")
Back to Journals » Medical Devices: Evidence and Research » Volume 16
Characterization of US Food and Drug Administration Class I Recalls from 2018 to 2022 for Moderate- and High-Risk Medical Devices: A Cross-Sectional Study
Authors Mooghali M , Ross JS , Kadakia KT, Dhruva SS
Received 16 March 2023
Accepted for publication 9 May 2023
Published 19 May 2023 Volume 2023:16 Pages 111—122
DOI https://doi.org/10.2147/MDER.S412802
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Maryam Mooghali,1,2 Joseph S Ross,1– 4 Kushal T Kadakia,5 Sanket S Dhruva6,7
1Department of Internal Medicine, Yale School of Medicine, New Haven, CT, USA; 2Yale Collaboration for Regulatory Rigor, Integrity, and Transparency (CRRIT), Yale School of Medicine, New Haven, CT, USA; 3Department of Health Policy and Management, Yale School of Public Health, New Haven, CT, USA; 4Center for Outcomes Research and Evaluation, Yale-New Haven Health System, New Haven, CT, USA; 5Harvard Medical School, Boston, MA, USA; 6Department of Medicine, UCSF School of Medicine, San Francisco, CA, USA; 7San Francisco Veterans Affairs Health Care System, San Francisco, CA, USA
Correspondence: Maryam Mooghali, Yale Center for Outcomes Research and Evaluation (CORE), 195 Church Street, New Haven, CT, 06510, USA, Tel +1 203 497-1239, Email [email protected]
Background: Medical device recalls are initiated in response to safety concerns. Class I (highest severity) recalls imply a reasonable likelihood of serious adverse events or death associated with device use. Recalled devices must be identified, assessed, and corrected or removed, upon which a recall can be terminated.
Objective: To characterize Class I medical device recalls and corresponding recalled devices.
Methods: This was a cross-sectional study of Class I recalls posted on the Food and Drug Administration’s annual log from January 1, 2018 to June 30, 2022 for moderate-risk and high-risk medical devices. Devices were categorized by therapeutic use, need for implantation, and life-sustaining designation; recalls were categorized by reason, status, and time elapsed.
Results: There were 189 unique Class I medical device recalls, including 151 (79.9%) for moderate-risk and 34 (18.0%) for high-risk devices. Sixty-five (34.4%) recalls were for cardiovascular devices, 36 (19.0%) for implanted devices, and 37 (19.6%) for life-sustaining devices. The median number of device units recalled in the US per recall notice was 4620 (interquartile range [IQR], 578– 42,591), with 11 (5.8%) recalls associated with more than 1 million device units. Overall, 125 (66.1%) devices had multiple recalls, with a median of 4 (IQR, 3– 11) recalls issued per recalled device. As of September 15, 2022, 50 (26.5%) recalls were terminated, with a median of 24 (IQR, 17.3– 30.8) months elapsed between recall initiation and termination. Recalls were terminated more commonly among devices recalled once compared to those recalled multiple times (36.2% vs 19.2%; p=0.02) and for recalls that recommended discontinuing further use of affected devices compared to those that recommended device assessment and/or education of affected population (31.8% vs 18.2%; p=0.04).
Conclusion: High-severity medical device recalls are common and affect millions of device units annually in the US. Recall termination takes a significant amount of time, putting patients at risk for serious safety concerns.
Keywords: medical devices recalls, device safety, Food and Drug Administration
Introduction
The Food and Drug Administration (FDA) is responsible for monitoring the safety and effectiveness of medical devices in the United States. FDA categorizes medical devices into three classes, based on the extent of regulatory evaluation needed to provide “reasonable assurance of safety and effectiveness”.1 While the highest-risk (Class III) devices must undergo premarket approval (PMA), requiring clinical data demonstrating device safety and effectiveness, most moderate-risk (Class II) devices are cleared through the premarket notification 510(k) pathway based on substantial equivalence to a legally marketed predicate device, rarely with clinical data.2 With limited premarket clinical testing before FDA authorization, even among the highest-risk devices,3,4 safety concerns are sometimes not identified until the post-market setting. Further, even when clinical data are available, rare adverse events or those that take time to develop may not be apparent at the time of FDA authorization.
FDA receives hundreds of thousands of safety-related reports annually from manufacturers, hospitals, clinicians, patients, and others concerning malfunctions, injuries, death, and other medical device-related adverse events.5 Additional safety concerns may be detected in post-market studies or investigations initiated by manufacturers, FDA, or independent researchers.6 To protect public health, these may lead to recalls, which are actions taken by the manufacturers or, rarely, by the FDA, to address safety issues with marketed products violating FDA regulations.7 After a recall is initiated, affected medical devices must be identified, assessed, and corrected or removed from the market with preventive measures implemented to prevent safety concerns from recurring, upon which a recall can be terminated. FDA categorizes recalls into three classes: Class I (highest severity), which indicate a reasonable likelihood of serious adverse events or death associated with the device, Class II (moderate severity), and Class III (low severity).8 Between 2018 and 2022, 13,623 medical devices were recalled,9 and these devices were associated with 5035 recall events, including 271 Class I, 4548 Class II, and 216 Class III recalls.9 Approximately 97% of recalls are for devices cleared through the 510(k) pathway.10 While Class I recalls represent only a fraction of recall events, they attract the most scrutiny from clinicians, manufacturers, and regulators due to their implications for patient safety.
Prior research on recalls has focused on devices used in specific clinical areas11 or the risk of recall associated with different regulatory pathways.10 The last global analysis of Class I recalls was published in 2018,12 after which time FDA issued new guidance intended to improve the timeliness of recall initiations.13 Medical device recalls are an increasingly important public health issue, with the number of Class I medical device recalls in 2022 being the highest in the past 15 years.14
Although some aspects of medical device recalls have been previously assessed,10–12 no study has comprehensively characterized recent Class I medical device recalls in the US for all medical devices across all clinical uses. Further, no study has examined the termination of medical device recalls. Accordingly, to inform regulatory and clinical efforts to protect patients from medical devices with serious safety concerns, we characterized all Class I medical device recalls and the profiles of recalled devices from January 2018 through June 2022, as well as their current recall status.
Materials and Methods
Data Sources and Study Sample
Using FDA’s publicly available annual log,15 we identified all Class I recalls posted on the FDA’s website from January 1, 2018 to June 30, 2022. Because nearly all Class I recalls are issued for medical devices classified as high-risk or moderate-risk,9 we focused on recalls for these two classes and excluded low-risk and unclassified devices. For each recall listed in the FDA’s annual log, we identified the corresponding database entry in the FDA’s Medical Device Recall database.16 Using these two data sources,15,16 we characterized recalls and the corresponding devices. All data were abstracted by one author, with a review of uncertain cases by two additional authors. Reasons for recall were identified by two authors.
The study did not require institutional review board approval because it was based on publicly available information, in accordance with 45 CFR §46. Informed consent was not needed because no patient data were used. This cross-sectional study adheres to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Characteristics of Recalls and Recalled Devices
Each recall is assigned a unique Recall Event ID by the FDA. We considered each unique Recall Event ID to be a separate recall. For all recalls, we abstracted the following information, as available and reported publicly by the FDA: recall event ID; recall reasons; units recalled in the US; quantity in commerce (ie, number of device units recalled around the world); number of complaints, injuries, and deaths (non-exclusive); recall initiation date; recall status (as of September 15, 2022); and recall termination date, if applicable. Building on prior literature,11 we determined the recall reasons by evaluating the “Reasons for Recall” in recall notices, as reported publicly by the FDA, as well as “Manufacturer Reason for Recall” in the corresponding recall database entries and classified them based on different stages of the device production cycle into one of the following seven categories: device design, processing, manufacturing, packaging/shipment, software, marketing, and others. The status of a recall can be open (ongoing), completed, or terminated. Following recall initiation, manufacturers are required to develop a recall strategy, which is reviewed by FDA. Upon completion of recall activities, manufacturers may submit a termination request to the responsible FDA district office.17 If the FDA district office determines that manufacturers have taken the necessary corrective and preventive actions, the recall status may be changed to completed.17 Next, FDA conducts its final evaluation to ensure that all reasonable efforts were taken by the manufacturer to prevent the recurrence of issues causing the recall before changing the recall status to terminated.17
We also characterized the recalled medical devices as follows: device name, implanted status, life-sustaining/supporting status, class, path to market entry (510(k), PMA, enforcement discretion, or exempt from FDA review), area of use (specialty), and dates of first manufacturing and distribution. For recalls associated with multiple devices, if at least one device was implanted or life-sustaining/supporting, we considered them as implanted or life-sustaining/supporting, respectively. We also evaluated whether there were multiple recalls associated with the recalled devices (using their PMA or 510(k) numbers) at any time until September 15, 2022 and recorded the number of recalls. For recalls associated with multiple devices, if at least one device had been recalled at any time in the past, we counted that device as having multiple recalls. Finally, using FDA’s database,18 we assessed whether any of the recalled devices had received a Breakthrough Devices Program designation.
Recommended Actions in Recall Notices
We categorized the actions recommended in recall notices into two mutually exclusive categories. If the manufacturer and/or FDA recommended discontinuing further use of the device, we classified this as “Stop further use”; if only further device assessment and/or education of the affected population was recommended, we classified that as “Evaluate/educate”. For implanted devices, we added a third category to denote if recalls recommended explanting affected devices. We also determined whether patient instructions were included in the recall notices for devices that would involve patient use. In recall notices informing clinicians about the instructions to be provided to patients, patient instructions were considered present.
Statistical Analysis
Descriptive statistics were used to characterize the sample. The data were recorded and summarized in Microsoft Excel 2018 software (Microsoft Corp). Using JMP Pro, Version 16.2 (SAS Institute Inc), we performed chi-squared tests to examine if there were associations between recall status (as of September 15, 2022) and device and recall characteristics, including device class, path to market entry, implanted status, life-sustaining/supporting status, clinical area of use (specialty), presence of multiple recalls, number of recalled device units in the US, recall reason, recommended action, availability of patient instructions in recall notice, and whether the device involved patient use. P-values less than 0.05 were considered statistically significant.
Results
Characteristics of Recalls and Recalled Devices
A total of 215 Class I medical device recalls were posted on FDA’s annual log between January 1, 2018 and June 30, 2022, of which 10 were for low-risk devices, 15 for unclassified devices, and 1 was a duplicate, leaving 189 unique recall IDs for our analysis. Fifty-six recall notices included multiple distinct authorized devices in the same class, and two included multiple distinct authorized devices in different classes. Among these 58 recalls, the median number of distinct authorized devices per recall was 2 (interquartile range [IQR], 2–3). Of the 189 recalls, 34 (18.0%) and 151 (79.9%) were associated with Class III (high-risk) and Class II (moderate-risk) devices, respectively (Table 1). Thirty-three (17.5%) devices, all of which were high-risk, had received PMA (one high-risk device had received 510(k) clearance), while 146 (77.2%) had been cleared through the 510(k) pathway. Approximately one-fifth of recalls were for implanted and life-sustaining devices (36 [19.0%] and 37 [19.6%], respectively). Recalled devices were mostly used in the cardiovascular specialty (65 [34.4%]), followed by anesthesiology (40 [21.2%]), and general hospital care (32 [16.9%]). One high-risk cardiovascular device, which was implanted and life-sustaining, had a Breakthrough designation. Of all recalls, 125 (66.1%) were for devices that had multiple recalls of any class (as of September 15, 2022), with a median of 4 (IQR, 3–11) recalls for each recalled device. Among these 125 recalls, 28 (22.4%) were for Class III (high-risk), 27 (21.6%) for implanted, and 24 (19.2%) for life-sustaining devices.
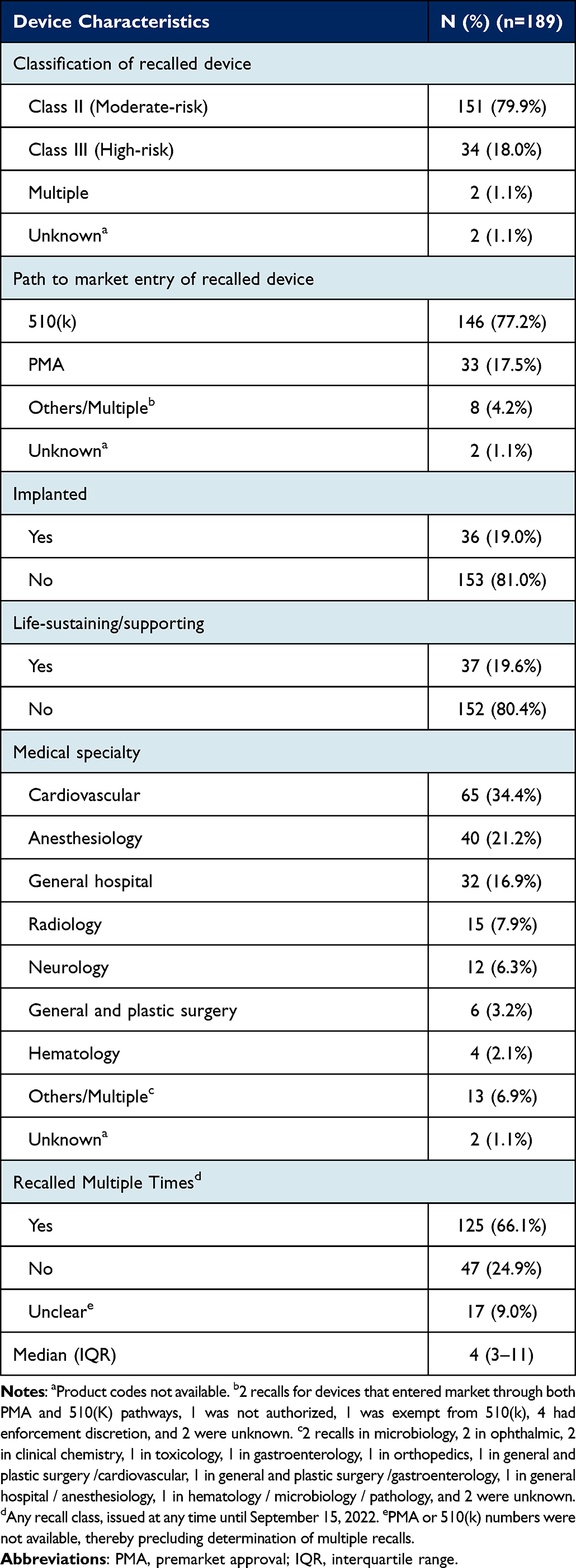 |
Table 1 Characteristics of Moderate and High-Risk Medical Devices with Class I Recalls from January 2018 to June 2022 |
Impact of Recalls
The median number of recalled device units in the US was 4620 (IQR, 578–42,591), with 11 (5.8%) recalls associated with more than 1 million device units (Table 2). The most common reason for recall was issues related to device design (103 [54.5%]), followed by manufacturing errors (25 [13.2%]) and processing errors (22 [11.6%]). The median duration between device distribution to recall initiation was 30.0 (IQR, 10.0–62.5) months.
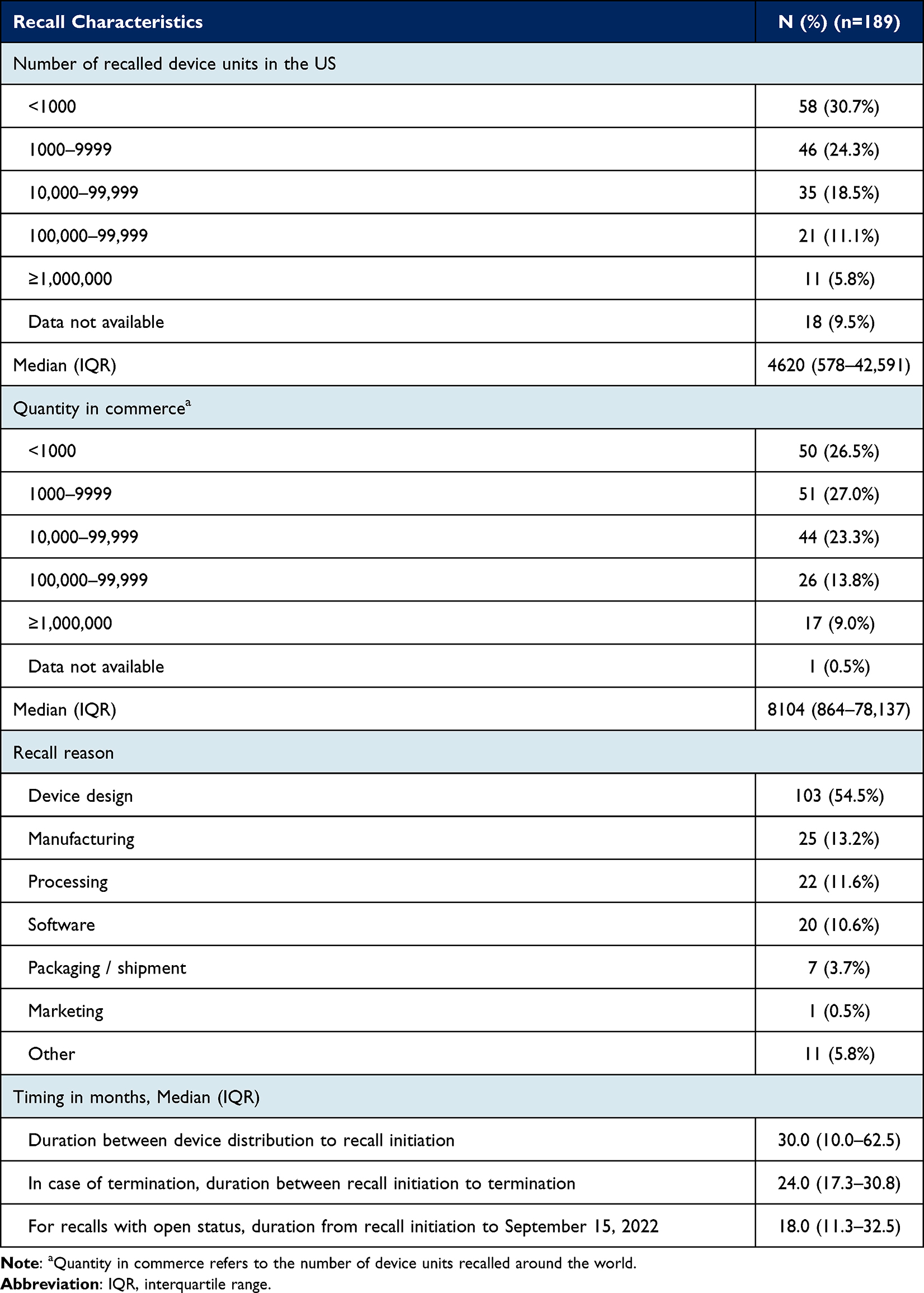 |
Table 2 Characteristics of Class I Medical Device Recalls from January 2018 to June 2022 |
Among all recalls, 82 (43.4%), 64 (33.9%), and 58 (30.7%) did not report the number of complaints, injuries, and deaths associated with the recalled device(s), respectively (Table 3). Of those that reported, the median number of complaints was 8 (IQR, 1–58). Seventeen (9.0%) recalls were associated with 100 or more complaints, and 24 (12.7%) had 10 or more reported injuries to patients. Thirty (15.8%) recalls had at least one patient death associated with the affected device(s).
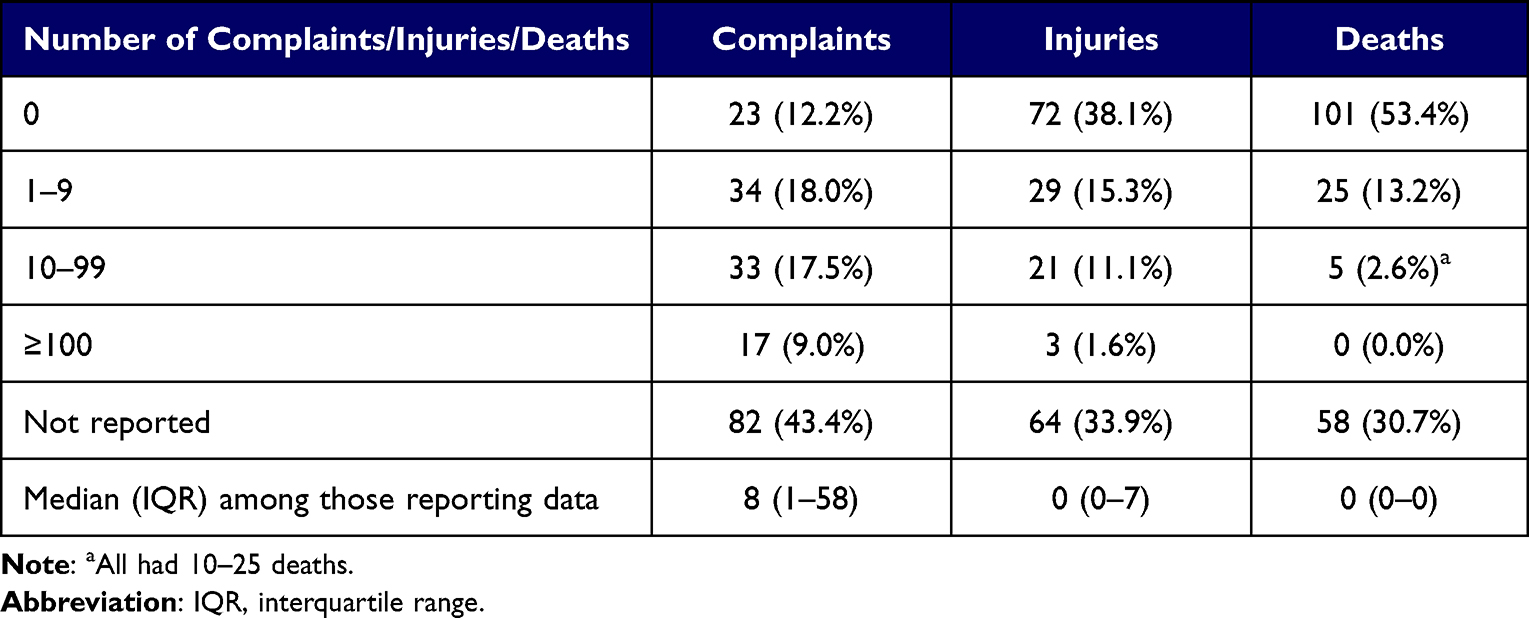 |
Table 3 Number of Complaints, Injuries, and Deaths Associated with Class I Medical Device Recalls from January 2018 to June 2022 |
Recall Status
As of September 15, 2022, 130 (68.8%) recalls were still open (ongoing), 9 (4.8%) were completed (ie, FDA district office determined that manufacturers had taken the necessary corrective and preventive actions), and 50 (26.5%) were terminated (ie, FDA completed its final evaluation after necessary corrective and preventive actions) (Figure 1). For terminated recalls, the median duration between recall initiation to termination was 24.0 (IQR, 17.3–30.8) months (Table 2). Four (8.0%) recalls were terminated in less than a year, while the time to termination for 10 (20.0% of terminated recalls) was more than 3 years. For recalls with open status, the median duration between recall initiation to our cut-off date, September 15, 2022, was 18.0 (IQR, 11.3–32.5) months.
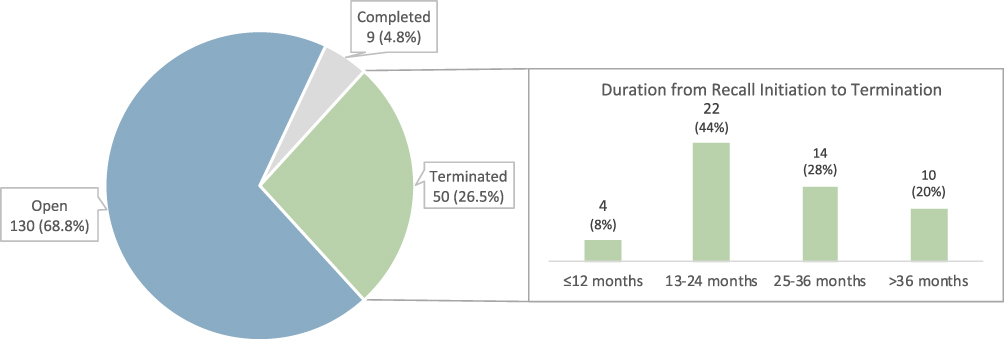 |
Figure 1 Recall status of Class I medical device recalls from January 2018 to June 2022, as of September 15, 2022. |
Recommended Actions in Recall Notices
For 92 Class II devices (60.9% of all Class II devices), the manufacturer and/or FDA recommended stopping further use of the recalled device (Table 4). Among Class III devices, the recommended action in recall notices was to explant the device in 2 (5.9%) recalls and to stop further use of the affected devices in 14 (41.2%) recalls. For all other affected devices, the recommendation was to evaluate and educate. Of 55 recalled devices that involve direct patient use, 30 (54.5%) recall notices did not include any patient instructions.
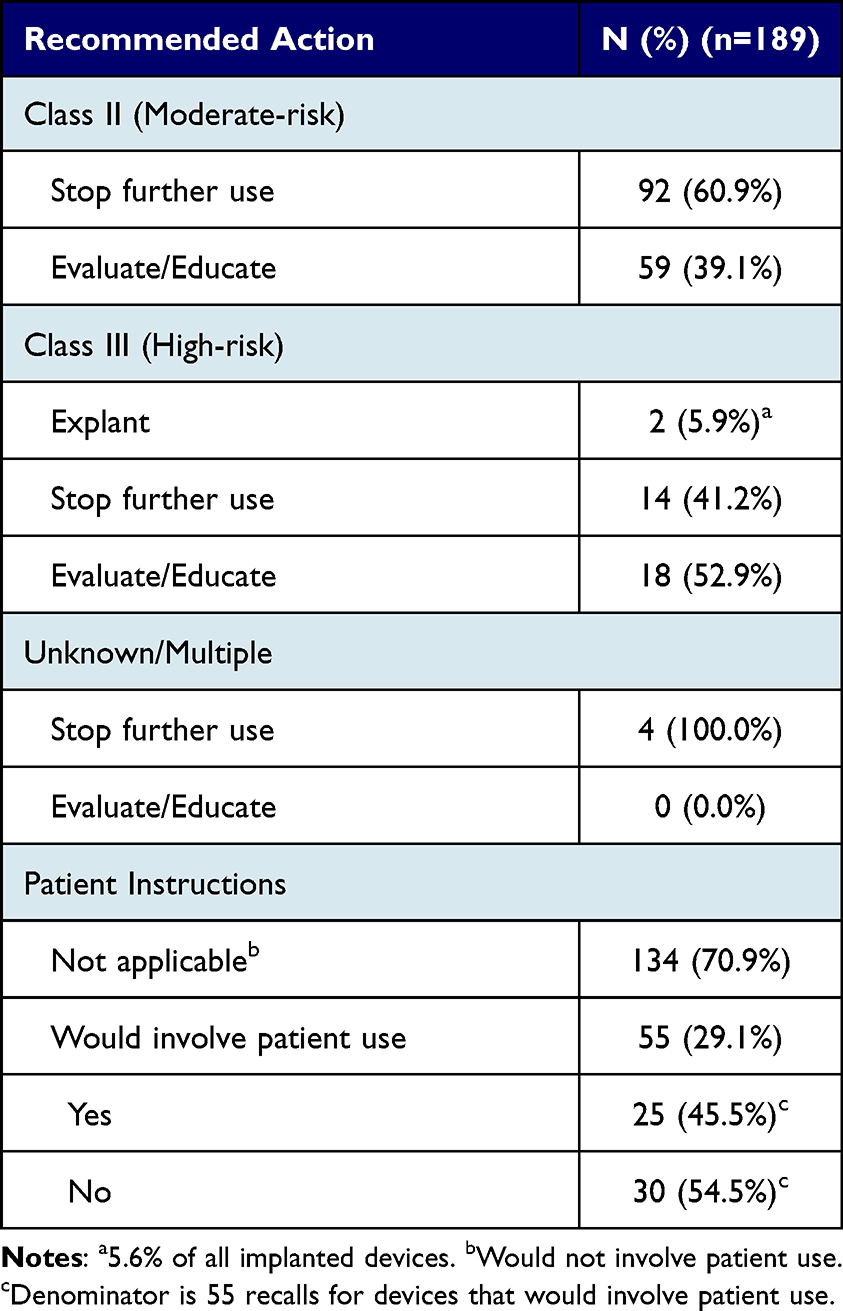 |
Table 4 Recommended Actions in Class I Medical Device Recall Notices from January 2018 to June 2022 |
Recall Status by Device and Recall Characteristics
Recalls were more commonly terminated for devices recalled only once compared to those recalled multiple times (36.2% vs 19.2%; p=0.02) (Table 5). Recall termination also occurred more often for recalls that recommended discontinuing further use of the affected device compared to those that recommended further device assessment and/or education of the affected population (31.8% vs 18.2%; p=0.04). We found no statistically significant differences in recall termination based on device class, path to market entry, implanted status, life-sustaining/supporting status, or clinical area of use. There were also no significant differences in terms of recall termination based on the number of recalled device units, recall reason, whether the device involved patient use, and whether patient instructions were included in the recall notice among devices that involved patient use.
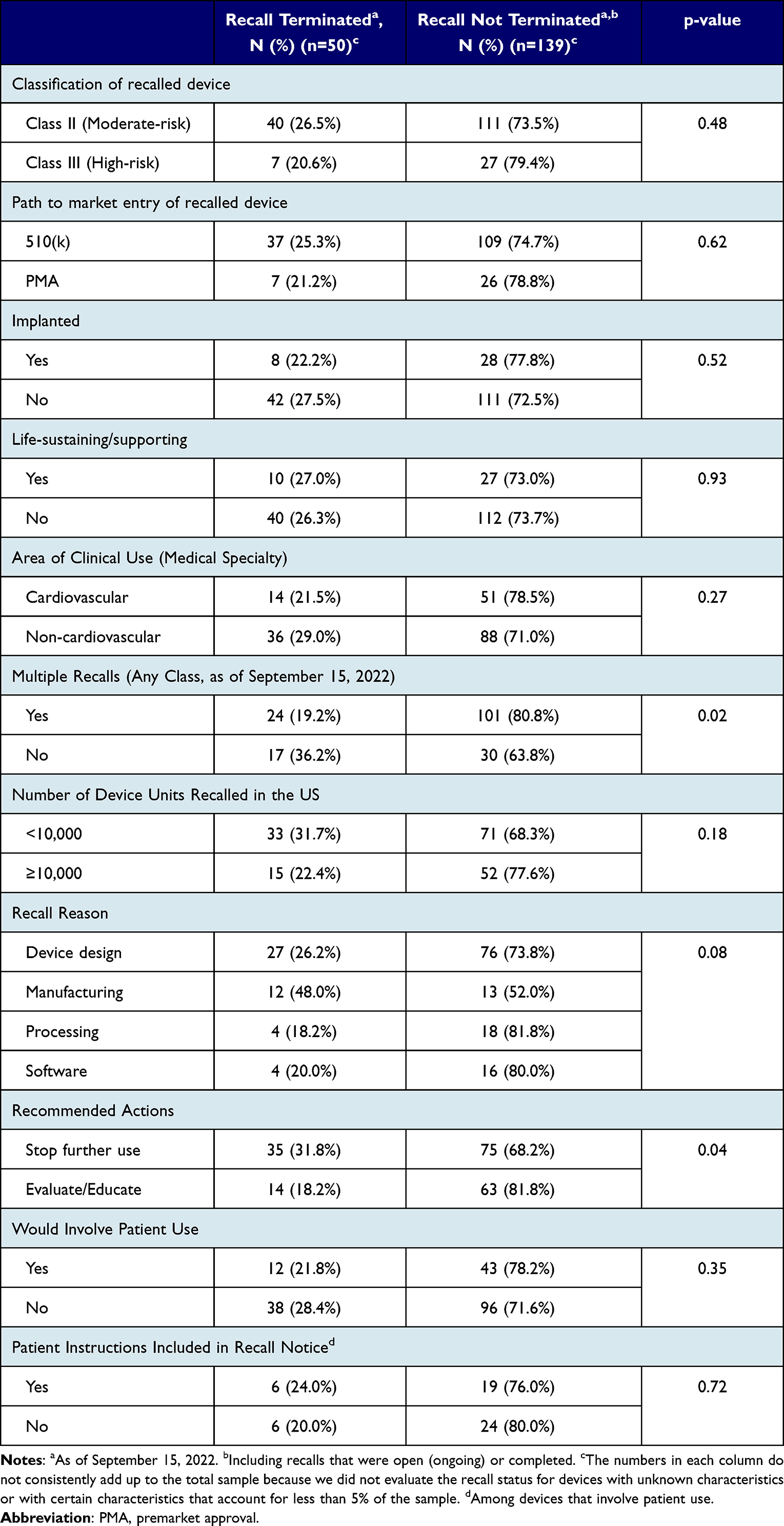 |
Table 5 Recall Status of Class I Medical Device Recalls from January 2018 to June 2022, as of September 15, 2022 |
Discussion
From 2018 to June 2022, there were 189 high-severity Class I recalls of high- and moderate-risk devices in the United States, affecting millions of device units. Two-thirds of the devices were recalled more than once. Only a small proportion of recalls were terminated, meaning that FDA completed its final evaluation after the manufacturers completed all necessary corrective and preventive actions. Notably, during this time when recalls are active but not yet terminated, recalled devices may be used for patient care, placing patients at risk.19 Even when recalls were terminated, they required a median of two years after recall initiation. These findings are consistent with the literature that implementing a recall could be a resource-intensive, costly, inefficient, and sometimes an incomplete process,17,20,21 highlighting the need to improve the medical device recall process to ensure patient safety.
A 2011 US Government Accountability Office (GAO) report noted that recalls were often delayed or incomplete because of challenges in locating recalled devices or users of devices.17 We found that the median duration from recall initiation to termination for the recalls issued in the past 5 years was approximately 50% longer than the average 516-day duration reported by the GAO in 2011,17 indicating that recall management is taking longer. A central motivation for FDA’s 2013 unique device identification system rule was to facilitate resolution of recalled devices, with correction or removal from the market, thereby more rapidly reducing or eliminating risk to patients.22 However, a recent study showed that information about unique device identifiers (UDIs) is inconsistently available in recall notices, likely compounding delays in addressing these recalls.23
We found that a significant proportion of recalls did not report the number of complaints, injuries, and deaths associated with the recalled devices. These data rely on adverse event reports, which have a multitude of reasons for inaccuracy and incompleteness.24 Integration of UDIs into electronic health records and claims forms would improve the accuracy of attributing safety events to medical devices.25,26 This would enable the inclusion of more comprehensive and accurate information about injuries and deaths in recall notices so patients and healthcare professionals could better understand the risk of recalled devices, especially as the majority of recall notices recommended clinicians evaluate risk and offer education with respect to the recalled devices. Furthermore, patient instructions were not included in most recall notices of devices that involved patient use. Because patient awareness is critical to recall management, FDA could develop a structured template to include patient instructions for applicable devices.
Other researchers have reported that approximately 40% of recalls have mischaracterized or vague FDA-determined causes.12 To address this concern, in our study, two independent reviewers categorized recall causes based on device production cycle steps. Consistent with previous studies,11 we found that design flaws and manufacturing errors are the two most common reasons for recalls. Thus, stricter regulation of device design and manufacturing processes could be helpful. Moreover, specific lots and manufacturing dates should be included in notices for these recalls to enable device identification and evaluation. Including UDIs in recall notices would facilitate this process.
Finally, our findings raise larger questions about medical device safety.27 Approximately three-fourths of recalls were for devices cleared through the 510(k) pathway. Studies have consistently demonstrated that most recalled devices were cleared through the 510(k) pathway;28–30 however, a study evaluating more than 5000 recall events found that while 97% of recalled devices were cleared through the 510(k) pathway, PMA authorized devices were nearly three times more likely to be recalled compared to those with 510(k) clearance.10
Our study should be considered in the context of its limitations. First, we only examined Class I recalls associated with moderate- and high-risk medical devices. While these are the highest severity and usually the most clinically significant recalls, other recalls will also benefit from effective and precise identification to ensure patient safety. Second, the assessed recalls relied on the FDA’s annual log of Class I medical device recalls, which might miss some recalls. Third, a small proportion of device recalls in our sample had a shorter period of follow-up, limiting opportunities for termination. Despite these limitations, our study evaluated the Class I medical device recalls in all specialties over the past 5 years, making the results generalizable and timely, and is the first study assessing factors associated with recall termination.
Conclusion
Class I medical device recalls are common and affect millions of device units in use in the US. Completing the required actions for recall termination takes a significant amount of time, posing serious safety concerns to patients for a longer period. Given the increasing frequency of recalls, the number of affected units per recall, and the widespread distribution of affected medical devices, more effort is needed to facilitate the timely and precise identification of affected devices to minimize patient harm.
Abbreviations
FDA, Food and Drug Administration; GAO, Government Accountability Office; IQR, interquartile range; PMA, premarket approval; STROBE, Strengthening the Reporting of Observational Studies in Epidemiology; UDI, unique device identifier.
Data Sharing Statement
Relevant data are available on reasonable request from the corresponding author.
Ethics Approval and Informed Consent Statement
Ethical approval was not required. Publicly available nonclinical datasets were used. Informed consent was not needed because no patient data were used.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Food and Drug Administration (FDA) of the US Department of Health and Human Services (HHS) as part of a financial assistance award [U01FD005938]. The contents are those of the authors and do not necessarily represent the official views of, nor an endorsement, by FDA/HHS, or the US Government.
Disclosure
Drs. Mooghali and Ross currently receive research support through Yale University from Arnold Ventures. Dr. Ross currently receives research support through Yale University from Johnson and Johnson to develop methods of clinical trial data sharing, from the Medical Device Innovation Consortium (MDIC) as part of the National Evaluation System for Health Technology (NEST), from the Food and Drug Administration for the Yale-Mayo Clinic Center for Excellence in Regulatory Science and Innovation (CERSI) program (U01FD005938), from the Agency for Healthcare Research and Quality (R01HS022882), and from the National Heart, Lung and Blood Institute of the National Institutes of Health (NIH) (R01HS025164, R01HL144644); in addition, Dr. Ross is an expert witness at the request of Relator’s attorneys, the Greene Law Firm, in a qui tam suit alleging violations of the False Claims Act and Anti-Kickback Statute against Biogen Inc. Mr. Kadakia reported having previously been employed and Cleveland Clinic London, Google (via Adecco), and the US Food and Drug Administration (FDA), and receiving consulting fees from the National Academy of Medicine outside the submitted work. Dr. Dhruva receives research support from the Department of Veterans Affairs Health Services Research and Development (1IK2HX003357), from the MDIC as part of the (NEST), and Arnold Ventures. Drs. Ross and Dhruva serve on the Medicare Evidence Development & Coverage Advisory Committee.
References
1. U.S. Food and Drug Administration. Regulatory controls. Available from: https://www.fda.gov/medical-devices/overview-device-regulation/regulatory-controls.
2. U.S. Food and Drug Administration. Premarket notification 510(k). Available from: https://www.fda.gov/medical-devices/premarket-submissions-selecting-and-preparing-correct-submission/premarket-notification-510k.
3. Rathi VK, Krumholz HM, Masoudi FA, Ross JS. Characteristics of clinical studies conducted over the total product life cycle of high-risk therapeutic medical devices receiving FDA premarket approval in 2010 and 2011. JAMA. 2015;314(6):604–612. doi:10.1001/jama.2015.8761
4. Dhruva SS, Bero LA, Redberg RF. Strength of study evidence examined by the FDA in premarket approval of cardiovascular devices. JAMA. 2009;302(24):2679–2685. doi:10.1001/jama.2009.1899
5. U.S. Food and Drug Administration. MDR data files. Available from: https://www.fda.gov/medical-devices/medical-device-reporting-mdr-how-report-medical-device-problems/mdr-data-files.
6. Tau N, Shepshelovich D. Assessment of data sources that support US food and drug administration medical devices safety communications. JAMA Intern Med. 2020;180(11):1420–1426. doi:10.1001/jamainternmed.2020.3514
7. U.S. Food and Drug Administration. Recalls, corrections and removals (devices). Available from: https://www.fda.gov/medical-devices/postmarket-requirements-devices/recalls-corrections-and-removals-devices.
8. U.S. Food and Drug Administration. Recalls background and definitions. Available from: https://www.fda.gov/safety/industry-guidance-recalls/recalls-background-and-definitions.
9. U.S. Food and Drug Administration. Recalls. Available from: https://datadashboard.fda.gov/ora/cd/recalls.htm.
10. Dubin JR, Simon SD, Norrell K, Perera J, Gowen J, Cil A. Risk of recall among medical devices undergoing US food and drug administration 510(k) clearance and premarket approval, 2008–2017. JAMA Netw Open. 2021;4(5):e217274. doi:10.1001/jamanetworkopen.2021.7274
11. Vajapey SP, Li M. Medical device recalls in orthopedics: recent trends and areas for improvement. J Arthroplasty. 2020;35(8):2259–2266. doi:10.1016/j.arth.2020.03.025
12. Sarkissian A. An exploratory analysis of U.S. FDA class I medical device recalls: 2014–2018. J Med Eng Technol. 2018;42(8):595–603. doi:10.1080/03091902.2019.1580778
13. U.S. Food and Drug Administration. Initiation of voluntary recalls under 21 CFR part 7, subpart C. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/initiation-voluntary-recalls-under-21-cfr-part-7-subpart-c.
14. Taylor NP. FDA class I recalls hit 15-year high in 2022. MedTech Dive. Available from: https://www.medtechdive.com/news/fda-class-i-recall-2022-ABT-BAX-GEHC-MDT-PHG/644072/.
15. U.S. Food and Drug Administration. Medical device recalls. Available from: https://www.fda.gov/medical-devices/medical-device-safety/medical-device-recalls.
16. U.S. Food and Drug Administration. Medical device recalls. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfRES/res.cfm.
17. U.S. Government Accountability Office (GAO). Medical devices: FDA should enhance its oversight of recalls. Available from: https://www.gao.gov/assets/a319569.html.
18. U.S. Food and Drug Administration. Breakthrough devices program. Available from: https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program.
19. Kadakia KT, Ross JS, Rathi VK. The Philips Respironics recall of ventilators and positive airway pressure machines—breakdowns in medical device surveillance. JAMA Intern Med. 2023;183(1):5–8. doi:10.1001/jamainternmed.2022.5141
20. Association for Healthcare Resource & Materials Management. Unique Device Identifier (UDI) impacts on recall management executive summary. Available from: https://www.ahrmm.org/system/files/media/file/2021/09/UDI-DI-Recall-Impact-Executive-Summary.pdf.
21. Kinard M, McGiffert L. Medical device tracking-how it is and how it should be. JAMA Intern Med. 2021;181(3):305–306. doi:10.1001/jamainternmed.2020.7797
22. U.S. Food and Drug Administration. Unique Device Identification System (UDI System). Available from: https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/unique-device-identification-system-udi-system.
23. Mooghali M, Ross JS, Kadakia KT, Dhruva SS. Availability of Unique Device Identifiers for Class I Medical Device Recalls From 2018 to 2022. JAMA Intern Med; 2023. doi:10.1001/jamainternmed.2023.0727
24. U.S. Food and Drug Administration. MAUDE - Manufacturer and User Facility Device Experience. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm.
25. Tomes M. Identification and market removal of risky medical devices. JAMA Intern Med. 2020;180(11):1426–1427. doi:10.1001/jamainternmed.2020.3512
26. Kadakia KT, Dhruva SS, Ross JS, Krumholz HM. Adding device identifiers to claims forms—a key step to advance medical device safety. BMJ. 2023;380:82. doi:10.1136/bmj.p82
27. Redberg RF, Dhruva SS. Moving from substantial equivalence to substantial improvement for 510(k) devices. JAMA. 2019;322(10):927–928. doi:10.1001/jama.2019.10191
28. Kadakia KT, Beckman AL, Ross JS, Krumholz HM. Renewing the call for reforms to medical device safety—the case of Penumbra. JAMA Intern Med. 2022;182(1):59–65. doi:10.1001/jamainternmed.2021.6626
29. Kadakia KT, Ross JS, Rathi VK. The Philips Respironics recall of ventilators and positive airway pressure machines - breakdowns in medical device surveillance. JAMA Intern Med. 2022. doi:10.1001/jamainternmed.2022.5141
30. Zuckerman DM, Brown P, Nissen SE. Medical device recalls and the FDA approval process. Arch Intern Med. 2011;171(11):1006–1011. doi:10.1001/archinternmed.2011.30
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.