")
Back to Journals » International Journal of Nanomedicine » Volume 19
Characterization of Drug with Good Glass-Forming Ability Loaded Mesoporous Silica Nanoparticles and Its Impact Toward in vitro and in vivo Studies
Authors Budiman A, Anastasya G, Handini AL, Lestari IN, Subra L, Aulifa DL
Received 7 December 2023
Accepted for publication 18 February 2024
Published 6 March 2024 Volume 2024:19 Pages 2199—2225
DOI https://doi.org/10.2147/IJN.S453873
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Arif Budiman,1 Gracia Anastasya,1 Annisa Luthfiyah Handini,1 Ira Novianty Lestari,2 Laila Subra,3 Diah Lia Aulifa2
1Department of Pharmaceutics and Pharmaceutical Technology, Universitas Padjadjaran, Bandung, Indonesia; 2Department of Pharmaceutical Analysis and Medicinal Chemistry, Universitas Padjadjaran, Bandung, Indonesia; 3Department of Pharmacy, Universiti Geomatika Malaysia, Kuala Lumpur, Malaysia
Correspondence: Diah Lia Aulifa, Department of Pharmaceutical Analysis and Medicinal Chemistry, Universitas Padjadjaran, Jl, Raya Bandung-Sumedang Km. 21, Bandung, 45363, Indonesia, Email [email protected]
Abstract: Solid oral dosage forms are mostly preferred in pharmaceutical formulation development due to patient convenience, ease of product handling, high throughput, low manufacturing costs, with good physical and chemical stability. However, 70% of drug candidates have poor water solubility leading to compromised bioavailability. This phenomenon occurs because drug molecules are often absorbed after dissolving in gastrointestinal fluid. To address this limitation, delivery systems designed to improve the pharmacokinetics of drug molecules are needed to allow controlled release and target-specific delivery. Among various strategies, amorphous formulations show significantly high potential, particularly for molecules with solubility-limited dissolution rates. The ease of drug molecules to amorphized is known as their glass-forming ability (GFA). Specifically, drug molecules categorized into class III based on the Taylor classification have a low recrystallization tendency and high GFA after cooling, with substantial “glass stability” when heated. In the last decades, the application of mesoporous silica nanoparticles (MSNs) as drug delivery systems (DDS) has gained significant attention in various investigations and the pharmaceutical industry. This is attributed to the unique physicochemical properties of MSNs, including high loading capacity, recrystallization inhibition, excellent biocompatibility, and easy functionalization. Therefore, this study aimed to discuss the current state of good glass former drug loaded mesoporous silica and shows its impact on the pharmaceutical properties including dissolution and physical stability, along with in vivo study. The results show the importance of determining whether mesoporous structures are needed in amorphous formulations to improve the pharmaceutical properties of drug with a favorable GFA.
Keywords: mesoporous silica nanoparticles, good glass-forming ability, characterization, dissolution, in vivo study
Graphical Abstract:
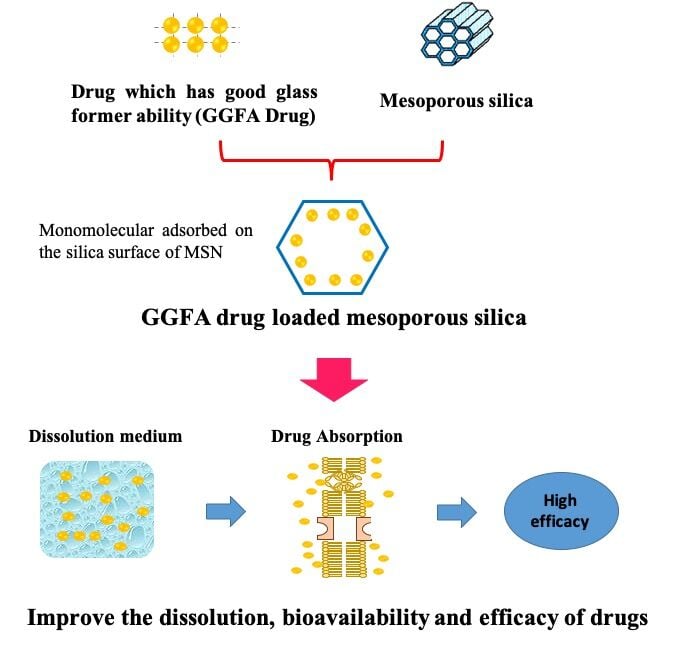
Introduction
Water solubility of drug is significantly important in drug development, as molecules absorption occurs after their dissolution in gastrointestinal fluids, directly impacting bioavailability.1 In drug dosage development, oral solid dosage forms are often selected due to the ease of product handling, convenience for use, low manufacturing cost, with good physical, and chemical stability.2 However, 75% of drug candidates are classified into Biopharmaceutics Classification System (BCS) classes II and IV.3 This makes a delivery system essential, as 84% of drug products in the market still require a proper and absorption system for the treatment of diseases.2
Amorphous formulations are increasingly becoming a popular method in drug discovery to enhance bioavailability, particularly for novel chemical entities with low water solubility.4 This significant increase has led to the study focusing on converting API into amorphous forms. For instance, during melt extrusion, the formulation is heated, converting the crystalline API to a molten state, and cooling to a solid state. The API can become kinetically stuck in a disordered, amorphous state or pass through a liquid to a solid phase, reverting into its original or another crystalline form throughout the cooling process. Physical stability is a crucial factor during processing and storage, determining the suitability of the API for amorphous formulation. However, current knowledge regarding the tendency of organic molecules to form crystals after cooling from the undercooled melt state and its connection with the long-term physical stability of the resulting glass is rudimentary. This shows that understanding important physicochemical factors affecting crystallization tendency would help design dosage forms for new chemical entities.5
A classification system of glass-forming ability (GFA) based on the recrystallization tendency of drug during cooling and heating cycles has previously been proposed.5 GFA class (I) refers to drug that recrystallize during cooling of the melt at a rate of 20 K/min. Meanwhile, class (II) did not recrystallize during the cooling of the melt at 20 K/min but recrystallized in the following heat cycle at 10 K/min. Finally, GFA class (III) drug is neither recrystallized during cooling of the melt at 20 K/min nor the following heat cycle at 10 K/min.5,6 Classes of GFA (1 to 3) represent poor, modest, and good glass formers, respectively. This classification system based on arbitrary categories was further substantiated, showing that drug can be classified according to their inherent crystallization tendency, regardless of the predefined categories.7 This shows that drug molecules in class (III) have both high GFA after cooling and elevated “glass stability” when heated above Tg.7,8
In recent years, nanomedicine has become the leading edge of nanotechnology, generating great expectations in the biomedical field.9 Silica nanoparticles with mesopores, referred to as MSNs, were also used due to uniform and tunable pore size, easy independent functionalization of the surface, internal, and external pores, including the gating mechanism of the pore opening, which characterized their distinctive and potential as drug carriers. Several investigations have been carried out on the use of these carriers for loading a variety of cargo ranging from drug to macromolecules such as proteins,10,11 DNA,12,13 and RNA.14,15 An exhaustive set of literature is available, with continuous study evaluating new methods for using mesoporous silica nanoparticles (MSNs) in drug delivery. Several reviews regarding MSNs in improving the solubility of drug,16,17 as well as controlled/sustained DDS,18 and applications in biomedicine19 have been published.
The incorporation of drug into MSNs is a promising strategy to stabilize its amorphous form.20 MSNs are thermodynamically stable and more favorable for drug to remain in a disordered rather than crystalline state inside the pore due to its small carrier size.21 Moreover, two mechanisms responsible for illuminating drug crystallization inhibition include (i) the molecular interaction between functional groups of drug molecules as well as the surface of MSNs, namely hydrogen bonding, and (ii) the nanoconfinement effect of MSNs, which suppresses nucleation and crystal growth as the pore size is smaller than the critical crystalline nuclei.22 Considering silica surface interaction, the large MSNs surface area has an additional free energy. Drug adsorption in the amorphous state is thermodynamically favorable due to its lower free energy state compared to the crystalline form.23,24 When drug molecules occupy all MSN surfaces, the excess drug no longer has direct contact but starts forming additional layers on the top of the initial monolayer.25,26 Consequently, the nanoconfinement effect of MSNs provides physical stabilization against the crystallization from the excess amount of amorphous drug. The surface area and pore volume also influence the loading capacity and crystallization.27
Several studies reported the incorporation of drug with good glass former ability (GGFA drug) into MSNs. Therefore, this study aimed to systematically summarize and discuss characterization of (GGFA drug) within MSNs, which affect their pharmaceutical properties, including solubility, dissolution, and physical stability, as well as in vivo analysis. In this review, we summarize the classification system of GFA, MSNs as drug delivery carrier systems, and the studies of GGFA loaded MSNs. Moreover, the review also elucidates the characterization of GGFA drug loaded MSNs, and their impact on in vitro and in vivo studies.
Drug with Good GFA (GGFA Drug)
A classification system of GFA based on the recrystallization tendency of drug during cooling and heating cycles has previously been proposed. The GGFA drug, categorized into class III based on Taylor’s classification,5 commonly showed neither crystallization after cooling nor reheating.28 The poorly water-soluble molecules categorized into class III based on Taylor’s classification are represented in Table 1
 |
Table 1 The Poorly Water-Soluble Molecules are Categorized into Class III Based on Taylor’s Classification |
Crystallization Tendency from Melt Screening Using Differential Scanning Calorimetry (DSC)
The crystallization tendency of molecules from the undercooled melt state was assessed using DSC screening method. For class (III) molecules, the result is the absence of crystallization or reheating at the melting point after chilling down below Tg. However, in the case of nilutamide and nimesulide, crystallization was observed when tested after reheating at a slow heating rate (2°C min-1).
DSC data obtained were used to make further fascinating observations. For example, local anesthetic procaine did not crystallize after cooling from the undercooled melt at a slow cooling rate (1°C min−1) and was resistant to crystallization on reheating. This observation exemplifies the significant variation in crystallization tendency between molecules of similar structures.5
Numerical Evaluation of GGFA Drug
Class (III) molecules show significant characteristics when cooled from the undercooled melt state, including high relative GFA. Compared to class (II) molecules, the majority of molecules did not crystallize after being heated at a slow pace (2°C min−1), and none of the systems showed crystallization at an intermediate rate (10°C min−1). After reheating at 2°C for one minute, nilutamide and nimesulide both crystallized but showed no significant effect in one of the triple runs. These results showed that class (III) molecules have strong “glass stability” after reheating above Tg and high glass transition temperatures (GFA) after cooling.
For each class (III) molecule, fragility and strength parameters were calculated. The range of values reflects moderately fragile or delicate liquids, which is equivalent to class (II) compounds. Since there was no crystallization during reheating, the GS parameters for class (III) molecules could not be calculated. Trg is not a perfect predictor of crystallization tendency, as evidenced by class (I) molecules which show the lowest average Trg values [0.69±0.02], and class (III) molecules with highest values [0.73±0.04].
In conclusion, inorganic systems have several data to support the correlation between GFA and crystallization behavior. However, these criteria are not the best in forecasting GFA of organic molecules due to the lack of a strong connection between the evaluated qualities and the observed crystallization behavior in this study, within the classes of molecules.5
Role of Physicochemical Properties
The propensity of molecules to crystallize depends on their inherent physicochemical characteristics. However, interpreting the observed crystallization tendency with these characteristics can be challenging due to the complexity of the process. Based on the PCA results, class (I) molecules have lower MW, simpler structures, and fewer compared to class (III) molecules, with more structural complexity. The crystalline lattice structure constrains the orientation of molecules for crystallization to take place. Additionally, class (I) molecules have greater average Tmelt, DHfus, DSfus, and lower (more negative) DGv values. A lower entropy of fusion suggests that the entropy of the liquid is relatively low or higher in the crystal. Meanwhile, a lower heat of fusion shows that less energy is required to fracture the crystalline lattice. The free energy differences between the crystalline and amorphous states are greater (more negative) for class (I) compounds and lower (less negative) for class (III) compounds, as estimated using the Hoffman equation.5
Since a higher melting material requires more energy to shatter the crystalline lattice, the observed tendency of class (I) compounds to have higher melting points relative to class (III) compounds is also considered. Therefore, it can be concluded that class (I) molecules possess a stronger driving force for nucleation and a higher inclination to crystallize compared to class (III). Despite the complexity of the crystallization process, the average values of the descriptors for class (I) and class (III) molecules showed several differences. This makes it impossible to predict the crystallization behavior after cooling from the undercooled melt.28
MSNs
MSNs are made through hydrolysis and condensation of silica precursors such as tetraethoxysilane (TEOS) around micelle templates. These micelles are produced by supramolecular self-assemblies of surfactant molecules, followed by template removal through calcination or solvent extraction, as shown in Figure 1. The two basic methods of synthesizing MSNs include liquid-crystal templating and cooperative self-assembly of micelle and silica sources.63 MSNs can also be made in a variety of sizes, from nano to microscale, with enormous surface areas between 700 and 1000 m2/g, as well as pore volumes between 0.6 and 1 cm2/g. Various morphologies such as spherical, rod, ellipsoid, and platelet are created suited for a variety of biological applications depending on the reaction conditions. Although the average pore sizes of MSNs are between 2 and 5 nm, it can be synthesized to 30 nm in size, allowing for the accommodation of bigger compounds such as proteins in addition to tiny molecules. Furthermore, silanol groups affect the surface characteristics of MSNs, ranging from hydrophobic to hydrophilic, which allow for optimal drug loading parameters and release profiles for various therapeutic compounds.
 |
Figure 1 Schematic illustration for the synthesis of MSNs. Reprinted from J Control Release, Volume 262, Maleki A, Kettiger H, Schoubben A, Rosenholm JM, Ambrogi V, Hamidi M. Mesoporous silica materials: from physico-chemical properties to enhanced dissolution of poorly water-soluble drugs. 329–347. Copyright 2017, with permission from Elsevier.17 |
Factors Affecting Pore Size
Small-molecule surfactants are used in the synthesis process, producing MSNs with pore sizes below 5 nm, such as MCM-41. However, its application as a carrier for large guest molecules such as proteins or nucleic acids may be restricted.64 This shows the necessity of developing large-pore MSNs (LPMSMs) to accommodate the large-size guest molecules including enzymes, enable the load of high amounts of different cargos, and promote drug dissolution rates. In this study, the impact of important parameters such as surfactant type, hydrothermal temperature, and introducing organic swelling agents (SAs) on controlling the pore size of MSNs was discussed.
The type and area of the hydrophobic groups in the templating surfactants mainly affect MSN pore size. Generally, tetra-alkylammonium salts are the most commonly used surfactant family in the preparation of ordered mesoporous silicas. Jana et al investigated the effect of different alkyl chain lengths of tetra-alkylammonium salts on pores diameter of MSNs. The results showed that an increase in surfactant chain length from C8 to C22 enlarged pore size from 1.6 to 4.2 nm.65 This showed the importance of surfactant chain length in pore size, where any alteration in the carbon chain length can modify the pore size of MSNs by approximately 4.1 nm.66,67
The addition of organic SAs is an effective method to increase the pore diameter of MSNs. Specifically, SAs can enter the hydrophobic core of micelles, causing an increase in size.68 In this context, benzene and its derivatives,65,69 linear hydrocarbons,70,71 cyclic hydrocarbons,72 and long-chain amines73 have been used as micelle expanders. For example, in acidic triblock copolymer templating systems, the pore diameters of MSNs might be increased by 40 nm using 1,3,5-trimethylbenzene (TMB) as a micelle expander, resulting in disordered mesostructures.69 This shows that using blends of two SAs to expand the pore size of MSNs can be an effective method, resulting in highly ordered MSNs with significantly more pore size augmentation.74,75
Factors Affecting Particle Size
Nano-sized MSNs have advantages compared to their bulk counterparts, such as high dispersibility and quick mass transit.76,77 Meanwhile, controlling particle size at micron scale is more challenging, facilitating the focus on nanoscale management. Since the hydrolysis rate of silane and siloxane bond condensation are both highly affected by pH, it is essential to control the particle size of MSNs.78 Previous investigations have shown that pH increases can lead to a higher TEOS hydrolysis rate. However, the TEOS condensation rate is not monotonic, reaching a maximum value of pH 8.4 and declining on both sides.76 This phenomenon leads to the preparation of MSNs with diameters ranging from tens to hundreds of nanometers using a quick pH-shifting method based on TEOS hydrolysate.79 Rapidly increasing the pH from 2 to 6.0–9.0 can enhance the simultaneous formation and growth of silica/surfactant nuclei. This results in controlled particle sizes within the nanoscale range due to the fast condensation of silica with a strong electrostatic attraction between silica and cationic surfactants.
Another essential component in the synthesis of MSNs is reaction temperature, which has a considerable impact on particle size. This is because higher reaction temperature increases the rates of hydrolysis and polymerization of silica precursors, resulting in particle expansion.80 Lv et al showed that changing the reaction temperature from 40 to 95°C increased the particle size of MSNs from 21 to 38 nm.81
MSNs as Drug Delivery Carrier Systems
The significant success of MSNs as DDS is based on their physical and chemical properties. Specifically, MSNs can provide a novel therapeutic armamentarium capable of addressing main challenges in conventional medicine, such as the lack of drug specificity, the narrow window of efficacy of some medicines, the possible low drug solubility and/or stability, the adverse pharmacokinetic profiles, and the side effects. Generally, nanoparticles used as DDS are required to fulfill some basic requirements, as illustrated in Figure 2. These include loading the maximum amount of cargo molecules, on-demand releasing to avoid premature release, precisely delivering the cargo to the diseased tissue and penetrating deep into the tumor in cancer treatment.
 |
Figure 2 Representative plot map from an MSNs platform to the preclinical studies, including the synthesis of MSNs, show of their stability, therapeutic cargo loading into the pores, quality assurance or characterization of loaded platform, smart release behavior, penetration into tumor mass, cellular uptake, and in vivo study. Reprinted from Manzano M, Vallet‐Regí M. Mesoporous silica nanoparticles for drug delivery. Adv Funct Mater. 2020;30(2). © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.82 |
Loading and Protecting the Therapeutic Cargo
The two different methods of loading drug into MSNs include in situ during the synthetic path or post-sorption (physisorption or chemisorption).64 Post-sorption is a separate step following particle synthesis, which allows independent optimization of loading conditions. The most common methods include physical adsorption from solution into the mesopores or the outer surface and covalent grafting. In any case, the larger surface area and pore volume of MSNs ensure higher drug loading compared to other nanoparticles. This allows the use of fewer nanoparticles of MSNs for the potential treatment of the same disease, which could be crucial in addressing toxicity issues. An additional benefit of using MSNs as drug carriers is the protection of cargo molecules loaded inside the mesopores from tough environmental factors in the systems such as degradation due to enzymatic or extreme pH.
Release Therapeutic Cargo on-Demand
A nanocarrier should have ability to release high local concentrations of the therapeutic cargo on-demand after the application of a stimulus to be considered ideal, leading to smart DDS.83 This method is essential, particularly when the cargo is cytotoxic, avoiding premature release of the transported cargo before reaching the targeted tissues, which improves the nanomedicine efficiency and reduces side effects. The stimuli-responsive method is essential for MSNs due to their peculiar and unique textural characteristics. Although it is relatively easy to load a therapeutic cargo into the mesopores of MSNs, preventing the diffusion of molecules from mesoporous channels is crucial. To address this limitation, there is a need to close the pore entrances with a cap after the cargo is loaded inside mesoporous network of cavities. The pore diameter of MSNs allows the use of large molecules to block the pore entrances. After the application of certain stimuli, the cap agents would detach from the pore entrances triggering the cargo on-demand. These capping systems can be divided into three main groups, namely reusable caps, based on a bulky ability to bind reversibly, completely reversible caps using the principle of reversal affinity of a ring-shaped macromolecule into a steam with two or more binding sites, and irreversible caps which function using a chemical bond cleavage of the capping molecule leading to a permanent separation from the pore entrance of MSNs.84 Internal and external stimuli have been widely used for triggering the release of therapeutic cargo from MSNs.85–87
GGFA Drug Loaded MSNs
The studies of GGFA loaded MSNs are represented in Table 2.
 |
Table 2 The Studies of GGFA Drug Loaded MSN |
Preparation of GGFA Drug Loaded MSNs
Based on previous studies, the loading of GGFA drug into MSNs can be accomplished through various methods, which are divided into solvent-free and solvent-based, as shown in Figure 3.
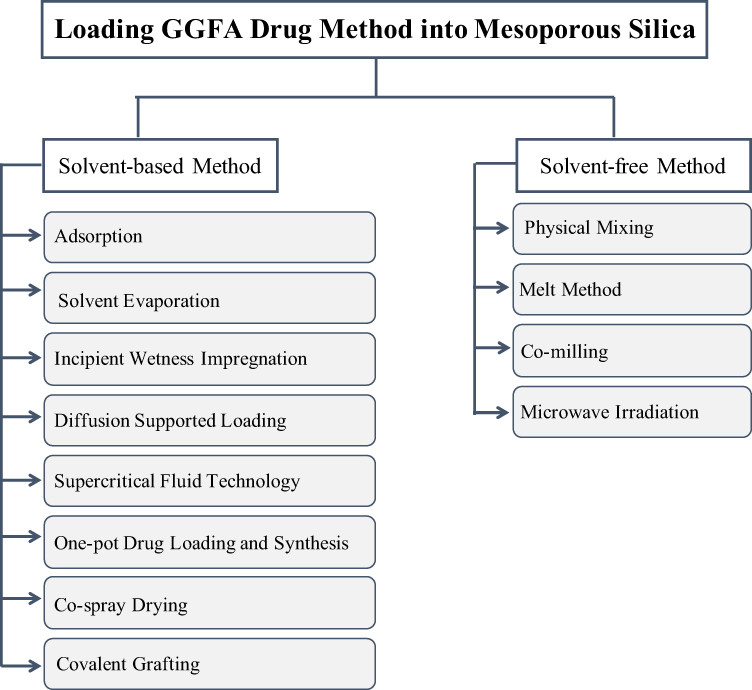 |
Figure 3 The different methods used to load GGFA drug into MSNs. Reprinted from Trzeciak K, Chotera-Ouda A, Bak-Sypien II, Potrzebowski MJ. Mesoporous silica particles as drug delivery systems—the state of the art in loading methods and the recent progress in analytical techniques for monitoring these processes. Pharmaceutics. 2021;13(7):950. Creative Commons.111 |
The solvent-based methods for loading GGFA drug into MSNs could be challenging. This could be due to the procedure requires multiple stages using large amounts of solvents, which are often difficult to control in terms of filling factor. The removal of the solvent is necessary to achieve the acceptable levels specified in the guidelines of the International Conference on Harmonization (ICH) Q3 (R5). Thus, using less toxic and safer solvents, such as ethanol, supercritical, or near-critical CO2, can be as an alternative in the loading drug process into MSNs or for human pharmaceutical applications. The selection of a solvent has also a significant impact on the loading GGFA drug process into MSNs. Moreover, the highest GGFA drug solubility in the solvent is not necessarily the best candidate for getting a high filling factor. Some methods were carried out to load GGFA drug into MSNs such as adsorption, solvent evaporation, incipient wetness impregnation, etc. Meanwhile, the solvent-free methods can generate a high degree of GGFA drug loading in MSN without consuming time. This method does not require an organic solvent for loading GGFA drug into MSN, thus this process is an environmentally friendly method. Moreover, the concentration of GGFA drug in MSN is easily predictable, as it is directly influenced by the ratio between GGFA drug and MSN.111
Characterization of GGFA Drug Loaded MSNs
Powder X-Ray Diffraction (PXRD)
The PXRD was conducted to characterize the amorphization of drug within MSNs. Kumar et al reported aceclofenac incorporated into MSNs, where sharp peaks for crystalline aceclofenac were observed at 22.043°, 24.305°, 25.724°, and 31.900°. Aceclofenac-loaded porous silicas showed typical mesoporous silica reflections in 2–8 with no significant difference in d (1 0 0) spacing, confirming the stability of the structure during loading. However, decreasing d100 peak intensities showed that drug was filling pores. The XRD of all drug-loaded formulations showed an amorphous halo with a drop in the intensity and number of crystalline aceclofenac peaks, showing a decrease in crystallinity or partial amorphization. A characteristic peak at 25.7° of reduced intensity suggested that the amorphization of drug was incomplete (not 100%). This condition also shows incomplete encapsulation in the pores of mesoporous matrix, as some crystalline drug was found on the surface of all silicas. Compared to loaded silicas, drug-loaded MS11/72 only indicated two reduced-intensity aceclofenac peaks at 24.3 and 25.7. This showed that the majority of drug molecules were trapped inside mesoporous channels of MS11/72 or finely precipitated as an amorphous substance.89 Elrahman et al reported the incorporation of ketoprofen into SBA-15, where the physical mixing of crystalline ketoprofen and SBA-15 samples showed reflexes associated with the presence of crystalline ketoprofen. Meanwhile, the absence of crystalline ketoprofen responses supported the amorphous nature of ketoprofen loaded into SBA-15-K and SBA15-AP(1:10)-K samples.105
The PXRD results of the RTV amorphization after preparation by solvent evaporation method were also reported in a previous study, as presented in Figure 4. The characteristic diffraction peaks of RTV crystal were observed in the PXRD pattern, while RTV prepared by solvent evaporation method showed halo patterns without any characteristic peaks. Similar results were observed in the RTV-loaded MSNs, where a halo pattern without any characteristic peaks was observed. This showed that the RTV amorphous was successfully formed by the solvent evaporation method in the RTV alone or after being incorporated into MSNs.112 Although the PXRD did not detect the presence of an amorphous form, it was discovered that the absence of crystallinity in the samples could not be used to confirm the amorphization of drug by spotting the halo pattern in the diffractogram.113
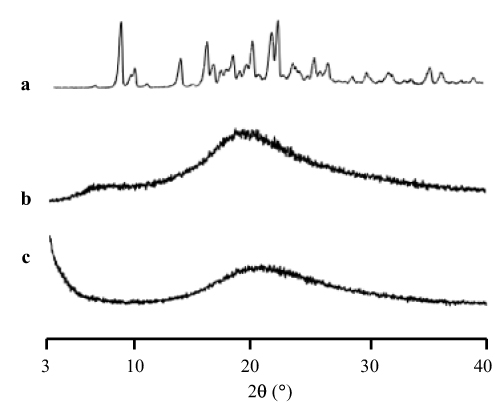 |
Figure 4 The PXRD patterns of (a) RTV crystal, (b) RTV amorphous, and (c) RTV/MSNs = 3:7. Reprinted from Budiman A, Aulifa DL. A comparative study of the pharmaceutical properties between amorphous drugs loaded-mesoporous silica and pure amorphous drugs prepared by solvent evaporation. Pharmaceuticals. 2022;15(6):730. Creative Commons.112 |
DSC Measurement
DSC is used to characterize the physical state and location of drug within MSNs by observing their glass transition temperature of melting point. Observations have shown that when drug is in the pores of MSNs, the melting point or Tg would be lower compared to the crystalline state.111 Moreover, DSC can also be used to determine the maximum loading of an amorphous drug.3 DSC analysis was used to investigate the physical state of Felodipine in mesoporous silica. Based on the results, the DSC curve of pure felodipine had a single endothermic peak at 143.7°C, corresponding to its intrinsic melting points. However, there was no melting peak of felodipine loaded in MSNs, showing a non-crystalline state (amorphous state). This showed that felodipine did not recrystallize during the encapsulation procedure and was present in its non-crystalline state within the pores.93
DSC scans of probucol-loaded mesoporous materials confirm the presence of crystalline drug at loading exceeding 100% pore filling, compared to amorphous probucol confinement within mesopores at lower drug loading. The existence of two endothermic peaks from DSC scans in samples (>40% wt%) suggested the presence of a polymorphic crystal combination. As shown in Figure 5, the ratio in the enthalpy of melting of crystalline probucol to drug loaded mesoporous materials can be used to assess the amount of crystalline material in the samples. For the larger pore materials SBA-15 and AMS 6, 40% loading resulted in 5% crystalline drug and 10% for MCM-41. Meanwhile, loading greater than 40% wt caused a rapid increase in the % crystallinity. A shift in the water desorption endotherm to a lower temperature suggested that crystalline probucol adsorption increased the hydrophobicity of the samples. Due to the increased amount of recrystallized, hydrophobic probucol in these samples, drug loading over full pore filling was predicted to produce a reduction in drug release kinetics.107
 |
Figure 5 The amount of crystalline material is determined by DSC as a percentage. Reprinted from Eur J Pharm Sci, Volume 138, Lau M, Giri K, Garcia-Bennett AE. Antioxidant Properties of Probucol Released from Mesoporous Silica. 105038. Copyright 2019, with permission from Elsevier.107 |
DSC analysis was conducted to determine the loading amount of cyclosporine A (CYP) into MSNs, as shown in Figure 6. Based on the results, the DSC curve of pure and CYP prepared by solvent evaporation showed an endothermic peak at 123.8°C and 123.2°C, respectively. This phenomenon was attributed to their glass transition temperature (Tg) without any melting peak, showing the absence of recrystallization of CYP after reheating. The ΔCp of glass transition event from CYP also decreased with a significant reduction in the concentration loaded in MSNs (CYP/MSN). The Tg of CYP was observed in the weight ratios of 7:3 and 5:5, suggesting the existence of CYP in an amorphous state outside of the mesopores. In contrast, the Tg of CYP/MSN1 = 3:7 was not detected in the DSC curve, showing monomolecular adsorption on silica surface of MSNs. Consequently, the loading of 30% CYP could be almost the maximum value to be loaded MSNs. The maximum amount of CYP within MSNs was also quantitatively determined by plotting the concentration of the CYP amorphous state and its ΔCp values. The results showed that the maximum amount of CYP/MSNs was 26.9%, with a linear coefficient of determination (R2) value of 0.98.3
 |
Figure 6 DSC curve of (a) CYP amorphous state, CYP/MSNs = (b) 7:3, (c) 5:5, and (d) 3:7. Reprinted from Budiman A, Aulifa DL. Characterization of drugs with good glass formers in loaded-mesoporous silica and its theoretical value relevance with mesopores surface and pore-filling capacity. Pharmaceuticals. 2022;15(1):93. Creative Commons.3 |
Fourier Transform Infrared (FTIR) Spectroscopy
FTIR spectroscopy is a universal study tool used to examine the chemical status of drug within MSNs and can be applied to confirm the interaction between drug and silica surface.114 The itraconazole has electronegative oxygen centers, contributing to the occurrence of the hydrogen bonding between the electrons lone pairs associated with nitrogen atoms and surface silanol groups in MSNs. After being loaded in MSNs, a free carbonyl vibration around 1700 cm−1 shifted to around 1675 cm−1, suggesting the hydrogen bonding between the carbonyl oxygen atom with the surface hydroxyl. The incorporation of itraconazole into MSNs caused a bathochromic shift of the silanol vibrations at 3750 cm−1 due to hydrogen bonding with carbonyl groups of itraconazole. This showed that surface interaction between itraconazole within MSNs can also affect the amorphization of itraconazole.115 FTIR spectra of the modified and non-modified SBA-15 carriers before and after ketoprofen adsorption were also reported. The elimination of the absorption band at 1690 cm−1 showed that carbonyl stretching vibrations in carboxylic groups in the adsorbed drug was caused by ketoprofen adsorption on modified SBA-15.116 Following drug adsorption on amino-modified SBA-15, a distinctive band at 1558 cm−1 was found, corresponding to stretching vibrations of ionized carboxylic groups, as shown in Figure 7,116 confirming the ionic nature of drug-carrier interaction.117 The formation of absorption bands at 1446 cm−1 and 1650 cm−1 in ketoprofen-loaded samples confirmed the presence of aromatic stretching C-C vibrations and carbonyl stretching vibrations associated with aromatic rings, respectively.118
 |
Figure 7 FTIR spectra of (A) the major absorption bands in 4000–400 cm−1 range: (a) SBA-15, (b) SBA-15-AP(1:10), (c) SBA-15-K, (d) SBA-15-AP(1:10)-K, and (e) ketoprofen. (B) The part of the FTIR spectrum showing the absorption band (stretching vibration) of carbonyl groups and ionized carboxylic groups. Reprinted from Appl Surf Sci, Volume 258(19), Moritz M, Łaniecki M. SBA-15 mesoporous material modified with APTES as the carrier for 2-(3-Benzoylphenyl)propionic acid. 7523–7529. Copyright 2012 with permission from Elsevier.118 |
Thermal Gravimetric Analysis (TGA)
Weight loss due to drug degradation followed by desorption of volatile components through a gradual increase in sample temperature using TGA can be used to determine the proportion of the total drug content. This is because MSNs carrier is more thermally stable compared to organic guest molecules.111 As shown in Figure 8, TGA analysis was carried out to determine the thermal stability of mesoporous silica samples KIT-6, SBA-15, procaine-loaded KIT-6 SO3H, and SBA-15-SO3H. Initially, all samples lost weight at around 150°C due to the physical absorption of water. Additionally, there was no weight loss caused by KIT-6 and SBA-15 disintegrating, showing that the samples were thermally stable KIT-6-SO3H and SBA-15-SO3H showed thermal degradation between the temperatures of 200 and 600°C. Compared to mesoporous silica, KIT-6, and SBA-15, the functionalized KIT-6-SO3H and SBA-15-SO3H indicated weight losses of approximately 30.6% and 31.2% approaching 650°C, respectively, due to PrSO3H group decomposition. Furthermore, between 300 and 500°C, a large exothermic peak related to the breakdown of organic moieties of procaine-loaded KIT-6-SO3H and SBA-15-SO3H was detected. The total weight loss for procaine medication was considerably increased at 650°C, reaching 51.5% and 44.2% for procaine-loaded KIT-6-SO3H and procaine-loaded SBA-15-SO3H, respectively. These TGA results significantly suggested that the procaine medication was successfully loaded.108
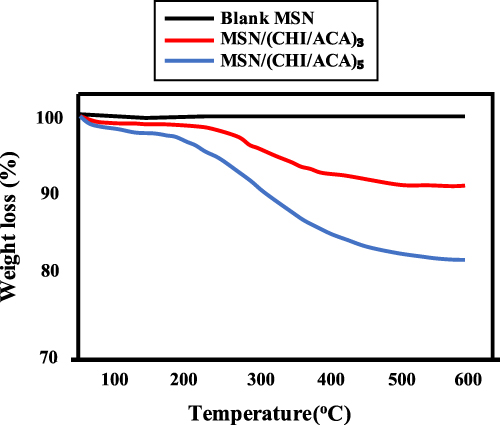 |
Figure 8 TGA patterns of MSNs, MSNs/(CHI/ACA)3 and MSNs/(CHI/ACA). Reprinted from Mater Sci Eng C, Volume 47, Hu L, Sun H, Zhao Q, et al. Multilayer encapsulated mesoporous silica nanospheres as an oral sustained drug delivery system for the poorly water-soluble drug felodipine. 313–324. Copyright 2015, with permission from Elsevier.93 |
Scanning Electron Microscope (SEM)
SEM is a powerful tool for investigating various nanostructured materials, including drug-loaded MSNs. This tool can create images of API within MSNs by scanning the surface with an electron beam, visualizing, and providing a 3D view.111 A previous study by Uejo et al has reported SEM images of fenofibrate-loaded MSNs. The SEM image of the physical mixture (PM) of fenofibrate-loaded MSNs showed both bulk fenofibrate powder and a fiber-like MSN structure. This showed that fenofibrate molecules interacted with MSNs during drug incorporation process and changed dispersion state compared to the PM sample. The micropores of MSNs served as an important factor in this particular dispersion state. Furthermore, the morphology of the sample with 66% fenofibrate-loaded showed particles that could be adsorbed onto the outer surfaces of MSNs fiber-like structure.96
In vitro Study
Dissolution Study
Dissolution study was conducted to evaluate the behavior of GGFA drug-loaded MSNs after dispersion. Lau et al reported dissolution profile of probucol-loaded MSNs, conducting in vitro dissolution tests were carried out on simulated intestinal fluid (SIF) under sink conditions.107 The burst release at drug loading of <30% wt was caused by amorphous PB from the pores of MSNs. This is because drug loading above complete pore filling can cause the recrystallization of probucol resulting in the decrease in drug release kinetics. Full release of probucol was achieved below this level except for MCM-41, where only 13.1% of probucol-loaded MSNs released the full amount of drug-loaded. This showed a strong dependency of release kinetics on the diffusion properties of dissolution media for the smaller pore material of MSNs.119 Furthermore, dose-dependent release profiles between 30 and 100 mg were evaluated to understand dissolution kinetics of probucol. At greater doses, a little increase in the amount of PB produced, followed by the absence of sink conditions, and the total percentage of medicine released declines. The formulation of SBA-15PB 29.9% only releases 67% of loaded PB after 24 hours at a dose of 100 mg. This tendency was observed across all mesoporous materials in this study, showing that PB reached its saturation state >30 wt% in dissolution media.120 Drug release patterns of the commercially available felodipine rapid-release and sustained tablets were also examined for comparison to confirm the practical application value of the produced formulation. The felodipine dissolving rate from felodipine-loaded MSNs was faster compared to the commercially available rapid-release felodipine tablet.93
Dissolution test of ritonavir-loaded MSNs was also carried out in 50 mM phosphate buffer pH 6.8 at 37°C under non-sink conditions, according to a previous study,3 as shown in Figure 9. Ritonavir crystal showed a slow dissolution rate to an RTV concentration of 0.24 μg/mL after 400 min, while dissolution of ritonavir amorphous was higher compared to the RTV crystal, at a concentration of 1.3 μg/mL. Specifically, ritonavir amorphous did not show the “spring-parachute” phenomenon but indicated rapid dissolution at the beginning of dissolution test, followed by a decrease in drug concentration due to its recrystallization. This phenomenon occurred because ritonavir amorphous prepared by solvent evaporation was not well dispersed in dissolution medium caused by the formation of large agglomerations, leading to a slower rate.121 In contrast, dissolution profile of ritonavir-loaded MSNs showed a spring-parachute phenomenon. The rapid dissolution of ritonavir amorphous was observed at the beginning of the test with a concentration of 2.22 μg/mL after 30 min, followed by a decrease in drug concentration due to associated nucleation and crystallization. Subsequently, their monomolecular dispersion of ritonavir amorphous within MSNs led to good dispersibility in dissolution medium at the beginning of dissolution test. Despite achieving a supersaturated solution, the release of ritonavir amorphous from MSNs was incomplete. The strong interaction between drug and the surface leads to an incomplete release of ritonavir from the pores of MSNs.122
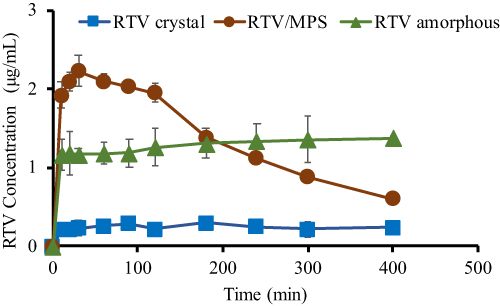 |
Figure 9 Dissolution profiles of ritonavir crystal, ritonavir amorphous, and ritonavir loaded MSN. Adapted from Budiman A, Aulifa DL. Characterization of drugs with good glass formers in loaded-mesoporous silica and its theoretical value relevance with mesopores surface and pore-filling capacity. Pharmaceuticals. 2022;15(1):93. Creative Commons.3 |
In vitro anti-Fungal Performance
The effect of drug loading on the antifungal activities was reported by Masood et al. In vitro, antifungal activities of pure salicylic acid (SA)/ketoconazole (KCZ) and SA/KCZ-loaded MSNs were observed on days 3, 5, 7, and 14. However, the differences in inhibition zone were observed between pure drug and drug-loaded MSNs. On day 3, the pure drug showed an inhibition zone of around 16.20 mm, while drug-loaded MSNs was 16.90 mm. After 14 days, the maximum value was observed at 16.70 and 17.90 mm for pure drug suspensions and drug-loaded MSNs, respectively. These results showed that the activity of SA/KCZ loaded in MSNs is a better formulation as compared to the pure SA/KCZ suspension.104
In vivo Study
Various biological effects including pharmacokinetics and anti-inflammatory pharmacodynamics were studied to evaluate the behaviors of nimesulide in MSNs system. The pharmacokinetic profiles and parameters are presented in Table 3. The result showed that nimesulide-loaded MSNs offered advantages in enhancing nimesulide bioavailability, as indicated by prolonged mean residence time (MRT), maximum concentration (Cmax), time to reach Cmax (Tmax), and area under the curve (AUC). The relative bioavailability of nimesulide-loaded MSNs was around 887.03%. By loading into MSNs, the oral bioavailability of nimesulide was enhanced due to the higher dissolution of the nimesulide amorphous within the carriers compared to crystalline NMS. Therefore, it was concluded that nimesulide-loaded MSNs showed a more effective drug delivery effect compared to crystalline state.106
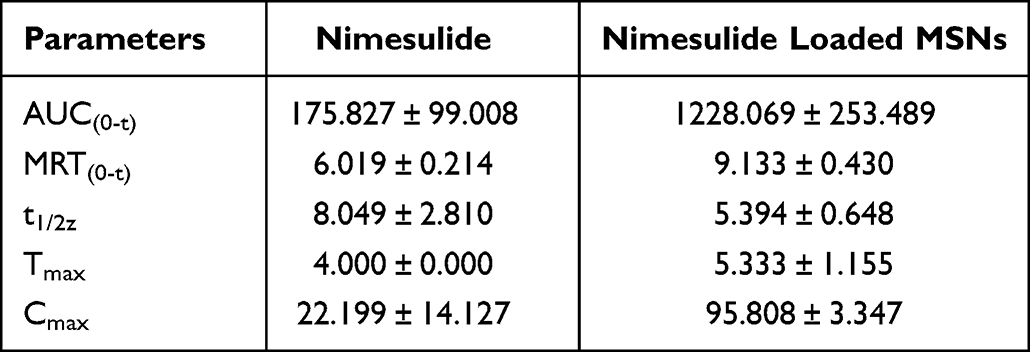 |
Table 3 The Pharmacokinetic Parameters of Nimesulide After Oral Administration of Nimesulide, and Nimesulide Loaded MSNs.106 |
Figure 10 shows the gradual increase in the paw edema degree in rats over time when administered normal saline, indicating the successful establishment of the model. The swelling rate profiles showed that nimesulide had an anti-inflammatory pharmacodynamic effect. Specifically, nimesulide-loaded MSNs showed a stronger anti-inflammatory pharmacodynamic effect compared to nimesulide crystal, indicating that the improved oral bioavailability led to a more effective therapeutic effect. The swelling inhibition rate of nimesulide-loaded MSNs was stronger, reaching a maximum multiple of 1.54 times compared to nimesulide crystal. This suggested that nimesulide-loaded MSNs delivered more nimesulide in response to in vivo environment, thereby presenting a stronger anti-inflammatory pharmacodynamic effect.106
 |
Figure 10 The anti-inflammatory effects of nimesulide-loaded MSN formulations on mouse ankle swelling. Reprinted from Microporous Mesoporous Mater, volume 294, Guo Y, Wu L, Gou K, et al. Functional mesoporous silica nanoparticles for delivering nimesulide with chiral recognition performance. 109862. Copyright 2020, with permission from Elsevier.106 |
The acceleration test was conducted to investigate the stability of the prepared nanoparticles. Figure 11 shows the confocal laser scanning microscopy (CLSM) pictures of different sections after 45 min. The cytomembrane and the nuclei were dyed red and blue, respectively, while the fluorescein isothiocyanate (FITC) presented a green fluorescence, which was formed through the superposition of three elements. In the control group, green fluorescence appeared in duodenum, as well as in jejunum and ileum of the dosing group. Consequently, the FITC solution was delivered into the intestinal tract, and FITC-labeled nanoparticles were also translated.93
 |
Figure 11 CLSM pictures of the duodenum, jejunum, and ileum after 45 min were given by control group and dosing group. Reprinted from Mater Sci Eng C, volume 47, Hu L, Sun H, Zhao Q, et al. Multilayer encapsulated mesoporous silica nanospheres as an oral sustained drug delivery system for the poorly water-soluble drug felodipine. 313–324. Copyright 2015, with permission from Elsevier.93 |
After 6 h, green fluorescence did not appear in any section for the control group, suggesting easy removal of FITC solution. However, for the dosing group, green fluorescence could be observed in the duodenum, jejunum, and ileum, showing that FITC-labeled nanoparticles had a stronger mucosa adhesive ability. After 12 h, for the dosing group light green fluorescence in the ileum was still observed, suggesting the strong mucosa adhesive ability of nanoparticles. Moreover, after 24 h, FITC-labeled nanoparticles were completely removed in the three sections of the intestine.93
Masood et al reported in vivo antifungal studies, wound healing studies, and histopathology studies on rabbits from the formulation of SA/KCZ-loaded MSNs. Antifungal studies were conducted to assess the efficacy of the optimized formulation of SA/KCZ-loaded MSNs in a rabbit model compared to SA/KCZ suspension. Fungal infection was induced in rabbits using isolated colonies of Candida albicans, showing that SA/KCZ-loaded MSNs indicated superior efficacy in eradicating infection, as none of the experimental animals presented a positive culture test. In contrast, in the case of SA/KCZ suspension, four out of six animals showed a positive culture test. Meanwhile, in the control group, all six animals showed a positive culture test with an average value of log CFU 4.24 ± 0.52. Rapid recovery from fungal infection was observed in the case of SA/KCZ-loaded MSNs, with no rabbits showing signs of a positive culture test. The significant effectiveness of SA/KCZ-loaded MSNs can be attributed to their improved bioadhesive nature, high occlusive property, enhanced oxygen supply to cells due to the porosity of silica nanoparticles, persistent keratolytic action along with antifungal properties, absence of bacterial growth based on silica particles, and continuous controlled availability of drug at the site of action.104
The wound healing studies of SA-KCZ loaded MSNs showed significant morphological changes on the 1st, 3rd, 7th, and 14th days. After treated drug-loaded MSNs, the edges of the wounded skin converged towards the center, appearing contracted and more healed compared to the control group. On the 3rd day after the injury, the superficial area of the wounded skin in both SA/KCZ suspension and SA-KCZ loaded MSNs was dried, with reduced inflammation around the wounded skin and a gradually forming pale yellow layer of crust. Although the wounded area decreased in both groups, the wound remained circular and larger in the control group. In contrast, the treated group showed a softer texture, and the wound was irregularly oval or circular with a smaller diameter. After 14 days, the wounded skin surface in the control group had transformed into hard crusts of a pale black appearance, with some having shed, showing skin tissue underneath. In the treated group, the wounded area had almost completely healed, with a small crust in the SA/KCZ suspension-treated group and no crust in MSN-treated group. As shown in Figure 12, the wounded skin treated with drug-loaded MSNs formulation showed rapid tissue repair.104
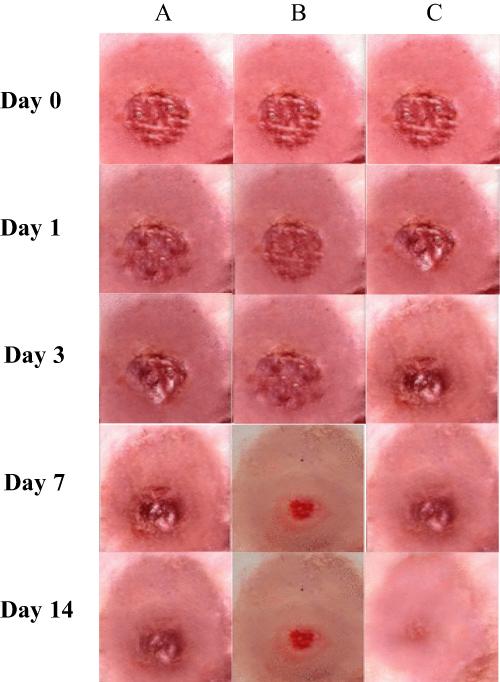 |
Figure 12 Wound healing effect in (A) control, (B) SA/KCZ suspension, and (C) SA/KCZ-loaded MSNSat day 1, day 3, day 7, and day 14. Reprinted from Masood A, Maheen S, Khan HU, et al. Pharmaco-technical evaluation of statistically formulated and optimized dual drug-loaded silica nanoparticles for improved antifungal efficacy and wound healing. ACS Omega. 2021;6(12):8210–8225. Copyright 2021; American Chemical Society. Creative Commons.104 |
Histopathology slides showed evidence of hemorrhage and the accumulation of extracellular matrix (ECM) in sheet-like formations in the control group on day 1. However, the control group showed significantly less macrophage accumulation compared to the two treated groups. In the SA-KCZ suspension-treated group, evidence of hemorrhage, accumulation of RBCs, and blood clots were visible. SA/KCZ-loaded MSNs showed evidence of hemorrhage, accumulation of RBCs, blood clots, and a smaller amount of sheet-like ECM appearance.
On day 14, sheet-like ECM and some wavy fibrous strands were found, but no appearance of follicular cell bases was observed in the control group. In the SA-KCZ suspension, most of the sheet-like extracellular structure had been replaced by wavy fibrous strand-like structures with evident infiltration by macrophages. This was attributed to the commencement of follicular base formation and trans-differentiation of cells. Comparatively, more differentiated cells compared to the control group and SA-KCZ suspension treated were found. The tissues in the SA/KCZ-loaded MSNs were healing rapidly, and skin was growing faster compared to the control and drug suspension. These results showed that the combination of drug-loaded MSNs showed a potential strategy to improve patient compliance by accelerating the healing of fungal infections.104
Author Perspective
This section discussed the speculated mechanism of drug dissolution from GGFA drug loaded MPS. Based on the results, dissolution of GGFA drug crystal was very low due to its poorly water-soluble drug. In the case of drug amorphous, drug gradually dissolved in dissolution medium, leading to low dissolution rate. This could be due to the interaction of water with the surface of the agglomeration of drug amorphous formed after dispersion into dissolution medium. The formation of agglomeration decreased the surface area of drug, leading to low wettability, and resulting in a low dissolution rate. In the case of drug-loaded MSNs, the GGFA drug was rapidly released and dissolved after dispersion into dissolution medium, leading to a high dissolution rate. This significantly high rate is attributed to the monomolecular dispersion of drug within MSNs, capable of improving the wettability of drug. According to the Noyes–Whitney/Nernst–Brunner equation, dissolution rate of drug was improved because of the surface area, which was wetted by dissolution medium.123
Regarding the bioavailability study, the absorption of poorly water-soluble drug, including GGFA drug, was enhanced only when drug was in a supersaturated state.124 Drug absorption commonly occurs by passive diffusion through movement from a higher concentration to the lower to achieve equilibrium. Based on absorption, the soluble drug in the body fluid, such as interstitial space, will move into the endothelium of blood vessels. Consequently, the solubility of drug in the aqueous compartment of the body plays a critical role in the absorption mechanism through passive diffusion. Drug absorption of drug loaded MSNs should be higher compared to crystal and amorphous. The high supersaturation of drug within MPS was achieved due to the monomolecular dispersion in dissolution medium. Meanwhile, absorption should be lower compared to drug-loaded MSNs due to their low wettability caused by agglomeration formation leading to a low dissolution rate.
Previous studies reported that higher concentrations of molecularly dissolved drug could significantly increase permeation rates.125–127This showed that the absorption of GGFA, poorly water-soluble drug, in MSNs formulation was considerably absorbed by the intestinal epithelium. Furthermore, the permeability could only be increased by high concentrations of molecularly dissolved drug. This showed that the absorption of GGFA-loaded MSNs could increase the concentration of molecularly dissolved API driven by passive diffusion. Improving the bioavailability of poorly water-soluble GGFA drug showed promising potential in enhancing their pharmacokinetics, efficacy, and safety by loading into MSNs. The speculated mechanism of bioavailability and efficacy improvement from GGFA drug in MSNs systems is summarized in Figure 13.
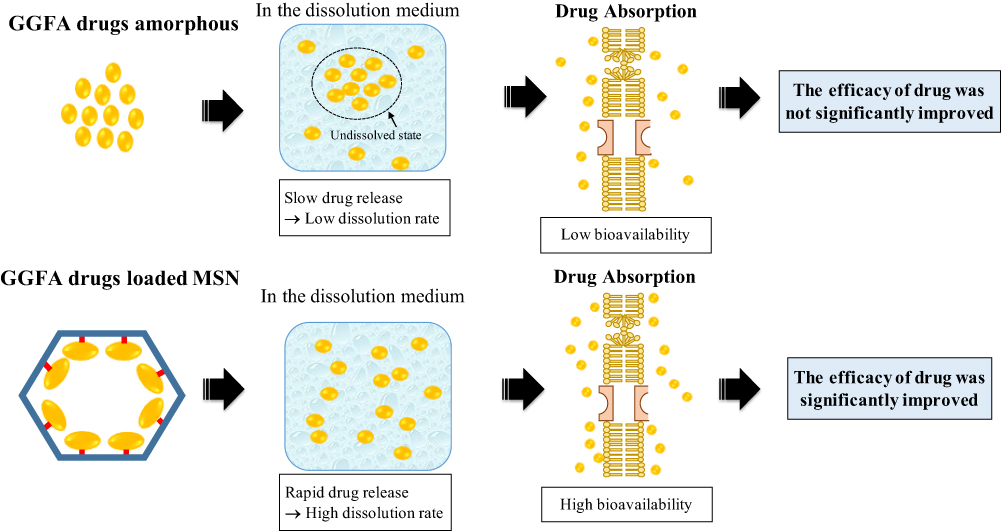 |
Figure 13 The speculated mechanism of bioavailability improvement from GGFA-loaded MSNs compared to its amorphous form alone. |
Conclusions
In conclusion, this study contributed to elucidating different mechanisms of GGFA drug with and without MSNs, including their impact on dissolution, bioavailability, and pharmacological activity. The results showed that the formation of agglomeration from GGFA drug after dispersion in the bulk medium led to the slow release at the beginning of dissolution test. The nanosized of drug within MSNs improved their dissolution profile, leading to the improvement of bioavailability as well as efficacy of GGFA drug. However, there were challenges in the preparation of the GGFA drug within the MPS system, particularly in selecting the preparation method and dissolution profile. Despite generating a supersaturated solution, the strong interaction between GGFA drug and the surface of MSNs resulted in the incomplete release of drug in the bulk medium, posing challenges for development. Moreover, this study provided fundamental insights into the GGFA drug within the MPS system, which was significant when formulating strategies to improve the pharmaceutical properties of poorly water-soluble drug.
Acknowledgments
We would like to thank the National Research and Innovation Agency (BRIN, RIIM3) and the Indonesia Endowment Funds for Education (LPDP) for supporting this work. We also would like to thank Universitas Padjadjaran for APC.
Funding
This research was funded by the National Research and Innovation Agency (BRIN, RIIM3) and the Indonesia Endowment Funds for Education (LPDP) to Diah Lia Aulifa (No.: 61/IV/KS/5/2023; No.: 2131/UN6.3.1/PT.00/2023).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Budiman A, Nurfadilah N, Muchtaridi M, Sriwidodo S, Aulifa DL, Rusdin A. The Impact of Water-Soluble Chitosan on the Inhibition of Crystal Nucleation of Alpha-Mangostin from Supersaturated Solutions. Polymers. 2022;14(20):4370. doi:10.3390/polym14204370
2. Zhang G. Phase transformation considerations during process development and manufacture of solid oral dosage forms. Adv Drug Deliv Rev. 2004;56(3):371–390. doi:10.1016/j.addr.2003.10.009
3. Budiman A, Aulifa DL. Characterization of drugs with good glass formers in loaded-mesoporous silica and its theoretical value relevance with mesopores surface and pore-filling capacity. Pharmaceuticals. 2022;15(1):93. doi:10.3390/ph15010093
4. Okada H, Ueda K, Yasuda Y, et al. Correlation between drug dissolution and resistance to water-induced phase separation in solid dispersion formulations revealed by solid-state NMR spectroscopy. Int J Pharm. 2020;577:119086. doi:10.1016/j.ijpharm.2020.119086
5. Baird JA, Van Eerdenbrugh B, Taylor LS. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J Pharm Sci. 2010;99(9):3787–3806. doi:10.1002/jps.22197
6. Blaabjerg LI, Lindenberg E, Löbmann K, Grohganz H, Rades T. Is there a correlation between the glass forming ability of a drug and its supersaturation propensity? Int J Pharm. 2018;538(1–2):243–249. doi:10.1016/j.ijpharm.2018.01.013
7. Blaabjerg LI, Lindenberg E, Löbmann K, Grohganz H, Rades T. Glass forming ability of amorphous drugs investigated by continuous cooling and isothermal transformation. Mol Pharm. 2016;13(9):3318–3325. doi:10.1021/acs.molpharmaceut.6b00650
8. Blaabjerg LI, Lindenberg E, Rades T, Grohganz H, Löbmann K. Influence of preparation pathway on the glass forming ability. Int J Pharm. 2017;521(1–2):232–238. doi:10.1016/j.ijpharm.2017.02.042
9. Narayan R, Nayak U, Raichur A, Garg S. Mesoporous silica nanoparticles: a comprehensive review on synthesis and recent advances. Pharmaceutics. 2018;10(3):118. doi:10.3390/pharmaceutics10030118
10. Slowing II, Trewyn BG, Lin VS-Y. Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins. J Am Chem Soc. 2007;129(28):8845–8849. doi:10.1021/ja0719780
11. Deodhar GV, Adams ML, Trewyn BG. Controlled release and intracellular protein delivery from mesoporous silica nanoparticles. Biotechnol J. 2017;12(1). doi:10.1002/biot.201600408
12. Cha W, Fan R, Miao Y, et al. Mesoporous silica nanoparticles as carriers for intracellular delivery of nucleic acids and subsequent therapeutic applications. Molecules. 2017;22(5):782. doi:10.3390/molecules22050782
13. Tao C, Zhu Y, Xu Y, Zhu M, Morita H, Hanagata N. Mesoporous silica nanoparticles for enhancing the delivery efficiency of immunostimulatory DNA drugs. Dalton Trans. 2014;43(13):5142–5150. doi:10.1039/C3DT53433B
14. Möller K, Müller K, Engelke H, Bräuchle C, Wagner E, Bein T. Highly efficient siRNA delivery from core–shell mesoporous silica nanoparticles with multifunctional polymer caps. Nanoscale. 2016;8(7):4007–4019. doi:10.1039/C5NR06246B
15. Hanafi-Bojd MY, Ansari L, Malaekeh-Nikouei B. Codelivery of anticancer drugs and siRNA by mesoporous silica nanoparticles. Ther Deliv. 2016;7(9):649–655. doi:10.4155/tde-2016-0045
16. Riikonen J, Xu W, Lehto VP. Mesoporous systems for poorly soluble drugs – recent trends. Int J Pharm. 2018;536(1):178–186. doi:10.1016/j.ijpharm.2017.11.054
17. Maleki A, Kettiger H, Schoubben A, Rosenholm JM, Ambrogi V, Hamidi M. Mesoporous silica materials: from physico-chemical properties to enhanced dissolution of poorly water-soluble drugs. J Control Release. 2017;262:329–347. doi:10.1016/j.jconrel.2017.07.047
18. Wang Y, Zhao Q, Han N, et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine. 2015;11(2):313–327. doi:10.1016/j.nano.2014.09.014
19. Mamaeva V, Sahlgren C, Lindén M. Mesoporous silica nanoparticles in medicine—recent advances. Adv Drug Deliv Rev. 2013;65(5):689–702. doi:10.1016/j.addr.2012.07.018
20. Czarnobaj K, Prokopowicz M, Greber K. Use of materials based on polymeric silica as bone-targeted drug delivery systems for metronidazole. Int J Mol Sci. 2019;20(6):1311. doi:10.3390/ijms20061311
21. Šoltys M, Zůza D, Boleslavská T, et al. Drug loading to mesoporous silica carriers by solvent evaporation: a comparative study of amorphization capacity and release kinetics. Int J Pharm. 2021;607:120982. doi:10.1016/j.ijpharm.2021.120982
22. Kinnari P, Mäkilä E, Heikkilä T, Salonen J, Hirvonen J, Santos HA. Comparison of mesoporous silicon and non-ordered mesoporous silica materials as drug carriers for itraconazole. Int J Pharm. 2011;414(1–2):148–156. doi:10.1016/j.ijpharm.2011.05.021
23. Qian KK, Bogner RH. Spontaneous crystalline-to-amorphous phase transformation of organic or medicinal compounds in the presence of porous media, part 1: thermodynamics of spontaneous amorphization. J Pharm Sci. 2011;100(7):2801–2815. doi:10.1002/jps.22519
24. Andersson J, Rosenholm J, Areva S, Lindén M. Influences of material characteristics on ibuprofen drug loading and release profiles from ordered micro- and mesoporous silica matrices. Chem Mater. 2004;16(21):4160–4167. doi:10.1021/cm0401490
25. Genina N, Hadi B, Löbmann K. Hot melt extrusion as solvent-free technique for a continuous manufacturing of drug-loaded mesoporous silica. J Pharm Sci. 2018;107(1):149–155. doi:10.1016/j.xphs.2017.05.039
26. Hempel NJ, Brede K, Olesen NE, Genina N, Knopp MM, Löbmann K. A fast and reliable DSC-based method to determine the monomolecular loading capacity of drugs with good glass-forming ability in mesoporous silica. Int J Pharm. 2018;544(1):153–157. doi:10.1016/j.ijpharm.2018.04.035
27. Bavnhøj CG, Knopp MM, Madsen CM, Löbmann K. The role interplay between mesoporous silica pore volume and surface area and their effect on drug loading capacity. Int J Pharm X. 2019;1:100008. doi:10.1016/j.ijpx.2019.100008
28. Kawakami K. Crystallization tendency of pharmaceutical glasses: relevance to compound properties, impact of formulation process, and implications for design of amorphous solid dispersions. Pharmaceutics. 2019;11(5):202. doi:10.3390/pharmaceutics11050202
29. Maulvi FA, Dalwadi SJ, Thakkar VT, Soni TG, Gohel MC, Gandhi TR. Improvement of dissolution rate of aceclofenac by solid dispersion technique. Powder Technol. 2011;207(1–3):47–54. doi:10.1016/j.powtec.2010.10.009
30. Iolascon G, Giménez S, Mogyorósi D. A review of aceclofenac: analgesic and anti-inflammatory effects on musculoskeletal disorders. J Pain Res. 2021;14:3651–3663. doi:10.2147/JPR.S326101
31. Balata G, Bakera R, Mahdi M. Improvement of solubility and dissolution properties of clotrimazole by solid dispersions and inclusion complexes. Indian J Pharm Sci. 2011;73(5):517. doi:10.4103/0250-474X.98995
32. Crowley PD, Gallagher HC. Clotrimazole as A pharmaceutical: past, present and future. J Appl Microbiol. 2014;117(3):611–617. doi:10.1111/jam.12554
33. Amr S. Formulation, characterization and In-Vitro release of oral felodipine self-nanoemulsifying drug delivery Systems. Al-Azhar J Pharm Sci. 2018;57(1):18–51. doi:10.21608/ajps.2018.46619
34. Fuhr LM, Marok FZ, Mees M, Mahfoud F, Selzer D, Lehr T. A physiologically based pharmacokinetic and pharmacodynamic model of the CYP3A4 substrate felodipine for drug–drug interaction modeling. Pharmaceutics. 2022;14(7):1474. doi:10.3390/pharmaceutics14071474
35. Vimalson DC, Parimalakrishnan S, Jeganathan NS, Anbazhagan S. Enhancement of Solubility and dissolution characteristics of fenofibrate by solid dispersion technique. Int Research J Pharm. 2018;9(10):145–150. doi:10.7897/2230-8407.0910242
36. Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98(19):2088–2093.
37. Friuli V, Bruni G, Musitelli G, Conte U, Maggi L. Influence of dissolution media and presence of alcohol on the in vitro performance of pharmaceutical products containing an insoluble drug. J Pharm Sci. 2018;107(1):507–511. doi:10.1016/j.xphs.2017.06.001
38. González-Barnadas A, Camps-Font O, Martín-Fatás P, Figueiredo R, Gay-Escoda C, Valmaseda-Castellón E. Efficacy and safety of selective COX-2 inhibitors for pain management after third molar removal: a meta-analysis of randomized clinical trials. Clin. Oral Investig. 2020;24(1):79–96.
39. Bahl D, Bogner RH. Amorphization alone does not account for the enhancement of solubility of drug co-ground with silicate: the case of indomethacin. AAPS Pharm Sci Tech. 2008;9(1):146–153. doi:10.1208/s12249-007-9013-9
40. Lucas S. The Pharmacology of Indomethacin. Headache. 2016;56(2):436–446.
41. Kozyra A, Mugheirbi NA, Paluch KJ, Garbacz G, Tajber L. Phase diagrams of polymer-dispersed liquid crystal systems of itraconazole/component immiscibility induced by molecular anisotropy. Mol Pharm. 2018;15(11):5192–5206. doi:10.1021/acs.molpharmaceut.8b00724
42. De Beule K, Van Gestel J. Pharmacology of itraconazole. Drugs. 2001;61 Suppl 1:27–37.
43. Pashazadeh‐Panahi P, Hasanzadeh M, Eivazzadeh‐Keihan R. Spectrophotometric study of ketoconazole binding with citrate capped silver nanoparticles and its monitoring in human plasma samples. J Mol Recognit. 2020;33(5). doi:10.1002/jmr.2830
44. Van Tyle JH; Ketoconazole. Mechanism of action, spectrum of activity, pharmacokinetics, drug interactions, adverse reactions and therapeutic use. Pharmacotherapy. 1984;4(6):343–373.
45. Gantiva M, Martínez F. Thermodynamic analysis of the solubility of ketoprofen in some propylene glycol+ water cosolvent mixtures. Fluid Phase Equilib. 2010;293(2):242–250.
46. Carbone C, Rende P, Comberiati P, Carnovale D, Mammí M, De Sarro G. The safety of ketoprofen in different ages. J Pharmacol Pharmacother. 2013;4(1_suppl):S99–S103. doi:10.4103/0976-500X.120967
47. Kundawala AJ, Patel PH. Solubility enhancement of loratadine by solid dispersion techniques. Int J Chemtech Res. 2017;10(7):207–217.
48. Baroody FM, Naclerio RM. Antiallergic effects of H1-receptor antagonists. Allergy. 2000;55(Suppl 64):17–27.
49. Shahzadi I, Masood MI, Chowdhary F, et al. Microemulsion Formulation for Topical Delivery of Miconazole Nitrate. Int J Pharm Sci Rev Res. 2014;24(2):30–36.
50. Mazzarino M, Comunità F, de la Torre X, Molaioni F, Botrè F. Effects of the administration of miconazole by different routes on the biomarkers of the “steroidal module” of the athlete biological passport. Drug Test Anal. 2021;13(10):1712–1726. doi:10.1002/dta.3121
51. Surov AO, Voronin AP, Drozd KV, et al. Polymorphic forms of antiandrogenic drug nilutamide: structural and thermodynamic aspects. Phys Chem Chem Phys. 2021;23(16):9695–9708. doi:10.1039/D1CP00793A
52. Rashid M, Shamshavali K, Chhabra M. Efficacy and safety of nilutamide in patients with metastatic prostate cancer who underwent orchiectomy: a systematic review and metaanalysis. Curr Clin Pharmacol. 2019;14(2):108–115. doi:10.2174/1574884714666190112151202
53. Piel G, Pirotte B, Delneuville I, et al. Study of the influence of both cyclodextrins and L-lysine on the aqueous solubility of nimesulide; isolation and characterization of nimesulide–L-lysine–cyclodextrin complexes. J Pharm Sci. 1997;86(4):475–480.
54. Vunnam N, Young MC, Liao EE, et al. Nimesulide, a COX-2 inhibitor, sensitizes pancreatic cancer cells to TRAIL-induced apoptosis by promoting DR5 clustering †. Cancer Biol Ther. 2023;24(1). doi:10.1080/15384047.2023.2176692
55. Kamisetti RR, Mekala M, Muvvala S, Penmatsa DB. Fast dissolving tablets of pimozide: design, optimization and in vitro characterization. Indones J Pharm. 2015;26(2):114. doi:10.14499/indonesianjpharm26iss2pp114
56. Ranjan A, Kaushik I, Srivastava SK. Pimozide suppresses the growth of brain tumors by targeting stat3-mediated autophagy. Cells. 2020;9(9):2141. doi:10.3390/cells9092141
57. Han L, Yang Q, Shen T, Qing J, Wang J. Lymphatic transport of orally administered probucol-loaded mPEG-DSPE micelles. Drug Deliv. 2015;1–7. doi:10.3109/10717544.2015.1028600
58. Liu HW, Luo Y, Zhou YF, Chen ZP. Probucol prevents diabetes-induced retinal neuronal degeneration through upregulating Nrf2. Biomed Res Int. 2020;2020:1–8. doi:10.1155/2020/3862509
59. Loftsson T. Degradation Pathways. In: Drug Stability for Pharmaceutical Scientists. Vol. 3.
60. Ford DJ, Raj PP, Singh P, Regan KM, Ohlweiler D. Differential peripheral nerve block by local anesthetics in the cat. Anesthesiology. 1984;60(1):28–33.
61. Morris JB, Tisi DA, Tan DCT, Worthington JH. Development and Palatability assessment of norvir® (Ritonavir) 100 mg powder for pediatric population. Int J Mol Sci. 2019;20(7):1718. doi:10.3390/ijms20071718
62. Loelius SG, Lannan KL, Blumberg N, Phipps RP, Spinelli SL. The HIV protease inhibitor, ritonavir, dysregulates human platelet function in vitro. Thromb Res. 2018;169:96–104.
63. Corma A. From microporous to mesoporous molecular sieve materials and their use in catalysis. Chem Rev. 1997;97(6):2373–2420. doi:10.1021/cr960406n
64. Knežević NŽ, Durand JO. Large pore mesoporous silica nanomaterials for application in delivery of biomolecules. Nanoscale. 2015;7(6):2199–2209. doi:10.1039/C4NR06114D
65. Jana SK, Nishida R, Shindo K, Kugita T, Namba S. Pore size control of mesoporous molecular sieves using different organic auxiliary chemicals. Microporous Mesoporous Mater. 2004;68(1–3):133–142. doi:10.1016/j.micromeso.2003.12.010
66. Widenmeyer M, Anwander R. Pore size control of highly ordered mesoporous silica MCM-48. Chem Mater. 2002;14(4):1827–1831. doi:10.1021/cm011273b
67. Zhang L, Qiao S, Jin Y, et al. Fabrication and size‐selective bioseparation of magnetic silica nanospheres with highly ordered periodic mesostructure. Adv Funct Mater. 2008;18(20):3203–3212. doi:10.1002/adfm.200800363
68. Kruk M. Access to ultralarge-pore ordered mesoporous materials through selection of surfactant/swelling-agent micellar templates. Acc Chem Res. 2012;45(10):1678–1687. doi:10.1021/ar200343s
69. Lettow JS, Han YJ, Schmidt-Winkel P, et al. Hexagonal to mesocellular foam phase transition in polymer-templated mesoporous silicas. Langmuir. 2000;16(22):8291–8295. doi:10.1021/la000660h
70. Feng P, Bu X, Stucky GD, Pine DJ. Monolithic mesoporous silica templated by microemulsion liquid crystals. J Am Chem Soc. 2000;122(5):994–995. doi:10.1021/ja992921j
71. Ulagappan N, Rao CNR. Evidence for supramolecular organization of alkane and surfactant molecules in the process of forming mesoporous Silica. Chem Comm. 1996;(24):2759. doi:10.1039/cc9960002759
72. Mandal M, Kruk M. Versatile approach to synthesis of 2-D hexagonal ultra-large-pore periodic mesoporous organosilicas. J Mater Chem. 2010;20(35):7506. doi:10.1039/c0jm01170c
73. Sayari A, Yang Y, Kruk M, Jaroniec M. Expanding the pore size of MCM-41 silicas: use of amines as expanders in direct synthesis and postsynthesis procedures. J Phys Chem B. 1999;103(18):3651–3658. doi:10.1021/jp984504j
74. Zhang J, Rosenholm JM, Gu H. Molecular confinement in fluorescent magnetic mesoporous silica nanoparticles: effect of pore size on multifunctionality. Chem Phys Chem. 2012;13(8):2016–2019. doi:10.1002/cphc.201100943
75. Zhang J, Li X, Rosenholm JM, chen GH. Synthesis and characterization of pore size-tunable magnetic mesoporous silica nanoparticles. J Colloid Interface Sci. 2011;361(1):16–24. doi:10.1016/j.jcis.2011.05.038
76. Wu SH, Mou CY, Lin HP. Synthesis of mesoporous silica nanoparticles. Chem Soc Rev. 2013;42(9):3862. doi:10.1039/c3cs35405a
77. Chiang YD, Lian HY, Leo SY, Wang SG, Yamauchi Y, Wu KCW. Controlling particle size and structural properties of mesoporous silica nanoparticles using the Taguchi method. J Phys Chem C. 2011;115(27):13158–13165. doi:10.1021/jp201017e
78. Qiao ZA, Zhang L, Guo M, Liu Y, Huo Q. Synthesis of mesoporous silica nanoparticles via controlled hydrolysis and condensation of silicon alkoxide. Chem Mater. 2009;21(16):3823–3829. doi:10.1021/cm901335k
79. Fowler CE, Khushalani D, Lebeau B, Mann S. Nanoscale materials with mesostructured interiors. Adv Mater. 2001;13(9):649–652. doi:10.1002/1521-4095(200105)13:9
80. Ma K, Sai H, Wiesner U. Ultrasmall Sub-10 nm near-infrared fluorescent mesoporous silica nanoparticles. J Am Chem Soc. 2012;134(32):13180–13183. doi:10.1021/ja3049783
81. Lv X, Zhang L, Xing F, Lin H. Controlled synthesis of monodispersed mesoporous silica nanoparticles: particle size tuning and formation mechanism investigation. Microporous Mesoporous Mater. 2016;225:238–244. doi:10.1016/j.micromeso.2015.12.024
82. Manzano M, Vallet‐Regí M. Mesoporous silica nanoparticles for drug delivery. Adv Funct Mater. 2020;30(2). doi:10.1002/adfm.201902634
83. Rosenholm M, Sahlgren J, Linden C. Multifunctional mesoporous silica nanoparticles for combined therapeutic, diagnostic and targeted action in cancer treatment. Curr Drug Targets. 2011;12(8):1166–1186. doi:10.2174/138945011795906624
84. Fuyuhiko T. Mesoporous Silica-Based Nanomaterials and Biomedical Applications - Part A.
85. Ambrogio MW, Thomas CR, Zhao YL, Zink JI, Stoddart JF. Mechanized silica nanoparticles: a new frontier in theranostic nanomedicine. Acc Chem Res. 2011;44(10):903–913. doi:10.1021/ar200018x
86. Lee JE, Lee N, Kim T, Kim J, Hyeon T. Multifunctional mesoporous silica nanocomposite nanoparticles for theranostic applications. Acc Chem Res. 2011;44(10):893–902. doi:10.1021/ar2000259
87. Peng F, Su Y, Zhong Y, Fan C, Lee ST, He Y. Silicon nanomaterials platform for bioimaging, biosensing, and cancer therapy. Acc Chem Res. 2014;47(2):612–623. doi:10.1021/ar400221g
88. Patil LD, Verma U, Patil UD, Naik JB, Narkhede JS. Inclusion of aceclofenac in mesoporous silica nanoparticles: drug release study and statistical optimization of encapsulation efficiency by response surface methodology. Mater Technol. 2019;34(12):751–763. doi:10.1080/10667857.2019.1624301
89. Kumar D, Sailaja Chirravuri SV, Shastri NR. Impact of surface area of silica particles on dissolution rate and oral bioavailability of poorly water soluble drugs: a case study with aceclofenac. Int J Pharm. 2014;461(1–2):459–468. doi:10.1016/j.ijpharm.2013.12.017
90. Gignone A, Manna L, Ronchetti S, Banchero M, Onida B. Incorporation of clotrimazole in ordered mesoporous silica by supercritical CO2. Microporous Mesoporous Mater. 2014;200:291–296. doi:10.1016/j.micromeso.2014.05.031
91. Le TT, Elzhry Elyafi AK, Mohammed AR, Al-Khattawi A. Delivery of Poorly Soluble Drugs via Mesoporous Silica: impact of Drug Overloading on Release and Thermal Profiles. Pharmaceutics. 2019;11(6):269. doi:10.3390/pharmaceutics11060269
92. Wu C, Zhao Z, Zhao Y, Hao Y, Liu Y, Liu C. Preparation of a push–pull osmotic pump of felodipine solubilized by mesoporous silica nanoparticles with a core–shell structure. Int J Pharm. 2014;475(1–2):298–305. doi:10.1016/j.ijpharm.2014.08.033
93. Hu L, Sun H, Zhao Q, et al. Multilayer encapsulated mesoporous silica nanospheres as an oral sustained drug delivery system for the poorly water-soluble drug felodipine. Mater Sci Eng C. 2015;47:313–324. doi:10.1016/j.msec.2014.10.067
94. Ahern RJ, Hanrahan JP, Tobin JM, Ryan KB, Crean AM. Comparison of fenofibrate–mesoporous silica drug-loading processes for enhanced drug delivery. Eur J Pharm Sci. 2013;50(3–4):400–409. doi:10.1016/j.ejps.2013.08.026
95. Van Speybroeck M, Mellaerts R, Mols R, et al. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur J Pharm Sci. 2010;41(5):623–630. doi:10.1016/j.ejps.2010.09.002
96. Uejo F, Limwikrant W, Moribe K, Yamamoto K. Dissolution improvement of fenofibrate by melting inclusion in mesoporous silica. Asian J Pharm Sci. 2013;8(6):329–335. doi:10.1016/j.ajps.2013.11.001
97. Wu C, Zhao Z, Zhao Y, Hao Y, Liu Y, Zhao W. Development of an oral push-pull osmotic pump of fenofibrate-loaded mesoporous silica nanoparticles. Int J Nanomed. 2015;1691. doi:10.2147/IJN.S76755
98. Charnay C, Bégu S, Tourné-Péteilh C, Nicole L, Lerner DA, Devoisselle JM. Inclusion of ibuprofen in mesoporous templated silica: drug loading and release property. Eur J Pharm Biopharm. 2004;57(3):533–540. doi:10.1016/j.ejpb.2003.12.007
99. Wang Y, Ke J, Gou K, et al. Amino functionalized mesoporous silica with twisted rod-like shapes: synthetic design, in vitro and in vivo evaluation for ibuprofen delivery. Microporous Mesoporous Mater. 2020;294:109896. doi:10.1016/j.micromeso.2019.109896
100. Braz WR, Rocha NL, de Faria EH, et al. Incorporation of anti-inflammatory agent into mesoporous silica. Nanotechnology. 2016;27(38):385103. doi:10.1088/0957-4484/27/38/385103
101. Zhang W, Zheng N, Chen L, et al. Effect of shape on mesoporous silica nanoparticles for oral delivery of indomethacin. Pharmaceutics. 2018;11(1):4. doi:10.3390/pharmaceutics11010004
102. Ferreira NH, Ribeiro AB, Rinaldi-Neto F, et al. Anti-melanoma activity of indomethacin incorporated into mesoporous silica nanoparticles. Pharm Res. 2020;37(9):172. doi:10.1007/s11095-020-02903-y
103. Ren X, Cheng S, Liang Y, et al. Mesoporous silica nanospheres as nanocarriers for poorly soluble drug itraconazole with high loading capacity and enhanced bioavailability. Microporous Mesoporous Mater. 2020;305:110389. doi:10.1016/j.micromeso.2020.110389
104. Masood A, Maheen S, Khan HU, et al. Pharmaco-technical evaluation of statistically formulated and optimized dual drug-loaded silica nanoparticles for improved antifungal efficacy and wound healing. ACS Omega. 2021;6(12):8210–8225. doi:10.1021/acsomega.0c06242
105. Abd-Elrahman AA, El Nabarawi MA, Hassan DH, Taha AA. Ketoprofen mesoporous silica nanoparticles SBA-15 hard gelatin capsules: preparation and in vitro / in vivo characterization. Drug Deliv. 2016;23(9):3387–3398. doi:10.1080/10717544.2016.1186251
106. Guo Y, Wu L, Gou K, et al. Functional mesoporous silica nanoparticles for delivering nimesulide with chiral recognition performance. Microporous Mesoporous Mater. 2020;294:109862. doi:10.1016/j.micromeso.2019.109862
107. Lau M, Giri K, Garcia-Bennett AE. Antioxidant Properties of Probucol Released from Mesoporous Silica. Eur J Pharm Sci. 2019;138:105038. doi:10.1016/j.ejps.2019.105038
108. Ayad MM, Salahuddin NA, Torad NL, El-Nasr AA. pH-responsive sulphonated mesoporous silica: a comparative drug release study. RSC Adv. 2016;6(63):57929–57940. doi:10.1039/C6RA07022A
109. Dening TJ, Taylor LS. Supersaturation potential of ordered mesoporous silica delivery systems. part 1: dissolution performance and drug membrane transport rates. Mol Pharm. 2018;15(8):3489–3501. doi:10.1021/acs.molpharmaceut.8b00488
110. Mahajan M, Rajput S. Development of mesoporous silica nanoparticles of ritonavir with enhanced bioavailability potential: formulation optimization, in-vitro and in-vivo evaluation. Int J Pharm Sci Res. 2018;9(10):4127–4137.
111. Trzeciak K, Chotera-Ouda A, Bak-Sypien II, Potrzebowski MJ. Mesoporous silica particles as drug delivery systems—the state of the art in loading methods and the recent progress in analytical techniques for monitoring these processes. Pharmaceutics. 2021;13(7):950. doi:10.3390/pharmaceutics13070950
112. Budiman A, Aulifa DL. A comparative study of the pharmaceutical properties between amorphous drugs loaded-mesoporous silica and pure amorphous drugs prepared by solvent evaporation. Pharmaceuticals. 2022;15(6):730. doi:10.3390/ph15060730
113. Dengale SJ, Ranjan OP, Hussen SS, et al. Preparation and characterization of co-amorphous ritonavir–indomethacin systems by solvent evaporation technique: improved dissolution behavior and physical stability without evidence of intermolecular interactions. Eur J Pharm Sci. 2014;62:57–64. doi:10.1016/j.ejps.2014.05.015
114. Acharya M, Mishra S, Sahoo N, Mallick R. Infrared spectroscopy for analysis of co-processed ibuprofen and magnesium trisilicate at milling and freeze drying. Acta Chim Slov. 2017;45–54. doi:10.17344/acsi.2016.2772
115. Budiman A, Aulifa DL. Encapsulation of drug into mesoporous silica by solvent evaporation: a comparative study of drug characterization in mesoporous silica with various molecular weights. Heliyon. 2021;7(12):e08627. doi:10.1016/j.heliyon.2021.e08627
116. Pretsch E, Bühlmann P, Affolter C. Structure Determination of Organic Compounds. Tables of Spectral Data.
117. Tang Q, Xu Y, Wu D, Sun Y. A study of carboxylic-modified mesoporous silica in controlled delivery for drug famotidine. J Solid State Chem. 2006;179(5):1513–1520. doi:10.1016/j.jssc.2006.02.004
118. Moritz M, Łaniecki M. SBA-15 mesoporous material modified with APTES as the carrier for 2-(3-Benzoylphenyl)propionic acid. Appl Surf Sci. 2012;258(19):7523–7529. doi:10.1016/j.apsusc.2012.04.076
119. McCarthy CA, Ahern RJ, Devine KJ, Crean AM. Role of Drug Adsorption onto the Silica Surface in Drug Release from Mesoporous Silica Systems. Mol Pharm. 2018;15(1):141–149. doi:10.1021/acs.molpharmaceut.7b00778
120. Kjellman T, Xia X, Alfredsson V, Garcia-Bennett AE. Influence of microporosity in SBA-15 on the release properties of anticancer drug dasatinib. J Mater Chem B. 2014;2(32):5265. doi:10.1039/C4TB00418C
121. Ilevbare GA, Taylor LS. Liquid–liquid phase separation in highly supersaturated aqueous solutions of poorly water-soluble drugs: implications for solubility enhancing formulations. Cryst Growth Des. 2013;13(4):1497–1509. doi:10.1021/cg301679h
122. Dening TJ, Zemlyanov D, Taylor LS. Application of an adsorption isotherm to explain incomplete drug release from ordered mesoporous silica materials under supersaturating conditions. J Control Release. 2019;307:186–199. doi:10.1016/j.jconrel.2019.06.028
123. Li W, Quan P, Zhang Y, et al. Influence of drug physicochemical properties on absorption of water insoluble drug nanosuspensions. Int J Pharm. 2014;460(1–2):13–23. doi:10.1016/j.ijpharm.2013.10.038
124. Buckley ST, Frank KJ, Fricker G, Brandl M. Biopharmaceutical classification of poorly soluble drugs with respect to “enabling formulations. Eur J Pharm Sci. 2013;50(1):8–16. doi:10.1016/j.ejps.2013.04.002
125. Fong SYK, Martins SM, Brandl M, Bauer-Brandl A. Solid phospholipid dispersions for oral delivery of poorly soluble drugs: investigation into celecoxib incorporation and solubility-in vitro permeability enhancement. J Pharm Sci. 2016;105(3):1113–1123. doi:10.1016/S0022-3549(15)00186-0
126. Jacobsen AC, Elvang PA, Bauer-Brandl A, Brandl M. A dynamic in vitro permeation study on solid mono- and diacyl-phospholipid dispersions of celecoxib. Eur J Pharm Sci. 2019;127:199–207. doi:10.1016/j.ejps.2018.11.003
127. Ueda K, Higashi K, Kataoka M, Yamashita S, Yamamoto K, Moribe K. Inhibition mechanism of hydroxypropyl methylcellulose acetate succinate on drug crystallization in gastrointestinal fluid and drug permeability from A supersaturated Solution. Eur J Pharm Sci. 2014;62:293–300. doi:10.1016/j.ejps.2014.06.007
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.