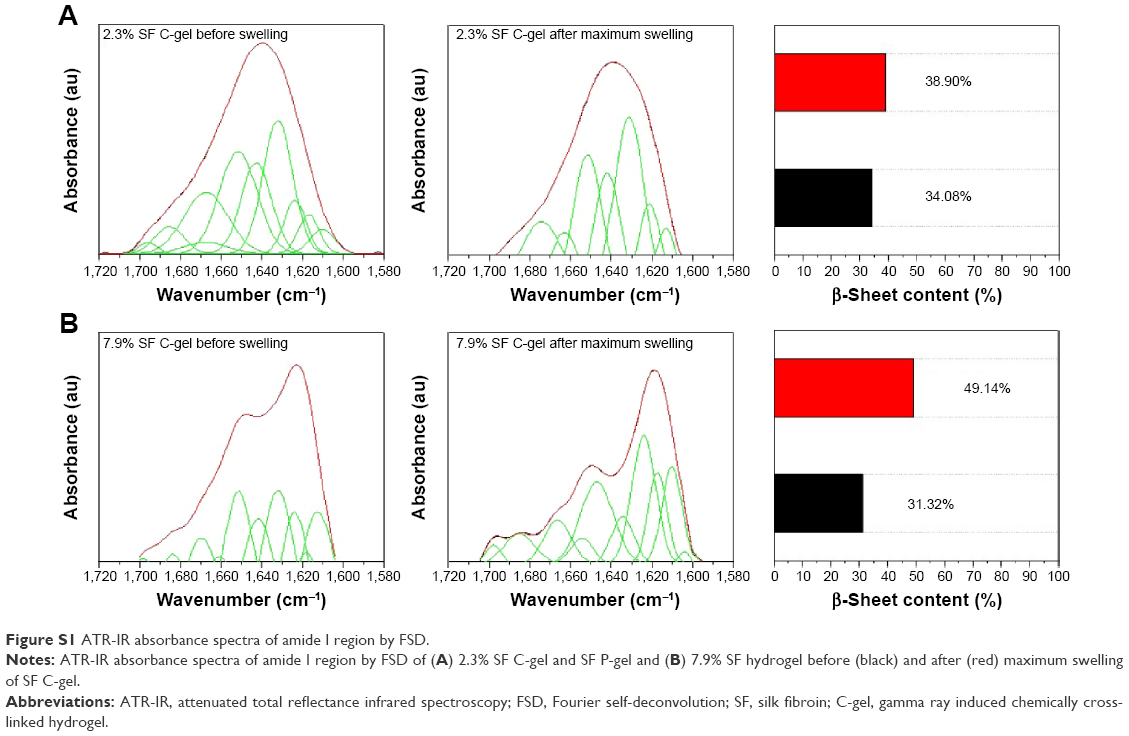 | Figure S1 ATR-IR absorbance spectra of amide I region by FSD. |
 | Table S2 Loading efficiency of model drugs in SF C-gel and SF P-gel |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.