Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases
Authors Bagyinszky E, Giau VV, Youn YC ![]() , An SSA, Kim SY
, An SSA, Kim SY ![]()
Received 13 February 2018
Accepted for publication 23 May 2018
Published 14 August 2018 Volume 2018:14 Pages 2067—2085
DOI https://doi.org/10.2147/NDT.S165445
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Taro Kishi
Eva Bagyinszky,1 Vo Van Giau,1 Young Chul Youn,2 Seong Soo A An,1 SangYun Kim3
1Department of Bionano Technology, Gachon Bionano Research Institute, Gachon University, Gyeonggi-do, South Korea; 2Department of Neurology, Chung-Ang University College of Medicine, Seoul, South Korea; 3Department of Neurology, Seoul National University College of Medicine & Neurocognitive Behavior Center, Seoul National University Bundang Hospital, Seongnam, South Korea
Abstract: Abnormal prion proteins are responsible for several fatal neurodegenerative diseases in humans and in animals, including Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker disease, and fatal familial insomnia. Genetics is important in prion diseases, but in the most cases, cause of diseases remained unknown. Several mutations were found to be causative for prion disorders, and the effect of mutations may be heterogeneous. In addition, different prion mutations were suggested to play a possible role in additional phenotypes, such as Alzheimer’s type pathology, spongiform encephalopathy, or frontotemporal dementia. Pathogenic nature of several prion mutations remained unclear, such as M129V and E219K. These two polymorphic sites were suggested as either risk factors for different disorders, such as Alzheimer’s disease (AD), variant CJD, or protease-sensitive prionopathy, and they can also be disease-modifying factors. Pathological overlap may also be possible with AD or progressive dementia, and several patients with prion mutations were initially diagnosed with AD. This review also introduces briefly the diagnosis of prion diseases and the issues with their diagnosis. Since prion diseases have quite heterogeneous phenotypes, a complex analysis, a combination of genetic screening, cerebrospinal fluid biomarker analysis and imaging technologies could improve the early disease diagnosis.
Keywords: genetics, mutation, prion, PRNP gene, Creutzfeldt–Jakob disease, Gerstmann–Sträussler–Scheinker disease, fatal familial insomnia, Alzheimer’s disease, diagnosis
Introduction
Prion diseases
Human transmissible spongiform encephalopathies or the prion diseases are fatal neurodegenerative disorders, based on the misfolding of prion protein (PrP).1 Their phenotypes are quite diverse, since several diseases can be distinguished, such as Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker (GSS) disease, fatal familial insomnia (FFI), or kuru.1 Prion diseases share several properties common with other neurodegenerative disorders, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) in terms of pathophysiology or morphology.2 However, they have a unique feature, since prion disorders could be transmissible. According to the etiology, prion disorders could be divided into three different categories: 1) unknown reasons of disease onset; 2) to have originated from an infection; and 3) a genetic mutation in prion (PRNP) gene.2 Approximately, 10%–15% of all prion diseases could be associated with genetic mutations with autosomal dominant inheritance pattern. Several de novo cases of genetic prion diseases have been observed, where family history was negative.1 Due to various clinical phenotypes, it was also suggested that prions may be involved in other types of neurodegenerative diseases.1
PRNP (NC_000020.11) is 16 kb long gene, located on chromosome 20 (4686151-4701588). It contains two exons, and the exon 2 carries the open reading frame,2 which encodes the 253 amino acid (AA) long PrP protein.3 Exon 1 is a noncoding exon, which may serve as transcriptional initiation site.4 The post-translational modifications result in the removal of the first 22 AA N-terminal fragment (NTF) and the last 23 AA C-terminal fragment (CTF). The NTF is cleaved after PrP transport to the endoplasmic reticulum (ER), while the CTF (glycosylphosphatidylinositol [GPI] signal peptide [GPI-SP]) is cleaved by the GPI anchor. GPI anchor could be involved in PrP protein transport. It may also play a role of attachment of prion protein into the outer surface of cell membrane.2–7 Normal PrP is composed of a long N-terminal loop (which contains the octapeptide repeat region), two short β sheets, three α helices, and a C-terminal region (which contains the GPI anchor; Figure 1). Cleavage of PrP results in a 208 AA long glyocoprotein, anchored in the cell membrane.
 | Figure 1 3D structure of normal PrP protein. Normal PrP contains 3α helices and two β sheets. |
The exact physiological role of PrP remained unclear, but it was suggested to be involved in several brain functions, such as neuronal protection, adhesion, and cell signaling, or in controlling the circadian system.8 Normal PrP may play a role in synaptic, especially the presynaptic, functions by interacting with synaptic release-associated proteins, ion channels, and metabotropic and ionotropic neurotransmitter receptors.5,8 Mouse models revealed that PrP could modulate the vesicles and enhance the strength of synaptic transmission. Two forms of PrP can be distinguished: the normal PrPc and the pathogenic PrPSc.8–10 PrPc is composed of ~42% α helices and ~3% β sheets and is thermodynamically stable.4 PrpSc contains ~42% β sheets and ~30% α helices. Significant increase of β sheets and the reduced level of α helices in PrPSc could result in protein assembly and neurodegeneration.11,12 The aggregation and accumulation of PrPSc are the key features of prion diseases in humans and in several animal species.2 In this review, we summarize all known prion mutations (Tables 1–4; Figure 2), and their disease phenotypes or possible disease association.
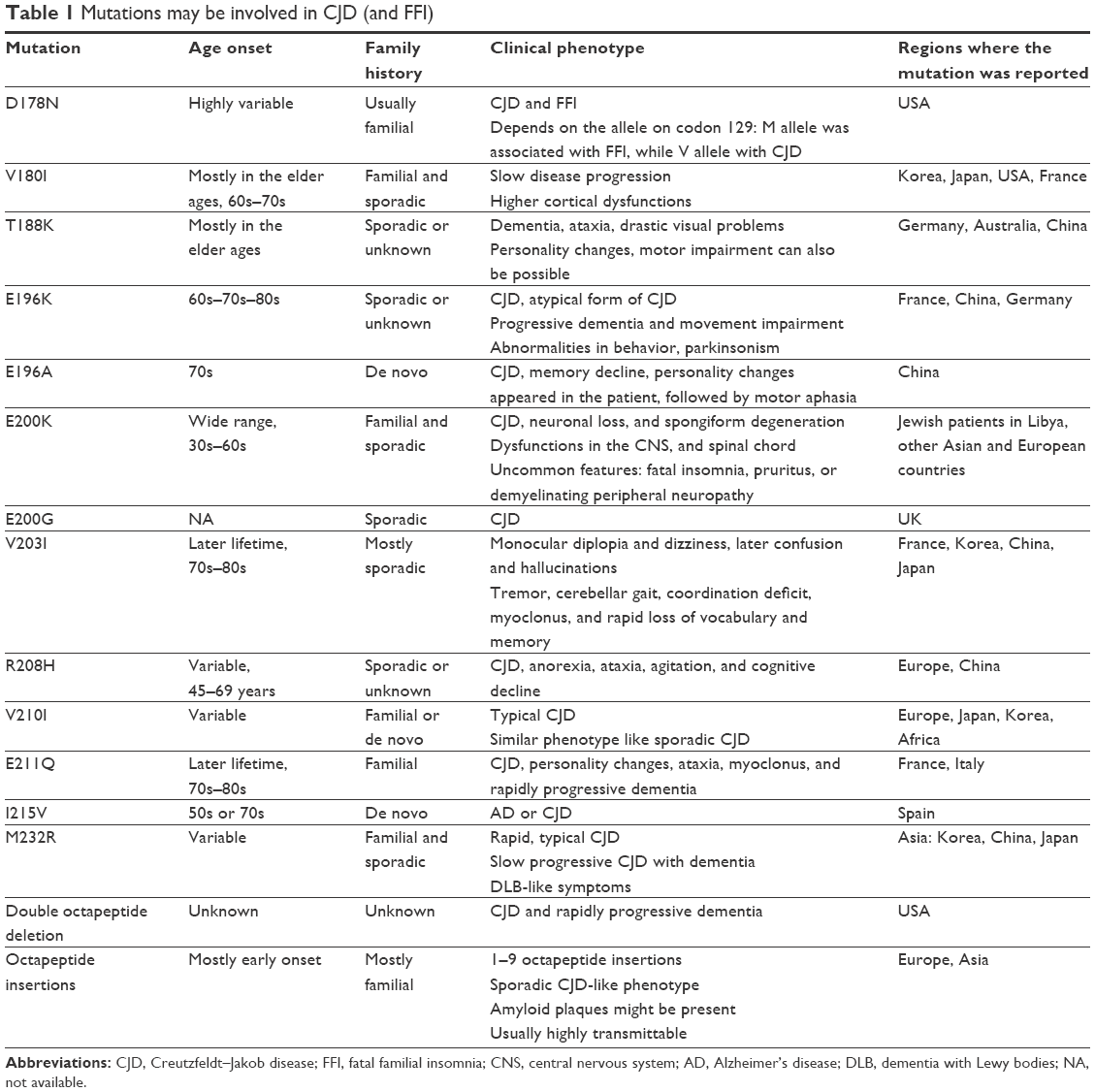 | Table 1 Mutations may be involved in CJD (and FFI) |
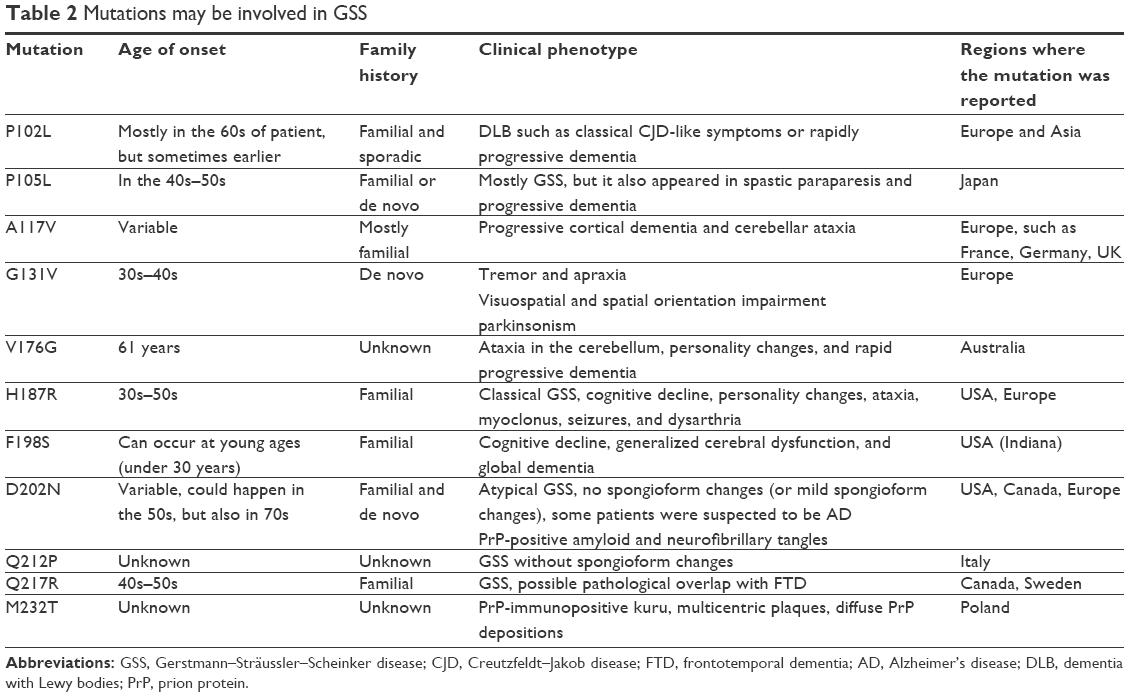 | Table 2 Mutations may be involved in GSS |
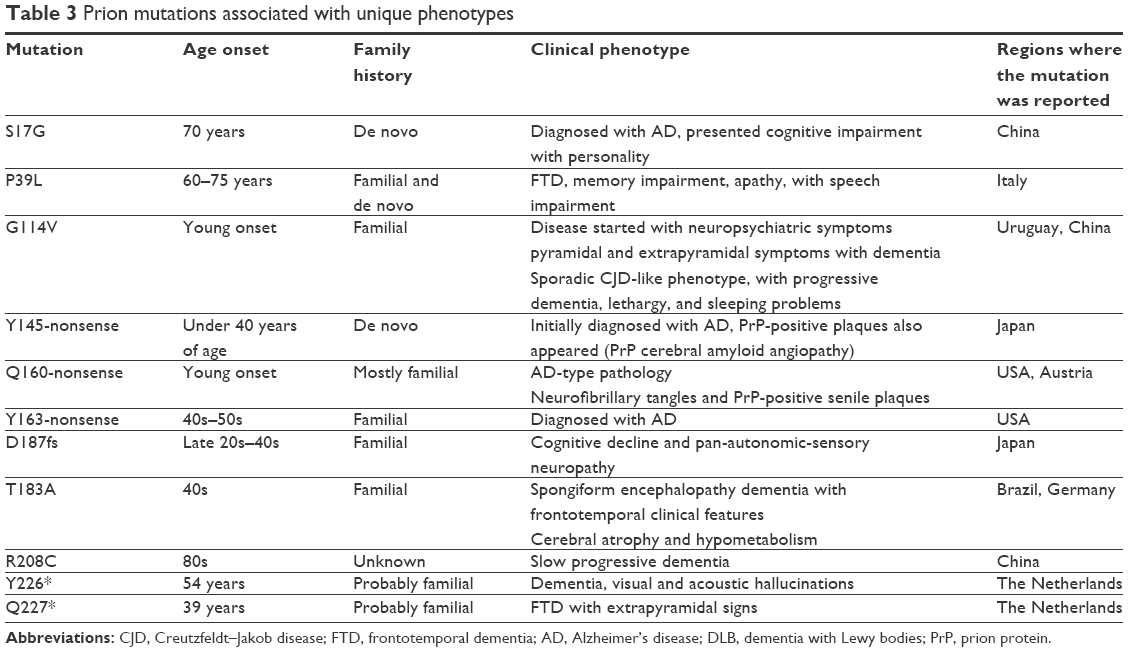 | Table 3 Prion mutations associated with unique phenotypes |
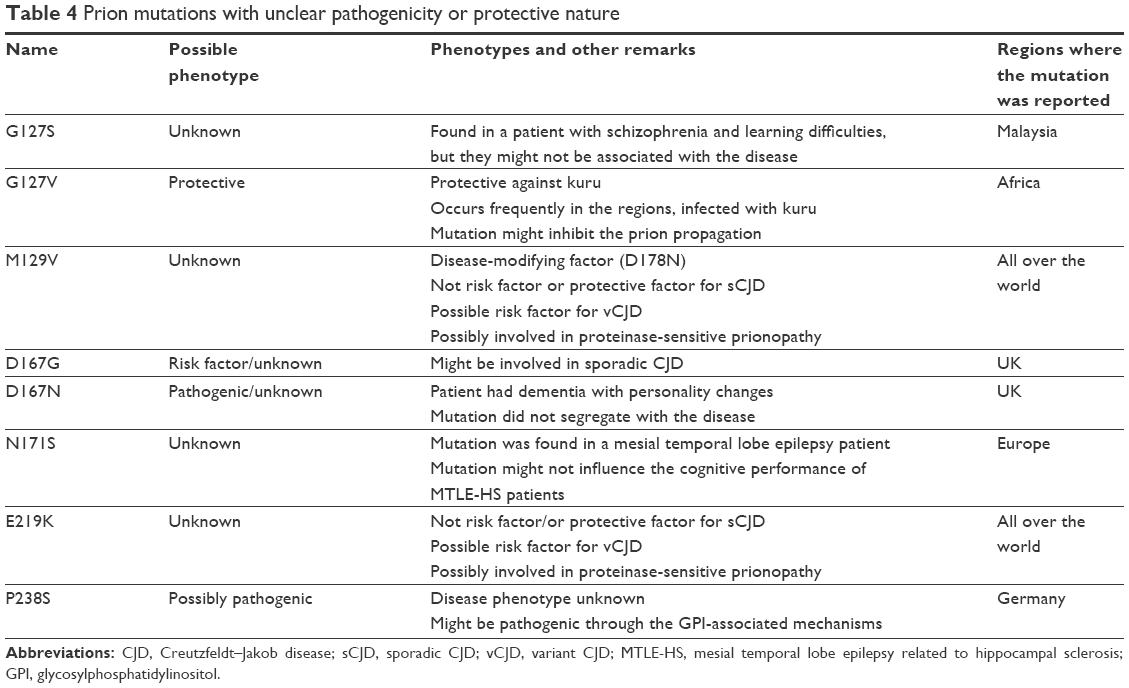 | Table 4 Prion mutations with unclear pathogenicity or protective nature |
 | Figure 2 Schematic representation of human PRNP gene with all known mutations and polymorphisms. |
Summary of prion mutations
Prion mutations, involved in CJD
In familial CJD (fCJD), progressive memory impairment, confusion, ataxia, and myoclonus were observed as the main symptoms. Patients in the final disease stages usually lose the ability to speak and move. Age of onset can be variable, since CJD may appear at young ages (in the 30s or 40s), but also in the later lifetime.3,4 Majority of all human prion diseases (75%) were identified as sporadic CJD (sCJD), associated with rapid disease progression, multifocal dementia, fatigue, insomnia, and depression.13 In the brain of patients, astrocytic gliosis, spongiform changes, and sometimes amyloid deposits have been observed (Table 1).5
V180I was found relatively frequently in East-Asian populations, but also in European (French) cases, and associated with fCJD and sCJD.14–19 fCJD with V180I usually occurs after 70 years of age; slow disease progression and cortical dysfunctions were observed in patients.17 The rate of 14-3-3 protein in fCJD patients with V180I was usually low.19,20 V180 is located in the hydrophobic core of PrP (inside the α2–α3 inter-helical interface),21 which is important for the stability of its globular domain. Simulations revealed that V180I may result in abnormal fluctuation of I184, which could disturb the contact between I184 and F198. V180I showed early misfolding and altered hydrophobic contacts.21 However, pathogenic role of PRNP V180I has been refuted by Beck et al in 2012, since it was also appeared in asymptomatic individuals.22
T188K was initially described by Finckh et al in a 59-year-old patient from Austria, who developed rapid progressive dementia and dysphasia, and family history was negative.23 T188K was also found in German patients by Roeber et al, but none of them presented positive family history.24 Later, mutation was also discovered in Chinese patients, who presented sCJD-like phenotypes, including progressive dementia, myoclonus, visual-, sleeping-, and cerebellar disturbances, dysfunctions in the pyramidal or extrapyramidal system, and mutism and were positive for 14-3-3 protein in cerebrospinal fluid (CSF).25,26 Disease duration was short, even <1 year.26 T188 is located in the C-terminal part of α2 helix, near the S2 loop. It is a highly conserved residue, suggesting that its mutations may result in significant dysfunctions of PrP.27 In silico models on T188K may result in the formation of a 310 helix at the S2-H2 loop and the loss of binding sites of different inhibitors for amyloid formation.27
T188R was found during a routine genetic testing of possible CJD patients, presenting dementia, ataxia, and drastic visual impairment. However, due to the lack of family history, its pathogenic nature has been questioned.22 Tartaglia et al reported this mutation in a sCJD case, where rapid progressive dementia, personality changes, and motor impairment appeared. The patient did not meet the criteria of sCJD according to the new European criteria of possible or probable sCJD, but met the University of California, San Francisco criteria of probable sCJD, and the family history remained unclear.26 Structure predictions on T188R revealed that this mutation could result in elevated β sheet content and the elongation of β strand in PrP.27,28
T188A was discovered in a female patient in her 80s, having CJD, without any family history of the disease.29 No significant structural changes were observed for T188A.29
E196K was associated with clinically heterogeneous neurodegenerative disease phenotypes.30 Mutation was discovered in a French patient, who was initially diagnosed with AD. She developed different personality changes, such as anorexia, emotional instability, or inappropriate giggling. Additional phenotypes were also present, such as progressive dementia and movement impairment.30 Tumani et al reported the mutation in a female patient with atypical CJD, where speech disturbance and gait ataxia were present. Later, she developed pyramidal and extrapyramidal signs, myoclonus, and mutism.31 Clerici et al discovered the mutation in an atypical CJD patient, and the history was unknown.32 Béjot et al found the mutation in an 80-year-old male patient, who showed rapid progressive behavioral problems and myoclonus after a stroke. He was diagnosed with CJD-like disease phenotype, but with the atypical form of disease.33 Eigenbrod et al found E196K in four German patients with probable CJD, who were initially diagnosed with PD. All of these patients were associated with nonspecific clinical phenotypes, abnormalities in behavior, parkinsonism, Wernicke encephalopathy, and brainstem infarction/internal hydrocephalus.34 Disease occurred in the late 60s or 70s. PrPSc was appeared in three areas of their brain: frontal cortex, cerebellum, and hippocampus.34 The spongiform changes may depend on codon 129 genotype, since V/V phenotype presented them in the deep cortical layers, hippocampus, and in the cerebellum. In patients with M/M or M/V genotype, spongiform changes appeared in all cortical layers.34,35 The mutation was also reported in China, in a 71-year-old female patient, who carried the homozygous M/M allele. She had nonspecific symptoms such as progressive impairment in movement, speech, and dementia.36 In silico predictions on this mutations revealed that E196 could form a salt bridge with R156, and the mutation may destroy this connection, resulting in protein destabilization and subdomain separation (between α1 and α2–α3 loops).37
E196A was discovered in three unrelated Chinese patients with CJD, aged between 54 and 76 years. Clinical symptoms were rapid progressive dementia, impairment in movement, loss of orbital reflex pressure, psychotic symptoms, and weakness in limbs.38 Their CSF was positive for 14-3-3 protein, which confirmed the diagnosis.38
E200K is one of the most common causes for fCJD worldwide, which could result in proteinase-K-resistant PrPSc and in abnormalities in the central and peripheral nervous system. Neuronal loss and spongiform degeneration were detected in the cerebellum, and the density was progressively decreased in the thalamus, as well as the temporal and frontal cortex.39 Western blot detected that the levels of protease-K PrPSc protein were correlated with the degree of lesion.39,40 This mutation was found in Jewish CJD patients in Lybia.41 The age onset ranged between 33 and 66 years.41 The phenotypes of the clinical features were heterogeneous, for example, dysfunctions in the CNS and spinal cord.42 Uncommon features were also reported, such as fatal insomnia, pruritus, or demyelinating peripheral neuropathy.42,43 Structure predictions on E200K mutation revealed that mutation may result in altered electrostatic potential. The altered surface charges could result in altered interactions of PrP with other chaperon proteins.44 This mutation could repress the ubiquitin-proteosome system, resulting in macroautophagy and formation of ubiquitin-positive body and aggregosome.45
E200G was discovered in a British CJD patient, where codon 129 carried the M/V allele. Neuronal loss was severe in the thalamus, with mild cerebellar involvement. PrPSc deposits appeared in synaptic boutons and in terminals of axons.46
V203I was first described by Peoc’h et al and was suggested to be involved in CJD. Proband patient was identified with monocular diplopia and dizziness, followed by confusion and hallucinations.30 Tremor, cerebellar gait, coordination deficit, and myoclonus also appeared.30 The second case of mutation was found in a Korean female patient, whose symptoms were similar to the first case, such as gait disturbance, rapidly progressing cognitive decline, tremor, rigidity, myoclonic jerks, and stupor. Both the cases were suggested to be associated with sCJD.47 Mutation also appeared in an 80-year-old Chinese CJD patient, who presented rapid loss of vocabulary and memory, dizziness, blurred vision, and ataxia. Tremor, myoclonus, and bilateral Babinski signs also appeared in him, and family history was negative.48 In Japan, this mutation was found in a homozygous stage, in a female patient with CJD. The patient developed progressive gait disturbances and cognitive dysfunctions, followed by akinetic mutism.49 The clinical phenotypes were similar to the previously described heterozygous cases, but the disease progression was more rapid.49 This mutation is located in the hydrophobic core of PrP. In silico modeling suggested that it could result in minor effects on prion conformation. This mutation could induce F198 to move out of the solvent, but it was restored.50 It could also affect the conformation of β1-α1 loop and disrupt the hydrophobic contact between α2 and α3 helices. The other suggestion was that the mutation could result in instability of the core.50
R208H was discovered in 1996, in a CJD patient without family history, but it appeared in a younger member of the pedigree, who probably was in the presymptomatic stage at the time of the analysis.51 The second case was described by Capellari in 2001, and the clinical phenotypes were similar in both the cases, such as anorexia, ataxia, agitation, and cognitive decline.52 Western blot analyses of brain homogenates revealed three types of PrPSc bands and an additional 17 kDa band. Postmortem studies found tau pathology and ballooned neurons in different areas, such as hippocampus and entorhinal cortex.53 R208H with homozygous V/V allele on codon 129 was associated with fCJD, and kuru plaques were also present.54 Matěj et al and Vita et al detected the mutation with the V/V allele of codon 129, associated with different phenotypes. Both the cases reported impairment in movement, and progressive supranuclear palsy (PSP)-like phenotype was also present.55,56 Mutation was also discovered in China in 2011, with similar phenotypes, described before.57 It was suggested that R208H could alter its dynamics of salt bridge between α1 and α2 helices (there may be salt bridge contact between D144/E146 and R208).58
V210I is a pathogenic CJD mutation, described in Europe, Africa,59,60 Korea, and Japan.61–63 Frequency of mutation was around 16.2% in EuroCJD. The mutation was identified in the Heidenhain (visual impairment-associated) form of variant CJD (vCJD).63 Similarly to V180I, V210I is located in the α2–α3 inter-helical interface of PrP, these two residues are located opposite to each other, and there is direct hydrophobic contact between them.64 The mutation might induce the rearrangement of hydrophobic core, resulting in alternations in the β2–α2 interaction, and leading to spontaneous generation of PrPSc.65
E211Q was discovered by Peoc’h et al in a patient with personality changes, ataxia, myoclonus, and rapidly progressive dementia.30 His siblings also developed clinical phenotypes of neurological impairments such as dementia, cerebellar syndrome, and movement abnormalities. Since the mutation is located on the surface of α3 helix in PrP, it was thought that it might not modify the PrP structure significantly.28 Second case of mutation was discovered in an Italian family, whose affected members developed similar phenotypes, in their 70s or 80s.66 This report suggested that mutations at codon 211 do not produce conservative AA exchange, and Glu211 is not a part of hydrophobic core.66 E211 is located in the α3 helix of PrP and could form a salt bridge with R208. Structure prediction of E211Q revealed that mutation did not change the dynamics of PrP significantly. However, this mutation could affect the folding pathway of PrP and increase its ability to aggregate.67
I215V was discovered in AD and CJD cases from two Spanish patients with probably de novo cases.69–74 The CJD cases carried the M/M allele for codon 129, while the AD patient had the heterozygous M/V allele, suggesting that codon 129 might influence the pathogenic nature of mutation, but further studies are needed to verify.68 Three dimensional model on this mutation revealed that I215V (located in the middle of α3 helix) has similar thermodynamic stability as in the normal PrP.68 However, additional modeling showed altered interactions with AAs (such as V176, C179, Q172, and N173), located in the helix-2 or with the β strand-2 (V161). The decreased hydrophobicity due to the isoleucine → valine exchange could result in reduced hydrophobic interactions between β strand-2, helix-2, and helix-3.68
M232R has been reported rarely, and majority of cases occurred in Korea, China, and Japan.69–74 The disease progression associated with M232R could be rapid, typical CJD progression, or slow progression of dementia, with atypical symptoms.71,72 This mutation was also reported in a Japanese patient with dementia with Lewy bodies, which suggests that M232R might be involved in forms of progressive dementia other than CJD.73 However, pathogenic nature of M232R has been refuted by Beck et al, suggesting that the mutation might rather be an uncommon polymorphism rather than a disease causative mutation.22 The pathogenic mechanism of M232R, leading to neuronal degeneration, could be associated with impairments of GPI functions.72 M232R, M232T, and P238S are located on the GPI signal peptide of PrP, which is cleaved and replaced from the mature protein by the GPI anchor, followed by translocation into the ER.74 M232R and M232T might increase the stability of PrP and enhance its transport to the plasma membrane. The post-translation modification of PrP could be inserted into the lipid bilayer in c-transmembrane (ctm) orientation, resulting in neurotoxic effects.74,75
PrP contains five copies of octapeptide repeats in the N-terminus region. Insertions and deletions in these octapeptide repeat region could be associated with pathogenic phenotypes. However, single octapeptide deletions may be benign, since they were reported in healthy European and Asian populations.76–79 Double octapeptide deletion was observed in patients diagnosed with CJD and rapidly progressive dementia, but it was missing in a large number of healthy individuals.77–79 Insertions of 1–9 octapeptide repeats were associated with pathogenic phenotype, which can be similar to sCJD.77 Insertions of octapeptide repeats were usually associated with early disease onset, and amyloid plaques were also observed in patients with more than six repeats. Brain materials from these patients are quite transmittable. These insertions can form protease resistant form PrP-amyloid.79
Prion mutations and FFI
FFI is a genetically inherited disorder, caused by the D178N mutation, depending on the genotype of codon 129 (Table 1).80 The disease also has a sporadic form (sporadic fatal insomnia). Clinical phenotype of the disease can be sleeping disturbances, autonomic hyper activation, and motor impairment. Neuronal loss and astrogliosis were observed in the anterior medial thalamus and inferior olives, which were also spread to the cerebral cortex and cerebellum.80–82 D178N can be involved in both CJD and FFI. Clinical phenotype of D178N depends on the polymorphism of codon 129 (M/V exchange), since FFI was associated with the M allele, and CJD was associated with V allele.82,83 Distinct phenotypes were identified in patients carrying the mutation with the heterozygous M/V allele for codon 129.80 Mutation in FFI was described first by Lugaresi et al in 1986. FFI is a rapidly progressive disease, where clinical phenotypes could be insomia, dysautonomia and motor signs.84 Additional phenotypes of D178N-129M genotype could also be possible, such as cerebellar ataxia without insomnia.85 This mutation can reduce the thermodynamic stability of PrP by destroying the salt bridge between D178 and R164. In addition, D178 is located near the disulfide bridge of PrP and could interfere with it.86,87
Prion mutations, involved in GSS
Onset of GSS could be between 30 and 60 years of age and associated with slow disease progression. Symptoms were ataxia in cerebellum, gait abnormalities, cognitive decline, dysarthria, ocular dysmetria, sleeping disturbances, myoclonus, spastic paraparesis, parkinsonism, and hyporeflexia or areflexia in the lower extremities. In the brain of GSS patients, astrocytic microgliosis, amyloid plaques, and neurofibrillary tangles are possible, but without spongiform changes (Table 2).1
P102L mutation was found all over the world, including in American, British, Korean,88,89 Chinese (Taiwanese),90 and in Japanese91,92 familial and sporadic cases. Progressive cerebellar syndrome was the most important clinical symptom of the mutation, followed by dementia and pyramidal dysfunctions.93 P102L can be associated with clinical heterogeneity, since additional phenotypes were also reported, such as classical fCJD-like symptoms or rapidly progressive dementia. Cognitive decline can appear in the late disease stages. Disease duration of GSS could range from 3 months to 13 years.93–95 P102L mutation could allow the scrapie-templated formation of amyloid–prion complexes, without any special conditions (such as neutral pH or without any cofactors). Four lysine residues are located nearby P102 (lysine cluster), which may be a critical component in PrP. P102L was suggested to be involved in the neutralization of the lysine cluster, which could be critical in prion functions.96–98 Disturbances of lysine cluster could be critical for prion folding and PrPSc formation.96–98
P105L was associated with familial or de novo cases of GSS, but it was also reported in spastic paraparesis.99 This mutation was found in several Japanese patients and was discovered by Kitamoto et al99 in five patients with spastic gait disturbance and progressive dementia, but without cerebellar signs (such as myoclonus and periodic synchronous discharges). Several amyloid plaques were present in the cerebral and motor cortex of patients. In the frontal lobe, neuronal loss and severe gliosis were detected.99 Itoh et al reported P105L in GSS in a male patient, who developed his early symptoms of clumsiness of his right hand in his early 40s, followed by slowly progressive spastic paraparesis, ataxia, dysarthria, memory disturbance, and apraxia.100 PrP-reactive amyloid plaques were found in his cerebral cortex, and several amorphous deposits were observed in the deep cortical layers, associated with neuronal loss. The mutation was co-existed with the heterozygous form of PRNP M129V variant.100 Kubo et al101 reported a familial case of mutation, where the clinical phenotypes can be weakness in the lower limbs and spastic, wide-based gait, followed by dementia, spastic quadriplegia, and pseudobulbar palsy. Atrophy was reported at the frontal and temporal lobes.101 Disease progression started before 50 years of age.101 Yamada et al also reported the mutation in spastic paraparesis, with additional symptoms such as ataxia, myoclonus, and dementia. In the brain, PrP-positive amyloid plaques appeared with tau-positive inclusions.102 Iwasaki et al103 reported the mutation in a GSS patient with ataxia. However, this patient did not represent any symptoms of spastic paraparesis, but extrapyramidal signs (bradykinesia and resting tremor) were present, which were missing in the previous cases of P105L. These reports suggested that the clinical phenotype of PRNP P105L may be variable.103 Similar to P102L, P105L could also be involved in the neutralization of lysine cluster.96–98
A117V is associated with inherited prion diseases, where the clinical phenotype is GSS. The first case of mutation was discovered in a French family, but it was also reported in families from Germany and the UK.104,105 Main clinical symptoms were progressive cortical dementia and cerebellar ataxia, in which the degree can be variable. Additional phenotypes were also observed in affected patients, such as dysarthria and incoordination, parkinsonism, myoclonus, or AD-like clinical symptoms.104,105 This mutation was initially suggested not to generate transmissible prion disease. Asante et al revealed that disease could be transmitted by brain tissues from A117V patients to mice.107 Transmissible property of mutation was confirmed in vole models. Both voles and mice presented prion disease pathology and PrPSc.106–108 The pathogenic mechanism of A117V might be associated with the abnormal ctm region of PrP protein and could cause stress in the ER.106–109
G131V was reported in GSS cases, occurred in the late 30s or early 40s. It was first described by Panegyres et al in a male patient, without any family history. Clinical symptoms, such as tremor and apraxia developed in his early 40s.110 Impairment appeared in his visuospatial skills and spatial orientation. Personality also changed, for example, he presented aggressive behavior and died at 51 years of age. PrP analysis revealed proteinase-resistant PrP in his brain.110 The second case of mutation was observed in a Dutch GSS patient, with slowly progressing cognitive decline. Later, he also developed ataxia and parkinsonism.111 In the cerebellum, PrP-positive amyloid plaques were observed, and tau-positive aggregates and amyloid deposits were also found in several areas in the brain, such as in the cerebral cortex, striatum, hippocampal formation, and midbrain.111 Structure predictions revealed that G131V may not affect the thermodynamic stability of PrP but could enhance the flexibility of several residues (112–130). Mutation could also increase the length of short β sheet.112
V176G was discovered in 2013, in a 61-year-old GSS female patient from Australia. Clinical phenotypes were cerebellar ataxia, personality changes, and rapid progressive dementia. Spongiform changes, tau filaments, and amyloid plaques appeared in her brain.113 Western blot analyses revealed proteinase-resistant and low molecular weight PrP bands. The significance of amyloid deposits associated with this mutation remained unclear.113
H187R was identified in a family with classical type of GSS.114 Affected patients were suffered in cognitive decline, personality changes, ataxia in the limbs, and dysarthria.114,115 Later, it was reported in an American family with prion encephalopathy, where the symptoms were progressive dementia, ataxia, myoclonus, and seizures. The disease occurred at young ages, between 33 and 50 years. In the brain, “curly” PrP deposits appeared, but without amyloid plaques or spongiosis.116 Colucci et al reported the mutation (combined with homozygous V/V allele at codon 129) in a family with GSS, where proteinase K-resistant PrPSc, “curly” PrP, and plaques were present, but the degree of spongiform degeneration was low.116,117 H187R can induce the formation of abnormal PrPSc and its increased ability of oligomerization.118 In vitro studies revealed that H187R could decrease the stability of PrP, by disturbing the salt bridge between E196 and R156. In addition, mutation could result in loss of tertiary structure of PrP by destroying several contacts (eg, between β1–α1–β2 and α2–α3).37
F198S was discovered in GSS patients from Indiana kindred119 and was associated with cognitive decline, indicating generalized cerebral dysfunction and global dementia.120 Nonglycosylated proteinase-K-resistant PrPSc was detected in patients with F198S, which seemed to be unique.120 Mutation may result in structural instability of PrP.120,121 Cell studies revealed that mutant PrP could fold properly, but after denaturation, it may be unable to revert into its native state. Mutation may also change the glycosylation pattern of PrP.122
D202N was involved in atypical GSS (without spongiform changes) and AD-like phenotypes. PrP-positive amyloid deposits appeared in the forebrain and cerebellum of the affected patient, and neurofibrillary tangles were present in his cerebral cortex.123,124 Later, D202N was discovered in a Canadian patient, who carried the M/V allele for codon 129. Postmortem analyses revealed mild spongiform changes and focal tangles in the neocortex region. Mutation was also found in an American patient, with the V/V allele at codon 129. This patient had PSP-like symptoms, such as akinetic rigidity, gait and postural disturbances, hyperreflexia, and dementia.124,125 In 2013, the mutation was reported in probably a de novo GSS case (Caucasian female from Germany) with parkinsonism and gaze impairment. The patient was homozygous for the V/V allele for codon 129.123 High levels of total- and phospho-tau, neuron-specific enolase, and S100β were present in her CSF, but she was negative for 14-3-3 protein. How codon 129 allele could affect the mutation phenotypes remained unclear.123 Pathogenic nature of D202N was suggested to be associated with α3-helix in PrP. α3-Helix is an anatomic helix, resulted by the conserved capping box and by the ionic bond, located between E200 and K204, and the mutation might result in decreased stability in this complex.125
Q212P was discovered by Piccardo et al and was suggested to be involved in CJD without spongiform changes, where the 21–30 kDa isoforms of PrP were not prominent.126 NMR approaches were performed on the mutation, and it was revealed that it could result in significant changes in the β2–α2 loop region and in the C-terminal end of PrP protein. Mutation could be involved in the change of hydrophobic interactions between the β2–α2 loop by destroying the interaction between Y225 and M166, which are critical residues in the loop position.127 The mutation results in altered interactions, such as Y225, could form interactions with the residues in the α3 helix and also increase the flexibility of C-terminal part of PrP.127
Q217R was discovered in a Swedish family with GSS, where tau-positive pathology was found with amyloid aggregates at the periphery of PrP plaques.128 Woulfe et al128 described the mutation in a patient initially diagnosed with frontotemporal dementia (FTD). Predictions on the mutation revealed that Q217R was located in the 3rd transmembrane (TM) region and that it could result in increased polar contribution and bigger conformational changes.128
M232T was discovered in a Polish GSS case. Mutation carrier patient had PrP-immunopositive kuru, multicentric plaques, and diffuse PrP depositions in his brain.129 Plaques could be found in different areas in the brain, such as the cerebral cortex, hippocampus, and the deep subcortical nuclei.129 Pathogenic nature of mutation may be associated with the abnormalities in the GPI-SP.74,75
Prion mutations, involved in other clinical phenotypes
Several prion mutations presented atypical disease phenotypes, which could be defined neither as CJD nor as FFI or GSS (Table 3). Prion diseases could share overlap with other neurodegenerative diseases, for example, AD, FTD, and PD or primary progressive aphasia.130 Pathologically, AD, FTD, and CJD are all neurodegenerative diseases or conformational disorders caused by a common pathogenesis of the excessive accumulation of abnormal, insoluble proteins, including the accumulation abnormal proteins, such as Aβ in AD, tau in FTD, and PrPc in CJD.131
S17G was found in a female AD patient through PRNP analysis of Chinese AD and FTD patients.132 Proband presented episodic memory loss, and impairment in daily routines and personality changes (irritability, depression, and apathy). Myoclonic jerks, seizures, extrapyramidal, or upper motor neuron signs were missing in her. Brain imaging showed diffuse cortical atrophy, enlargement of the cerebral ventricle and cistern of the whole brain, especially in the frontotemporal lobe, and hippocampus.132 However, studies on mouse PrP revealed that N-terminal signal peptide could transport large hydrophilic proteins through the cell membrane. This peptide may play a role in the cellular transport of PrP too. In addition, this region has high tendency of β structure formation, which may also be important in prion dysfunctions.133
P39L appeared in frontotemporal lobar degeneration patients. Patients developed this disease in their late 60s or 70s. It was suggested that P39L may result in less-rigid N-terminus of PrP, and it could permit abnormal cell–protein interactions.134–136
G114V was initially discovered in a family from Uruguay, where the affected members were diagnosed with prion disease.137 Clinical phenotypes appeared at young ages and started with neuropsychiatric symptoms. The disease had had long duration in the later disease stage, and prominent pyramidal and extrapyramidal symptoms were appeared with dementia.137 This mutation was also discovered in a Chinese patient, who presented a sCJD-like phenotype, with progressive dementia, lethargy, and sleeping problems.138 Neurological screening revealed additional phenotypes, such as myoclonus in lower limbs and hyperreflexia. Proteinase K-resistant PrPSc appeared in several regions of the brain, especially in the gray matter.138 Family history was positive, and the clinical phenotypes and disease duration were similar to those in the Uruguayan family.138
Y145* was discovered in a Japanese patient with AD-type pathology.139 Her disease symptoms started before her 40s, and her family history was negative. In her brain, no spongioform changes were found, but senile plaques, kuru plaques, and neurofibrillary tangles were detected. Instead of amyloid peptides, PrP was found in the plaques. Truncated PrP appeared in the kuru plaques. Later, this patient was suspected to have GSS.139 Ghetti et al named this phenotype as PrP cerebral amyloid angiopathy (PrP-CAA).140 Y145* mutation was suggested to induce the misfolding of PrP into proteinase-resistant form of PrP.141
Q160* was discovered in patients with AD-type pathology. The first case of mutation was reported by Finckh et al, but no information on the clinical phenotype was available.23 Jayadev et al reported the mutation in a patient diagnosed with AD in her early 40s, with positive family history.142 The patient developed depression and short-term memory problems. Neurofibrillary tangles and senile plaques appeared in her, but these plaques were PrP-positive and amyloid-negative.142 The third case of mutation was reported by Guerreiro et al; the patient had similar symptoms and was clinically diagnosed with EOAD and with probable positive family history.143 Fong et al144 also reported this mutation in an atypical case of disease. Patient had slow progressive dementia, with impairment in language and behavior.144 Additional symptoms were orbitofrontal syndrome, cyclic diarrhea, and peripheral neuropathy in his late 20s. He was suspected of having behavioral variant FTD, but did not meet the criteria. Several of his family members developed early onset dementia or autonomic/peripheral neuropathy; however, penetrance of mutation was reduced.144
Y163* mutation discovered by Mead et al145 showed a unique phenotype of chronic diarrhea with autonomic neuropathy. Cognitive decline and seizures also appeared in the patients.145 Prion-positive amyloid deposits were found in the peripheral organs such as bowel and peripheral nerves. In the late disease stages, the prion deposits were reported in the cortical amyloid plaques with CAA and tauopathy.145 Pathogenic nature of mutation may be associated with the truncation protein or the missing GPI anchor.145
D178fs could result in a premature stop codon at codon 195 by a computed tomography (CT) deletion in codon 178.146 A patient developed cognitive impairment with pan-autonomic failure and sensory neuropathy with frequent vomiting and diarrhea in her late 20s. Tau and 1-4-33 and enolase levels were elevated in her CSF, with positive family history, since her mother and maternal grandfather also developed similar phenotypes.146
T183A was described first in a Brazilian family, diagnosed with dementia and frontotemporal clinical features, with spongiform changes, neuronal loss.147 The second case of mutation was characterized by dementia, cerebral atrophy, and hypometabolism.148 Later, ataxia in the cerebellum and electroencephalogram (EEG) abnormalities were also reported. Symptoms started in the early 40s of the patient and disease duration was 4 years. T183A mutation may affect the glycosylation sites in PrP, since mutation could result in the elimination of one of this site from the two consensus sites.147,148 T183A was suggested to reduce the stability of H2H3 subdomain, resulting in higher propensity for intra- and inter-β-sheets.149
R208C was discovered in a Chinese patient with slow progressive dementia in his 80s.130 Moderate cerebral atrophy was detected, and the patient was clinically diagnosed with AD, with no family history of the disease.130
Patient with Y226* was diagnosed with dementia, visual and acoustic hallucinations, and disease occurred at the age of 54 years.150,151 PrP deposits PrP-CAA, focal tau accumulations, and mild focal spongiosis were observed in her brain, and the CSF test for 1-4-33 was positive.150,151 Family history was probably positive, since her mother developed probable CJD. Structure predictions, performed by Kovač et al revealed that this mutation could break an α4 helix and that the reduced C-terminal could result in altered hydrophobic interactions of α3 helix and β2–α2 loop.152 The missing GPI anchor could also be important in this mutation.150–152
Q227* was found in a 42-year-old female patient, who was diagnosed with FTD and presented extrapyramidal signs.151 One of her aunts was affected with similar disease phenotypes. PrP-positive amyloid deposits and tangles were found without spongiosis in her brain.151 GSS-like phenotype was present with multicentric amyloid plaques and neurofibrillary tangles, without PrP-CAA, in the cerebral gray matter.151
Prion mutations with complicated pathogenic nature or protective mutations
Several variants were reported in PRNP gene, whose pathogenic nature was not clarified (Table 4). The most common and well-known variants are M129V and E219K.153 Additional mutations were discovered, which were not confirmed as disease-associated variants. In addition, G127V mutation was suggested as protective variant against kuru.154
M129V and E219K are relatively common polymorphisms of PRNP gene, and their pathogenic nature is complicated. They were suggested as neutral or protective variants, but could also be risk- or disease-modifying factors for different disorders.155 M129V was suggested to play a role in sCJD, iatrogenic CJD (iCJD), and classical CJD (cCJD).155 Homozygous (M/M) form of mutation was associated with increased risk of sCJD and vCJD in Caucasians, but not in Asians. In the UK, vCJD cases were described with M129V homo- or heterozygous exchange. However, patients with M/V allele might have a longer incubation period than those with V/V.155 No correlation was found between PRNP codon 129 and sporadic AD or vascular dementia in Korea.156–158 Protease-sensitive prionopathy (PSPr), a novel prion disease, whose neuropathological changes could be minimal gliosis, microplaques (cerebellum), PrP aggregates, and/or spongiform changes.159 PRNP codon 129 might be involved in PSPr, since patients were described with VV allele.160–162 Later, PSPr cases were found in V/V, M/V or M/M genotypes, and significant clinical differences were found in clinical symptoms, age of onset, and disease duration. Since proteinase-K sensitivity for PrPSc was lower in case of M/V or M/M genotype than in V/V, name of the disease was changed to variable PSPr.161,162 Multiple system atrophy (MSA) is a neurological disease, characterized by dysfunctions in the extrapyramidal, pyramidal, cerebellar system. V/V allele at codon 129 was associated with the disorder, suggesting that PrP could be involved in MSA.163 E219K was reported in 6% of healthy Japanese individuals. Japanese and Korean studies suggested that E219K on the PRNP gene might be protective against sCJD.164–166 However, PRNP E219K may be involved in vCJD in the UK. Data from mouse experiments suggest that mice with the heterozygous E219K allele have longer incubation time to vCJD than those with homozygous K219 genotype. The heterozygous E219K allele might not protect against vCJD; in addition, it could be associated with vCJD.164–167
G127S was found in a Malaysian patient, having epilepsy and learning difficulties. However, since other family members with similar symptoms were negative for the mutation, these phenotypes might not be associated with G127S. It is unknown, how G127S could affect the prion conformation. This mutation is at the same location like G127V, which could be protective against kuru.168
G127V is located in the highly conserved region of PrP, and at the kuru-affected areas, mutation occurred frequently. In the mutant protein, valine was suggested to increase the stability of PrP.154 G127V may be the result of positive evolutionary selection during the kuru epidemics. Mouse experiments revealed that the mutation could inhibit the prion propagation. However, additional studies are needed to understand the structural basis, associated with the protective mechanism of G127V.169
D167G was suggested as a susceptibility factor for sCJD, but no detailed reports are available on the variant.170 A D167N was also found in the same residue (D167), but its pathogenic nature was not established yet either.171 Proband patient developed memory impairment and personality changes such as emotional instability and aggressive behavior. Later, incontinence, severe non-fluent expressive dysphasia, and impairment in movement also appeared in him. MRI revealed atrophy in the frontotemporal cortex. Family history was unknown, since her mother also carried the same mutation, but did not show any kind of disease phenotype.171
N171S was found to be prevalent in mesial temporal lobe epilepsy related to hippocampal sclerosis (MTLE-HS), especially with the epileptogenesis.172 However, Coimbra et al reported that N171S (and M129V) might not influence the cognitive performance of MTLE-HS patients.173
P238S was first described by Windl et al, but the disease phenotype associated with the mutation is unknown.174 P238S is the third mutation, described in the GPI sequence. Studies showed that P238S did not reveal any impairment of GPI-anchor mechanisms.74,75 However, P238S seemed to increase the cleavage of GPI sequence from mature PrP, compared with the wild-type PrP. In addition, GPI-SP degradation was prevented due to mutation, and the signal peptide was accumulated in the cells, which could result in neurodegeneration.74,75
Diagnosis, markers, and diagnostic criteria for prion diseases
Prion diseases are different from the infectious, virus-, or bacteria-associated diseases, since PrP has no DNA or RNA.175 In addition, the misfolded PrP cannot be recognized by the immune system, and no immune reaction and inflammation occur in the infected host.161 Misfolding and aggregation of PrP could cause different fatal neurodegenerative disorders. Disease diagnosis is quite complicated, and several patients are diagnosed with prion disease, when severe clinical symptoms appear. Definite diagnosis can be performed only after histopathological analysis and brain biopsy/autopsy. However, several new diagnostic tools are under development, which could provide more accurate disease diagnosis.175,176
The PrP detection is critical in prion disease diagnosis. The PrPSc could be in the peripheral tissues and body fluids, and it was suggested that they might also be useful biomarkers for the disease in the presymptomatic stage. Blood-based PrP can be approached with protein misfolding cyclic amplification, matrix capture technology and immunoassay, or multiple assays.177
14-3-3 Proteins are abundant proteins in brain. They are adaptor proteins, which bind specific substrates through their phosphor-serine or threonine motifs. Diverse functions of 14-3-3 proteins were described as follows: they can be involved in several brain functions, such as development, migration, and survival of neurons. They could also be involved in the regulation of ion channels. The 14-3-3 proteins can play a role in different neurodegenerative diseases, including CJD.178 14-3-3 Protein in CSF was found as the useful reliable marker for CJD diagnosis. Total 14-3-3 levels may be higher in patients, diagnosed with definite or probable CJD, compared with the patients with other neurodegenerative diseases. 14-3-3 Protein may play a role in CJD pathogenesis. In case of fCJD, several mutations such as E200K and V210I were also associated with the elevated levels of 14-3-3 protein. However, this protein is less prominent in GSS or FFI patients, and it may not be useful for the early stages of iCJD.179,180 CSF-tau was also suggested as a marker for prion diseases. Ratio of pTau and tTau might be less effective marker than 14-3-3 protein. Combination of tau and 14-3-3 may improve the diagnosis of the prion diagnosis.181–183
The World Health Organization (WHO) and the Center of Disease Control and Prevention (https://www.cdc.gov/prions/cjd/diagnostic-criteria.html) established the diagnostic criteria for cCJD forms, including genetic CJD, sCJD, or iCJD. Psychical tests, brain biopsy, and imaging methods are available for the CJD diagnosis. Definitely, sCJD could be confirmed by neuropathological test and immunohistochemistry, when proteinase-resistant PrPSc and/or scrapie-associated fibrils are present. Probable CJD patients present rapid progressive dementia and at least of four different symptoms, including myoclonus, visual cerebellar signs, pyramidan/extrapyramidal signs, or akinetic mutism. EEG sign could present periodic sharp wave complexes, and CSF tests should be positive for 14-3-3 protein, in patients whose disease duration is <2 years. Neuroimaging technologies play an important role in the neurodegenerative disease, including prion disease diagnosis.180,184–187 Imaging tools such as CT, MRI, and PET are currently used in the differential diagnosis of different types of prion disorders, and imaging should reflect the alterations in the gray and white matter. In probable CJD patients, high signal abnormalities could be seen in caudate nucleus and/or putamen.180,184–187 In possible CJD, progressive dementia may be present with at least two clinical symptoms described above. However, patients may be negative for EEG, CSF test protein, or imaging, and disease duration should be >2 years. In genetic CJD, patients should have disease-specific PRNP mutation and present the signs of definite or probable CJD. iCJD could be possible if patients had history of exposure to some disease risk factor, for example, dura mater graft transplantation and blood transfusion. vCJD could be emerged from bovine spongiform encephalopathy (BSE) contaminated food consumption, such as beef or beef products. The vCJD patients could have diffusely slow EEG and be negative for 14-3-3 protein. Spongioform changes could be dense with possible dense amyloid plaques.180,184–187 Diagnosis of GSS is based on the combination of different parameters, including multiple amyloid plaques in brain, pathogenic disease causing mutations in PRNP gene, and different specific disease symptoms such as ataxia, memory dysfunctions, abnormal eye movement, or spasticity.189 FFI diagnosis is based on different parameters: patients should have D178N mutation with M/M genotype at codon 129, with positive family history. Patients should have abnormal sleep patterns and thalamic hypometabolism on PET scan, and different symptoms such as dementia, mood changes, ataxia, and sleep disturbances.190
Conclusion
Prion diseases could occur mostly (~85%–90%) because of unknown risk factors, while about 10%–15% of the diseases were caused by genetic mutations. PRNP was established as the only causative gene for different prion diseases, since several pathogenic and possibly pathogenic variants were discovered in its coding region. These mutations were associated with different clinical phenotypes of neurodegenerative diseases, such as CJD, GSS, FFI, or other types of dementia. The main problem with genetic prion diseases is that the phenotype could be heterogeneous. M129V and E219K are the mutations with the most complicated pathogenic nature, since several studies revealed that they might be disease modifiers, risk factors for vCJD, and protective against sCJD. Pathogenic nature of V180I and M232R was refuted, since they also appeared in healthy controls.1,171,190 The aim of this study was to introduce prion mutations, and their clinical phenotypes and disease phenotypes were also summarized. In addition, their possible pathogenic mechanisms were also discussed, based on in silico or in vitro studies. Genetic prion disease could occur in different ages: several patients present CJD symptoms at young ages (40s or even earlier), while others present disease phenotypes at later lifetime (in their 70s or 80s). There may be several factors that could affect the age of onset, for example, possible additional disease modifier genes, environmental factors, or lifestyle.188
Similar to AD and FTD, prion diseases are based on the aggregation of abnormal protein aggregates. The abnormal protein assembly could result in a cascade of neurodegenerative pathways.193 In addition, pathological overlap may occur between these disorders. These findings suggested that there may be a common disease pathway between different neurodegenerative diseases; however, the exact mechanism remained unclear yet.191 Normal PrP could interact with several proteins in brain and play a role in several mechanisms including amyloid β cleavage. Amyloid peptides could interact with PrP, which may be important in neurodegenation and in the pathology of both prion disease and AD.192
In PRNP gene, several variants were found, whose pathogenic nature was not established yet. The Exome Aggregation Consortium Browser (Exome Aggregation Consortium [ExAC, http://exac.broadinstitute.org/]) is an exome sequencing database, which was established in early 2015. In this project, genomes of >60,000 unaffected and unrelated individuals were screened, for the disease-associated genes and candidates.193 This database could be useful in disease-association and population genetics study. Beyond the published mutations, several novel missense and silent variants were discovered in these individuals in PRNP gene. Further investigations are needed on the novel missense variants, if they could be associated with any kinds of disease progression or if the carrier individuals would develop prion disease in the future. Several novel variants represented low frequency (singleton mutation), which suggested that their clinical significance might not be ignored in the future. In addition, the 23andMe Inc. (Mountain View, CA, USA) studied the prion mutations more extensively. It compared the mutations in 16,025 patients affected with prion disease, 60,706 population controls, and 531,575 individuals genotyped by 23andME Inc. This study may re-evaluate the role of PRNP mutations in disease progression, since some missense variants may be benign, while several others have a penetrance from f<0.1% to 100%. This analysis revealed four mutations (V180I, R208H, V210I, and M232R) in controls and suggested that they could not be categorized as benign or Mendelian variants. These mutations may have low penetrance and could occupy a risk continuum. These approaches in prion diseases may provide an estimation for risk of asymptomatic individuals, who carried incompletely penetrant mutations in PRNP. Large reference databases could be helpful in genetic counseling, diagnosis, and also in therapies.193
The most important issue with prion diseases is that the disease remains unrecognized. Prion disorders are clinically heterogeneous and share several common features with other neurodegenerative diseases, such as possible genetic background, assembly of misfolded proteins, or neuronal loss.194 These patients were later confirmed by autopsy or by postmortem analyses of having CJD.195 For early dementia patients, a more complex genetic screening is needed due to the pathologic similarities between the early stages of different disorders. There is no effective treatment against the neurodegenerative disorders, but the possible therapeutic strategies might be more useful before the pre-symptomatic stage. Combination of protein biomarkers, imaging, and genetic screening could be useful for more accurate diagnosis of neurodegenerative diseases. We believe that genetic counseling and genetic screening could improve the accuracy of differential diagnosis. Recently, next generation sequencing technologies have been developed, which could provide faster, more accurate genetic profiling for patients with different types of dementia.195–198
Acknowledgments
This research was supported by a National Research Foundation (NRF) of Korea Grants awarded by the Korean government (Ministry of Education, Science & Technology [MEST], no. 2017R1A2B4012636) and by the MD-PhD grant (FRD2016-19) through Gachon University GilYa Lee Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
Brown K, Mastrianni JA. The prion diseases. J Geriatr Psychiatry Neurol. 2010;23:277–298. | ||
Imran M, Mahmood S. An overview of animal prion diseases. Virol J. 2011;8:493. | ||
Kübler E, Oesch B, Raeber AJ. Diagnosis of prion diseases. Br Med Bull. 2003;66:267–279. | ||
Puckett C, Concannon P, Casey C, Hood L. Genomic structure of the human prion protein gene. Am J Hum Genet. 1991;49:320–329. | ||
Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. PNAS. 2001;98:14955–14960. | ||
Steinert JR. Prion protein as a mediator of synaptic transmission. Commun Integr Biol. 2015;8:e1063753. | ||
Shen L, Ji HF. Mutation directional selection sheds light on prion pathogenesis. Biochem Biophys Res Commun. 2011;410:159–163. | ||
Roucou X, Gains M, LeBlanc AC. Neuroprotective functions of prion protein. J Neurosci Res. 2004;75:153–161. | ||
Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15:34. | ||
Castle AR, Gill AC. Physiological functions of the cellular prion protein. Front Mol Biosci. 2017;4:19. | ||
van Rheede T, van Rheede T, Smolenaars MM, Madsen O, de Jong WW. Molecular evolution of the mammalian prion protein. Mol Biol Evol. 2003;20:111. | ||
Lloyd S, Mead S, Collinge J. Genetics of prion disease. Top Curr Chem. 2011;305:1–22. | ||
Mackenzie G, Will R. Creutzfeldt-Jakob disease: recent developments. F1000Res. 2017;6:2053. | ||
Chasseigneaux S, Haik S, Laffont-Proust I, et al. V180I mutation of the prion protein gene associated with atypical PrPSc glycosylation. Neurosci Lett. 2006;408:165–169. | ||
Iwasaki Y, Iwasaki Y, Mori K, et al. An autopsied case of V180I Creutzfeldt-Jakob disease presenting with panencephalopathic-type pathology and a characteristic prion protein type. Neuropathology. 2011;31:540–548. | ||
Nixon R, Camicioli R, Jamison K, Cervenakova L, Mastrianni JA. The PRNP-V180I mutation is associated with abnormally glycosylated PrPCJD and Intracellular PrP accumulations. Presented at XIVth International Congress of Neuropathology Scientific Programme. Brain Pathol. 2000;10:670. | ||
Yang TI, Jung DS, Ahn BY, et al. Familial Creutzfeldt-Jakob disease with V180I mutation. J Korean Med Sci. 2010;25:1097–1100. | ||
Mutsukura K, Satoh K, Shirabe S, et al. Familial Creutzfeldt-Jakob disease with a V180I mutation: comparative analysis with pathological findings and diffusion-weighted images. Dement Geriatr Cogn Disord. 2009;28:550–557. | ||
Jin K, Shiga Y, Shibuya S, et al. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology. 2004;62:502–555. | ||
Shi Q, Shen XJ, Zhou W, et al. Rare V180I mutation in PRNP gene of a Chinese patient with Creutzfeldt-Jakob disease. Prion. 2014;8:411–444. | ||
van der Kamp MW, Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J Mol Biol. 2010;404:732–748. | ||
Beck J, Collinge J, Mead S. Prion protein gene M232R variation is probably an uncommon polymorphism rather than a pathogenic mutation. Brain. 2012;135(Pt 2):e209. | ||
Finckh U, Müller-Thomsen T, Mann U, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000;66:110–117. | ||
Roeber S, Grasbon-Frodl EM, Windl O, et al. Evidence for a pathogenic role of different mutations at codon 188 of PRNP. PLoS One. 2008;3(5):e2147. | ||
Shi Q, Gao C, Zhou W, et al. Human prion disease with a T188K mutation in Chinese: a case report. Cases J. 2009;2:7820. | ||
Chen C, Shi Q, Zhou W, et al. Clinical and familial characteristics of eight Chinese patients with T188K genetic Creutzfeldt-Jakob disease. Infect Genet Evol. 2013;14:120–124. | ||
Guo J, Ning L, Ren H, Liu H, Yao X. Influence of the pathogenic mutations T188K/R/A on the structural stability and misfolding of human prion protein: insight from molecular dynamics simulations. Biochim Biophys Acta. 2012;1820:116–123. | ||
Tartaglia MC, Thai JN, See T, et al. Pathologic evidence that the T188R mutation in PRNP is associated with prion disease. J Neuropathol Exp Neurol. 2010;69:1220–1227. | ||
Collins S, Boyd A, Fletcher A, et al. Novel prion protein gene mutation in an octogenarian with Creutzfeldt-Jakob disease. Arch Neurol. 2000;57:1058–1063. | ||
Peoc’h K, Manivet P, Beaudry P, et al. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat. 2000;15:482. | ||
Tumani H, Windl O, Kretzschmar HA, Ludolph AC. Clinically atypical CJD: diagnostic relevance of cerebrospinal fluid markers and molecular genetic analysis? Dtsch Med Wochenschr. 2002;127:318–320. | ||
Clerici F, Elia A, Girotti F, et al. Atypical presentation of Creutzfeldt-Jakob disease: the first Italian case associated with E196K mutation in the PRNP gene. J Neurol Sci. 2008;275:145–147. | ||
Béjot Y, Osseby GV, Caillier M, Moreau T, Laplanche JL, Giroud M. Rare E196K mutation in the PRNP gene of a patient exhibiting behavioral abnormalities. Clin Neurol Neurosurg. 2010;112:244–247. | ||
Eigenbrod S, Frick P, Giese A, Schelzke G, Zerr I, Kretzschmar HA. Comprehensive neuropathologic analysis of genetic prion disease associated with the E196K mutation in PRNP reveals phenotypic heterogeneity. J Neuropathol Exp Neurol. 2011;70:192–200. | ||
Schelzke G, Eigenbrod S, Romero C, et al. Genetic prion disease with codon 196 PRNP mutation: clinical and pathological findings. Neurobiol Aging. 2011;32:756.e1–756.e9. | ||
Shi Q, Chen C, Song XN, et al. A Chinese Creutzfeldt-Jakob disease patient with E196K mutation in PRNP. Prion. 2011;5:117–120. | ||
Hadži S, Ondračka A, Jerala R, et al. Pathological mutations H187R and E196K facilitate subdomain separation and prion protein conversion by destabilization of the native structure. FASEB J. 2015;29:882–893. | ||
Shi Q, Zhou W, Chen C, et al. Rare E196A mutation in PRNP gene of 3 Chinese patients with Creutzfeldt-Jacob disease. Prion. 2016;10:331–337. | ||
Antoine JC, Laplanche JL, Mosnier JF, Beaudry P, Chatelain J, Michel D. Demyelinating peripheral neuropathy with Creutzfeldt-Jakob disease and mutation at codon 200 of the prion protein gene. Neurology. 1996;46:1123–1127. | ||
Jeong BH, Ju WK, Huh K, et al. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J Korean Med Sci. 1998;13:234–240. | ||
Chapman J, Brown P, Goldfarb LG, Arlazoroff A, Gajdusek DC, Korczyn AD. Clinical heterogeneity and unusual presentations of Creutzfeldt-Jakob disease in Jewish patients with the PRNP codon 200 mutation. J Neurol Neurosurg Psychiatry. 1993;56:1109–1112. | ||
Korczyn AD. Creutzfeldt-Jakob disease among Libyan Jews. Eur J Epidemiol. 1991;7:490–493. | ||
Kim MO, Cali I, Oehler A, et al. Genetic CJD with a novel E200G mutation in the prion protein gene and comparison with E200K mutation cases. Acta Neuropathol Commun. 2013;1:80. | ||
Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sönnichsen FD. Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. J Biol Chem. 2000;275:33650–33654. | ||
Kovacs GG, Molnár K, Keller E, Botond G, Budka H, László L. Intraneuronal immunoreactivity for the prion protein distinguishes a subset of E200K genetic from sporadic Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol. 2012;71:223–232. | ||
Kim MO, Cali I, Oehler A, et al. Genetic CJD with a novel E200G mutation in the prion protein gene and comparison with E200K mutation cases. Acta Neuropathol Commun. 2013;1:80. | ||
Jeong BH, Jeon YC, Lee YJ, et al. Creutzfeldt-Jakob disease with the V203I mutation and M129V polymorphism of the prion protein gene (PRNP) and a 17 kDa prion protein fragment. Neuropathol Appl Neurobiol. 2010;36:558–563. | ||
Shi Q, Chen C, Wang XJ, et al. Rare V203I mutation in the PRNP gene of a Chinese patient with Creutzfeldt-Jakob disease. Prion. 2013;7:259–262. | ||
Komatsu J, Sakai K, Hamaguchi T, Sugiyama Y, Iwasa K, Yamada M. Creutzfeldt-Jakob disease associated with a V203I homozygous mutation in the prion protein gene. Prion. 2014;8:336–338. | ||
van der Kamp MW, Daggett V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J Mol Biol. 2010;404:732–748. | ||
Mastrianni JA, Iannicola C, Myers RM, DeArmond S, Prusiner SB. Mutation of the prion protein gene at codon 208 in familial Creutzfeldt-Jakob disease. Neurology. 1996;47:1305–1312. | ||
Mitrova E, Belay G, Zakova D, Sterlzer M. The first case of genetic Creutzfeldt-Jakob disease with the rare mutation R208H, methionine/valine heterozygous at codon 129 of the prion protein gene. Clin Med J. 2015;1:101–105. | ||
Roeber S, Krebs B, Neumann M, et al. Creutzfeldt-Jakob disease in a patient with an R208H mutation of the prion protein gene (PRNP) and a 17-kDa prion protein fragment. Acta Neuropathol. 2005;109:443–448. | ||
Basset-Leobon C, Uro-Coste E, Peoc’h K, et al. Familial Creutzfeldt-Jakob disease with an R208H-129V haplotype and Kuru plaques. Arch Neurol. 2006;63:449–452. | ||
Matěj R, Kovacs GG, Johanidesová S, et al. Genetic Creutzfeldt-Jakob disease with R208H mutation presenting as progressive supranuclear palsy. Mov Disord. 2012;27:476–479. | ||
Vita MG, Gaudino S, Di Giuda D, et al. R208H-129VV haplotype in the prion protein gene: phenotype and neuroimaging of a patient with genetic Creutzfeldt-Jakob disease. J Neurol. 2013;260:2650–2652. | ||
Chen C, Shi Q, Tian C, et al. The first Chinese case of Creutzfeldt-Jakob disease patient with R208H mutation in PRNP. Prion. 2011;5:232–234. | ||
Bamdad K, Naderi-Manesh H. Contribution of a putative salt bridge and backbone dynamics in the structural instability of human prion protein upon R208H mutation. Biochem Biophys Res Commun. 2007;364:719–724. | ||
Ripoll L, Laplanche JL, Salzmann M, et al. A new point mutation in the prion protein gene at codon 210 in Creutzfeldt-Jakob disease. Neurology. 1993;43:1934–1938. | ||
Pocchiari M, Salvatore M, Cutruzzolá F, et al. A new point mutation of the prion protein gene in Creutzfeldt-Jakob disease. Ann Neurol. 1993;34:802–807. | ||
Furakawa H, Kitamoto T, Hashiguchi H, Tateishi J. A Japanese case of Creutzfeldt-Jakob disease with a point mutation in the prion protein gene at codon 210. J Neurol Sci. 1996;141:120–122. | ||
Shyu W, Hsu Y, Kao M, Tsao W. Panencephalitic Creutzfeldt-Jakob disease in a Chinese family: unusual presentation with PrP codon 210 mutation and identification by PCR-SSCP. J Neurol Sci. 1996;143:176–180. | ||
Tajima Y, Satoh C, Mito Y, Kitamoto T. Creutzfeldt-Jakob disease with a codon 210 mutation: first pathological observation in a Japanese patient. Intern Med. 2014;53:483–487. | ||
Imbriani P, Marfia GA, Marciani MG, et al. Heidenhain variant in two patients with inherited V210I Creutzfeldt-Jakob disease. Int J Neurosci. 2016;126:381–383. | ||
Biljan I, Ilc G, Giachin G, et al. Toward the molecular basis of inherited prion diseases: NMR structure of the human prion protein with V210I mutation. J Mol Biol. 2011;412:660–673. | ||
Ladogana AR, Almonti S, Petraroli R, et al. Mutation of the PRNP gene at codon 211 in familial Creutzfeldt-Jakob disease. Am J Med Genet. 2001;103:133–137. | ||
Peoc’h K, Levavasseur E, Delmont E, et al. Substitutions at residue 211 in the prion protein drive a switch between CJD and GSS syndrome, a new mechanism governing inherited neurodegenerative disorders. Hum Mol Genet. 2012;21:5417–5428. | ||
Muñoz-Nieto M, Ramonet N, López-Gastón JI, et al. A novel mutation I215V in the PRNP gene associated with Creutzfeldt-Jakob and Alzheimer’s diseases in three patients with divergent clinical phenotypes. J Neurol. 2013;260:77–84. | ||
Nishimoto YS, Ito D, Suzuki S, Shimizu T, Kitamoto T, Suzuki N. Slow-progressive ataxia with a methionine-to-arginine point mutation in codon 232 in the prion protein gene (PRNP). Clin Neurol Neurosurg. 2011;113:696–698. | ||
Choi BY, Kim SY, Seo SY, et al. Mutations at codons 178, 200-129, and 232 contributed to the inherited prion diseases in Korean patients. BMC Infect Dis. 2009;9:132. | ||
Kon T, Miki Y, Arai A, et al. Creutzfeldt-Jakob disease with homozygous M232R mutation: A case report. J Neurol Sci. 2015;352(1–2):108–109. | ||
Shiga Y, Satoh K, Kitamoto T, et al. Two different clinical phenotypes of Creutzfeldt-Jakob disease with a M232R substitution. J Neurol. 2007;254:1509–1517. | ||
Koide T, Ohtake H, Nakajima T, et al. A patient with dementia with Lewy bodies and codon 232 mutation of PRNP. Neurology. 2002;59:1619–1621. | ||
Shimizu H, Yamada M, Matsubara N, et al. Creutzfeldt–Jakob disease with an M232R substitution: report of a patient showing slowly progressive disease with abundant plaque-like PrP deposits in the cerebellum. Neuropathology. 2009;29:735–743. | ||
Guizzunti G, Zurzolo C. The fate of PrP GPI-anchor signal peptide is modulated by P238S pathogenic mutation. Traffic. 2014;15:78–93. | ||
Gu Y, Singh A, Bose S, Singh N. Pathogenic mutations in the glycosylphosphatidylinositol signal peptide of PrP modulate its topology in neuroblastoma cells. Mol Cell Neurosci. 2008;37:647–656. | ||
Beck JA, Mead S, Campbell TA, et al. Two-octapeptide repeat deletion of prion protein associated with rapidly progressive dementia. Neurology. 2001;57:354–356. | ||
Capellari S, Parchi P, Wolff BD, et al. Creutzfeldt-Jakob disease associated with a deletion of two repeats in the prion protein gene. Neurology. 2002;59:1628–1630. | ||
Moore RA, Herzog C, Errett J, et al. Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein Sci. 2006;15:609–619. | ||
Petersen RB, Parchi P, Richardson SL, Urig CB, Gambetti P. Effect of the D178N mutation and the codon 129 polymorphism on the metabolism of the prion protein. J Biol Chem. 1996;271:12661–12668. | ||
Marcon G, Indaco A, Di Fede G, et al. Panencephalopathic Creutzfeldt-Jakob disease with distinct pattern of prion protein deposition in a patient with D178N mutation and homozygosity for valine at codon 129 of the prion protein gene. Brain Pathol. 2014;24:148–151. | ||
Mead S, Joiner S, Desbruslais M, et al. Creutzfeldt-Jakob disease, prion protein gene codon 129VV, and a novel PrPSc type in a young British woman. Arch Neurol. 2007;64:1780–1784. | ||
Harder A, Gregor A, Wirth T, et al. Early age of onset in fatal familial insomnia. Two novel cases and review of the literature. J Neurol. 2004;251:715–724. | ||
Lugaresi E, Medori R, Montagna P, et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med. 1986;315:997–1003. | ||
Goldfarb LG, Petersen RB, Tabaton M, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–808. | ||
Choi BY, Kim SY, Seo SY, et al. Mutations at codons 178, 200-129, and 232 contributed to the inherited prion diseases in Korean patients. BMC Infect Dis. 2009;9:132. | ||
Swietnicki W, Petersen RB, Gambetti P, Surewicz WK. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J Biol Chem. 1998;273:31048–31052. | ||
Hsiao K, Dlouhy SR, Farlow MR, et al. Mutant prion proteins in Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Nat Genet. 1992;1:68–71. | ||
Park MJ, Jo HY, Cheon SM, Choi SS, Kim YS, Kim JW. A case of Gerstmann-Sträussler-Scheinker disease. J Clin Neurol. 2010;6:46–50. | ||
Chi NF, Lee YC, Lu YC, Wu HM, Soong BW. Transmissible spongiform encephalopathies with P102L mutation of PRNP manifesting different phenotypes: clinical, neuroimaging, and electrophysiological studies in Chinese kindred in Taiwan. J Neurol. 2010;257:191–197. | ||
Takazawa T, Ikeda K, Ito H, et al. A distinct phenotype of leg hyperreflexia in a Japanese family with Gerstmann-Sträussler-Scheinker syndrome (P102L). Intern Med. 2010;49:339–342. | ||
Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in Gerstmann-Sträussler syndrome with mutant PrP plaques. Biochem Biophys Res Commun. 1993;192:525–531. | ||
Wadsworth JD, Joiner S, Linehan JM, et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129:1557–1569. | ||
Hsiao K, Baker HF, Crow TJ, et al. Linkage of a prion protein missense variant to Germann-Sträsler syndrome. Nature. 1989;338:342–345. | ||
Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H. Mutations of the prion protein gene phenotypic spectrum. J Neurol. 2002;249:1567–1582. | ||
Kraus A, Raymond GJ, Race B, et al. PrP P102L and nearby lysine mutations promote spontaneous in vitro formation of transmissible prions. J Virol. 2017;91:e01276-17. | ||
Kraus A, Anson KJ, Raymond LD, et al. Prion protein prolines 102 and 105 and the surrounding lysine cluster impede amyloid formation. J Biol Chem. 2015;290:21510–21522. | ||
Groveman BR, Kraus A, Raymond LD, et al. Charge neutralization of the central lysine cluster in prion protein (PrP) promotes PrP(Sc)-like folding of recombinant PrP amyloids. J Biol Chem. 2015;290:1119–1128. | ||
Kitamoto Y, Amano N, Terao Y, et al. A new inherited prion disease (PrP-P105L mutation) showing spastic paraparesis. Ann Neurol. 1993;34:808–813. | ||
Itoh Y, Yamada M, Hayakawa M, et al. A variant of Gerstmann-Sträussler-Scheinker disease carrying codon 105 mutation with codon 129 polymorphism of the prion protein gene: a clinicopathological study. J Neurol Sci. 1994;127:77–86. | ||
Kubo M, Nishimura T, Shikata E, Kokubun Y, Takasu T. A case of variant Gerstmann-Sträussler-Scheinker disease with the mutation of codon P105L. Rinsho Shinkeigaku. 1995;35:873–877. | ||
Yamada M, Itoh Y, Inaba A, et al. An inherited prion disease with a PrP P105L mutation: clinicopathologic and PrP heterogeneity. Neurology. 1999;53:181–188. | ||
Iwasaki Y, Kizawa M, Hori N, Kitamoto T, Sobue G. A case of Gerstmann-Sträussler-Scheinker syndrome with the P105L prion protein gene mutation presenting with ataxia and extrapyramidal signs without spastic paraparesis. Clin Neurol Neurosurg. 2009;111:606–660. | ||
Mallucci GR, Campbell TA, Dickinson A, et al. Inherited prion disease with an alanine to valine mutation at codon 117 in the prion protein gene. Brain. 1999;122:1823–1837. | ||
Tranchant C, Sergeant N, Wattez A, Mohr M, Warter JM, Delacourte A. Neurofibrillary tangles in Gerstmann-Sträussler-Scheinker syndrome with the A117V prion gene mutation. J Neurol Neurosurg Psychiatry. 1997;63:240–246. | ||
Mastrianni JA, Curtis MT, Oberholtzer JC, et al. Prion disease (PrP-A117V) presenting with ataxia instead of dementia. Neurology. 1995;45:2042–2050. | ||
Asante EA, Linehan JM, Smidak M, et al. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog. 2013;9:e1003643. | ||
Pirisinu L, Di Bari MA, D’Agostino C, et al. Gerstmann-Sträussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep. 2016;6:20443. | ||
Nonno R, Angelo Di Bari M, Agrimi U, Pirisinu L. Transmissibility of Gerstmann-Sträussler-Scheinker syndrome in rodent models: New insights into the molecular underpinnings of prion infectivity. Prion. 2016;10:421–433. | ||
Panegyres PK, Toufexis K, Kakulas BA, et al. A new PRNP mutation (G131V) associated with Gerstmann-Sträussler-Scheinker disease. Arch Neurol. 2001;58:1899–1902. | ||
Jansen C, Parchi P, Capellari S, et al. A second case of Gerstmann-Sträussler-Scheinker disease linked to the G131V mutation in the prion protein gene in a Dutch patient. J Neuropathol Exp Neurol. 2011;70:698–702. | ||
Lundberg P, Magzoub M, Lindberg M, et al. Cell membrane translocation of the N-terminal (1-28) part of the prion protein. Biochem Biophys Res Commun. 2002;299:85–90. | ||
Simpson M, Johanssen V, Boyd A, et al. Unusual clinical and molecular-pathological profile of gerstmann-Sträussler-Scheinker disease associated with a novel PRNP mutation (V176G). JAMA Neurol. 2013;70:1180–1185. | ||
Cervenáková L, Buetefisch C, Lee HS, et al. Novel PRNP sequence variant associated with familial encephalopathy. Am J Med Genet. 1999;88:653–656. | ||
Bütefisch CM, Gambetti P, Cervenakova L, Park KY, Hallett M, Goldfarb LG. Inherited prion encephalopathy associated with the novel PRNP H187R mutation: a clinical study. Neurology. 2000;55:517–522. | ||
Colucci M, Moleres FJ, Xie ZL, et al. Gerstmann-Sträussler-Scheinker: a new phenotype with “curly” PrP deposits. J Neuropathol Exp Neurol. 2006;65:642–651. | ||
Hosszu LL, Tattum MH, Jones S, et al. The H187R mutation of the human prion protein induces conversion of recombinant prion protein to the PrP(Sc)-like form. Biochemistry. 2010;49:8729–8738. | ||
Zhong L. Exposure of hydrophobic core in human prion protein pathogenic mutant H187R. J Biomol Struct Dyn. 2010;28:355–361. | ||
Uflacker A, Doraiswamy PM, Rechitsky S, See T, Geschwind M, Tur-Kaspa I. Preimplantation genetic diagnosis (PGD) for genetic prion disorder due to F198S mutation in the PRNP gene. JAMA Neurol. 2014;71:484–486. | ||
Unverzagt FW, Farlow MR, Norton J, Dlouhy SR, Young K, Ghetti B. Neuropsychological function in patients with Gerstmann-Sträussler-Scheinker disease from the Indiana kindred (F198S). J Int Neuropsychol Soc. 1997;3:169–178. | ||
Piccardo P, Liepnieks JJ, William A, et al. Prion proteins with different conformations accumulate in Gerstmann-Sträussler-Scheinker disease caused by A117V and F198S mutations. Am J Pathol. 2001;158:2201–2207. | ||
Zaidi SI, Richardson SL, Capellari S, et al. Characterization of the F198S prion protein mutation: enhanced glycosylation and defective refolding. J Alzheimers Dis. 2005;7:159–171. | ||
Plate A, Benninghoff J, Jansen GH, et al. Atypical parkinsonism due to a D202N Gerstmann-Sträussler-Scheinker prion protein mutation: first in vivo diagnosed case. Mov Disord. 2013;28:241–244. | ||
Gallo M, Paludi D, Cicero DO, et al. Identification of a conserved N-capping box important for the structural autonomy of the prion alpha 3-helix: the disease associated D202N mutation destabilizes the helical conformation. Int J Immunopathol Pharmacol. 2005;18:95–112. | ||
Gu Y, Verghese S, Bose S, Mohan M, Singh N. Mutant prion protein D202N associated with familial prion disease is retained in the endoplasmic reticulum and forms “curly” intracellular aggregates. J Mol Neurosci. 2007;32:90–96. | ||
Piccardo P, Dlouhy SR, Lievens PM, et al. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57:979–988. | ||
Ilc G, Giachin G, Jaremko M, et al. NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. PLoS One. 2010;5:e11715. | ||
Woulfe J, Kertesz A, Frohn I, Bauer S, George-Hyslop PS, Bergeron C. Gerstmann-Sträussler-Scheinker disease with the Q217R mutation mimicking frontotemporal dementia. Acta Neuropathol. 2005;110:317–319. | ||
Bratosiewicz J, Barcikowska M, Cervenakowa L, Brown P, Gajdusek DC, Liberski PP. A new point mutation of the PRNP gene in Gerstmann-Sträussler-Scheinker case in Poland. Folia Neuropathol. 2000;38:164–166. | ||
Zheng L, Longfei J, Jing Y, et al. PRNP mutations in a series of apparently sporadic neurodegenerative dementias in China. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:938–944. | ||
Kudo W, Lee HP, Zou WQ, et al. Cellular prion protein is essential for oligomeric amyloid-β-induced neuronal cell death. Hum Mol Genet. 2012;21:1138–1144. | ||
Zhang W, Jiao B, Xiao T, et al. Mutational analysis of PRNP in Alzheimer’s disease and frontotemporal dementia in China. Sci Rep. 2016;6:38435. | ||
Lundberg P, Magzoub M, Lindberg M, et al. Cell membrane translocation of the N-terminal (1-28) part of the prion protein. Biochem Biophys Res Commun. 2002;299:85–90. | ||
Bernardi L, Cupidi C, Bruni AC. Pathogenic mechanisms of the prion protein gene mutations: a review and speculative hypotheses for pathogenic potential of the Pro39Leu mutation in the associated FTD-like phenotype. J Neurol Neurosci. 2017;8:208. | ||
Oldoni E, Fumagalli GG, Serpente M, et al. PRNP P39L variant is a rare cause of frontotemporal dementia in Italian population. J Alzheimers Dis. 2016;50:353–357. | ||
Bernardi L, Cupidi C, Frangipane F, et al. Novel N-terminal domain mutation in prion protein detected in 2 patients diagnosed with frontotemporal lobar degeneration syndrome. Neurobiol Aging. 2014;35:2657.e7–2657.e11. | ||
Rodriguez MM. A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. Neurology. 2005;64:1455–1457. | ||
Ye J, Han J, Shi Q, et al. Human prion disease with a G114V mutation and epidemiological studies in a Chinese family: a case series. J Med Case Rep. 2008;2:331. | ||
Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in Gerstmann-Sträussler syndrome with mutant PrP plaques. Biochem Biophys Res Commun. 1993;192:525–531. | ||
Ghetti B, Piccardo P, Spillantini MG, et al. Vascular variant of prion protein cerebral amyloidosis with t-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A. 1996;93:744–748. | ||
Abdallah A, Wang P, Richt JA, Sreevatsan S. Y145Stop is sufficient to induce de novo generation prions using protein misfolding cyclic amplification. Prion. 2012;6:81–88. | ||
Jayadev S, Nochlin D, Poorkaj P, et al. Familial prion disease with Alzheimer disease-like tau pathology and clinical phenotype. Ann Neurol. 2011;69(4):712–720. | ||
Guerreiro R, Brás J, Wojtas A, Rademakers R, Hardy J, Graff-Radford N. A nonsense mutation in PRNP associated with clinical Alzheimer’s disease. Neurobiol Aging. 2014;35:2656.e13–2656.e16. | ||
Fong JC, Rojas JC, Bang J, et al. Genetic prion disease caused by PRNP Q160X mutation presenting with an orbitofrontal syndrome, cyclic diarrhea, and peripheral neuropathy. J Alzheimers Dis. 2017;55:249–258. | ||
Mead S, Gandhi S, Beck J, et al. A novel prion disease associated with diarrhea and autonomic neuropathy. N Engl J Med. 2013;369:1904–1914. | ||
Matsuzono K, Ikeda Y, Liu W, et al. A novel familial prion disease causing pan-autonomic-sensory neuropathy and cognitive impairment. Eur J Neurol. 2013;20:e67–e69. | ||
Nitrini R, Rosemberg S, Passos-Bueno MR, et al. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol. 1997;42:138–146. | ||
Grasbon-Frodl E, Lorenz H, Mann U, Nitsch RM, Windl O, Kretzschmar HA. Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol. 2004;108:476–484. | ||
Chebaro Y, Derreumaux P. The conversion of helix H2 to beta-sheet is accelerated in the monomer and dimer of the prion protein upon T183A mutation. J Phys Chem B. 2009;113:6942–6948. | ||
Kovač V, Čurin Šerbec V. Prion proteins without the glycophosphatidylinositol anchor: potential biomarkers in neurodegenerative diseases. Biomark Insights. 2018;13:1177271918756648. | ||
Jansen C, Parchi P, Capellari S, et al. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010;119:189–197. | ||
Kovač V, Zupančič B, Ilc G, Plavec J, Čurin Šerbec V. Truncated prion protein PrP226* – A structural view on its role in amyloid disease. Biochem Biophys Res Commun. 2017;484:45–50. | ||
Erginel-Unaltuna N, Peoc’h K, Komurcu E, Acuner TT, Issever H, Laplanche JL. Distribution of the M129V polymorphism of the prion protein gene in a Turkish population suggests a high risk for Creutzfeldt-Jakob disease. Eur J Hum Genet. 2011;9:965–968. | ||
Mead S, Whitfield J, Poulter M, et al. A novel protective prion protein variant that colocalizes with kuru exposure. N Engl J Med. 2009;361:2056–2065. | ||
Kaski D, Mead S, Hyare H, et al. Variant CJD in an individual heterozygous for PRNP codon 129. Lancet. 2009;2374:2128. | ||
Jeong BH, Lee KH, Lee YJ, et al. Lack of association between PRNP 1368 polymorphism and Alzheimer’s disease or vascular dementia. BMC Med Genet. 2009;10:32. | ||
Ahn K, Kim E, Kwon YA, Kim DK, Lee JE, Jo SA. No association of prion protein gene polymorphisms with Alzheimer’s disease in Korean population. Exp Mol Med. 2006;38:727–731. | ||
Jeong BH, Na HR, Bae JC, et al. Absence of association between codon 129 and 219 polymorphisms of the prion protein gene and vascular dementia. Dement Geriatr Cogn Disord. 2007;24:86–90. | ||
Head MW, Lowrie S, Chohan G, Knight R, Scoones DJ, Ironside JW. Variably protease-sensitive prionopathy in a PRNP codon 129 heterozygous UK patient with co-existing tau, α synuclein and Aβ pathology. Acta Neuropathol. 2008;120:821–823. | ||
Gambetti P, Dong Z, Yuan J, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. | ||
Rodríguez-Martínez AB, Garrido JM, Zarranz JJ, et al. A novel form of human disease with a protease-sensitive prion protein and heterozygosity methionine/valine at codon 129: case report. BMC Neurol. 2010;10:99. | ||
Zou WQ, Puoti G, Xiao X, et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010;68:162–172. | ||
Shibao C, Garland EM, Gamboa A, et al. PRNP M129V homozygosity in multiple system atrophy vs. Parkinson’s disease. Clin Auton Res. 2008;18:13–19. | ||
Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. Protective prion protein polymorphisms against sporadic Creutzfeldt-Jakob disease. Lancet. 1998;351:419. | ||
Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1998;43:826–828. | ||
Jeong BH, Lee KH, Kim NH, et al. Association of sporadic Creutzfeldt-Jakob disease with homozygous genotypes at PRNP codons 129 and 219 in the Korean population. Neurogenetics. 2005;6:229–232. | ||
Lukic A, Beck J, Joiner S, et al. Heterozygosity at polymorphic codon 219 in variant Creutzfeldt-Jakob disease. Arch Neurol. 2010;67:1021–1023. | ||
Ch’ng GS, An SS, Bae SO, Bagyinszky E, Kim SY. Identification of two novel mutations, PSEN1 E280K and PRNP G127S, in a Malaysian family. Neuropsychiatr Dis Treat. 2015;11:2315–2322. | ||
Asante EA, Smidak M, Grimshaw A, et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature. 2015;522:478–481. | ||
Bishop MT, Pennington C, Heath CA, Will RG, Knight RSG. PRNP variation in UK sporadic and variant Creutzfeldt Jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med Genet. 2009;10:146. | ||
Beck JA, Poulter M, Campbell TA, et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC prion unit. Hum Mutat. 2010;31:E1551–E1563. | ||
Walz R, Castro RM, Velasco TR, et al. Surgical outcome in mesial temporal sclerosis correlates with prion protein gene variant. Neurology. 2003;61:1204–1210. | ||
Coimbra ER, Rezek K, Escorsi-Rosset S, et al. Cognitive performance of patients with mesial temporal lobe epilepsy is not associated with human prion protein gene variant allele at codons 129 and 171. Epilepsy Behav. 2006;8:635–642. | ||
Windl O, Giese A, Schulz-Schaeffer W, et al. Molecular genetics of human prion diseases in Germany. Hum Genet. 1999;105:244–252. | ||
Kübler E, Oesch B, Raeber AJ. Diagnosis of prion diseases. Br Med Bull. 2003;66:267–279. | ||
Jackson GS, Mead S, Collinge J. Developing early diagnostics for prion diseases. Neurodegener Dis Manag. 2013;3:53–60. | ||
Letourneau-Guillon L, Wada R, Kucharczyk W. Imaging of prion diseases. J Magn Reson Imaging. 2012;35:998–1012. | ||
Shimada T, Fournier AE, Yamagata K. Neuroprotective function of 14-3-3 proteins in neurodegeneration. Biomed Res Int. 2013;2013:564534. | ||
Macfarlane RG, Wroe SJ, Collinge J, Yousry TA, Jäger HR. Neuroimaging findings in human prion disease. J Neurol Neurosurg Psychiatry. 2007;78:664. | ||
Zerr I, Poser S. Clinical diagnosis and differential diagnosis of CJD and vCJD. With special emphasis on laboratory tests. APMIS. 2002;110:88–98. | ||
Wood H. Cerebrospinal fluid tau levels – a potential diagnostic biomarker for Creutzfeldt–Jakob disease. Neuropsychiatr Dis Treat. 2015;11:2315–2322. | ||
Skinningsrud A, Stenset V, Gundersen AS, Fladby T. Cerebrospinal fluid markers in Creutzfeldt-Jakob disease. Cerebrospinal Fluid Res. 2008;5:14. | ||
Hamlin C, Puoti G, Berri S, et al. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2012;79:547–552. | ||
Nozaki T, Hamaguchi T, Sanjo N, et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010;133:3043–3057. | ||
Knight R. The Diagnosis of Prion Diseases. Cambridge: Cambridge University Press; 1999:3–11. | ||
Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(Pt 10):2659–2668. | ||
Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39:E2. | ||
Mastrianni JA. Genetic Prion Diseases. GeneReviews; 2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1229/. Accessed July 25, 2018. | ||
Forloni G, Tettamanti M, Lucca U, et al. Preventive study in subjects at risk of fatal familial insomnia: innovative approach to rare diseases. Prion. 2015;9:75–79. | ||
Glatzel M, Stoeck K, Seeger H, Lührs T, Aguzzi A. Human prion diseases: molecular and clinical aspects. Arch Neurol. 2005;62:545–552. | ||
Collinge J, Palmer MS. Prion diseases in humans and their relevance to other neurodegenerative diseases. Dementia. 1993;4:178–185. | ||
Kang M, Kim SY, An SS, Ju YR. Characterizing affinity epitopes between prion protein and β-amyloid using an epitope mapping immunoassay. Exp Mol Med. 2013;45:e34. | ||
Minikel EV, Vallabh SM, Lek M. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8:322ra9. | ||
Maddox RA, Blasé JL, Mercaldo ND, et al. Clinically unsuspected prion disease among patients with dementia diagnoses in an Alzheimer’s disease database. Am J Alzheimers Dis Other Dement. 2015;30:752–755. | ||
Dupiereux I, Zorzi W, Quadrio I, et al. Creutzfeldt-Jakob, Parkinson, Lewy body dementia and Alzheimer diseases: from diagnosis to therapy. Cent Nerv Syst Agents Med Chem. 2009;9:2–11. | ||
Geldmacher DS, Whitehouse PJ. Differential diagnosis of Alzheimer’s disease. Neurology. 1997;48:2–9. | ||
Manuelidis EE, Manuelidis L. Suggested links between different types of dementias: Creutzfeldt-Jakob disease, Alzheimer disease, and retroviral CNS infections. Alzheimer Dis Assoc Disord. 1989;3:100–110. | ||
Giau VV, An SS, Bagyinszky E, Kim SY. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol Cell Toxicol. 2015;11:89–143. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.