Back to Journals » Drug Design, Development and Therapy » Volume 12
Characterization of clinical grade CD19 chimeric antigen receptor T cells produced using automated CliniMACS prodigy system
Authors Zhang W, Jordan KR, Schulte B, Purev E
Received 24 May 2018
Accepted for publication 9 August 2018
Published 5 October 2018 Volume 2018:12 Pages 3343—3356
DOI https://doi.org/10.2147/DDDT.S175113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Wei Zhang,1 Kimberly R Jordan,2 Brian Schulte,3 Enkhtsetseg Purev1
1Division of Hematology, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA; 2Division of Immunology, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA; 3Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA
Background: Chimeric antigen receptor (CAR) T-cell therapy is highly effective for treating acute lymphoblastic leukemia and non-Hodgkin’s lymphoma with high rate complete responses. However, the broad clinical application of CAR T-cell therapy has been challenging, largely due to the lack of widespread ability to produce and high cost of CAR T-cell products using traditional methods of production. Automated cell processing in a closed system has emerged as a potential method to increase the feasibility of producing CAR T cells locally at academic centers due to its minimal reliance on experienced labor, thereby making the process less expensive and more consistent than traditional methods of production.
Method: In this study, we describe the successful production of clinical grade CD19 CAR T cells using the Miltenyi CliniMACS Prodigy Automated Cell Processor at University of Colorado Anschutz Medical Campus in a rapid manner with a high frequent CD19 CAR expression.
Results: The final CAR T-cell product is highly active, low in immune suppression, and absent in exhaustion. Full panel cytokine assays also showed elevated production of Th1 cytokines upon IL-2 stimulation when specifically killing CD19+ target cells.
Conclusion: These results demonstrate the feasibility of producing CAR T cells locally in a university hospital setting using automated cell processor for future clinical applications.
Keywords: automated decentralized cell production, CD19, chimeric antigen receptor, immunophenotype, activation status, cytokine panel
Introduction
The field of cellular immunotherapy continues to demonstrate significant promise in the treatment of advanced hematologic malignancies.1–4 Genetically modified T cells expressing chimeric antigen receptors (CARs) are an effective therapy against many types of malignancies, including acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia, B-cell non-Hodgkin lymphoma (NHL), acute myeloid leukemia, multiple myeloma (MM), Hodgkin’s lymphoma, T-cell lymphoma and some solid tumors.5–20 The success of CD19 CAR T-cell trials has led to the FDA approval of tisagenlecleucel (Kymriah; Novartis International AG, Basel, Switzerland) and axicabtagene ciloleucel (Yescarta; Kite, Los Angeles, CA, USA) for the treatment of B-cell ALL and NHL, respectively. CARs are synthesized constructs and enable recognition and killing of target cells in a highly specific and MHC-independent manner when introduced into T cells. This is accomplished through an antigen recognition motif that is typically derived from a target-specific antibody that is fused to an intracellular CD3 signaling domain (CD3ζ), termed “first-generation” CAR. Incorporation of a T-cell costimulatory domain (eg, CD28, 4-1BB) was shown to dramatically increase the potency of CAR T cells such that the majority of current clinical trials use these “second-generation” constructs.21 “Third-generation” CARs that incorporate multiple costimulatory domains have also been tested, but it remains unclear whether these constructs provide advantages over second-generation constructs. The engineered nature of CARs allows for a high degree of flexibility and provides opportunities for further modifications to modulate CAR T-cell behavior and survival.22
Currently, the production of a majority of clinical grade CAR T cells is expensive. The prices of commercially available CD19 CAR T-cell therapies are $475,000 for Kymriah and $373,000 for Yescarta. The high prices are at least partially due to the complex process of production of this personalized cell product which requires numerous Good Manufacture Practice (GMP) instruments and technicians with significant and highly specific expertise (Figure 1).23,24 The process begins with harvesting a patient’s lymphocytes by apheresis. Following collection, CD3+ T cells, CD4+ and CD8+ T cells, or T central memory (TCM) cells are selected depending on the protocol’s specific production platform, activated, and then transduced with a viral vector expressing a CAR transgene. Cells are then expanded with protocol-specific cytokine cocktails that promote activated CAR T-cell proliferation and maintenance.25,26 Once desired number of CAR T-cells are achieved, cells are harvested and evaluated using vigorous quality control.23,24 Current techniques for producing CAR T cells require at least 12–17 days.24,26 While significant hurdles have been overcome in the production process, an average 10%–20% of patients who undergo leukapheresis do not receive CAR T-cell therapy due to failure of production and/or disease progression during the therapeutic cell production.
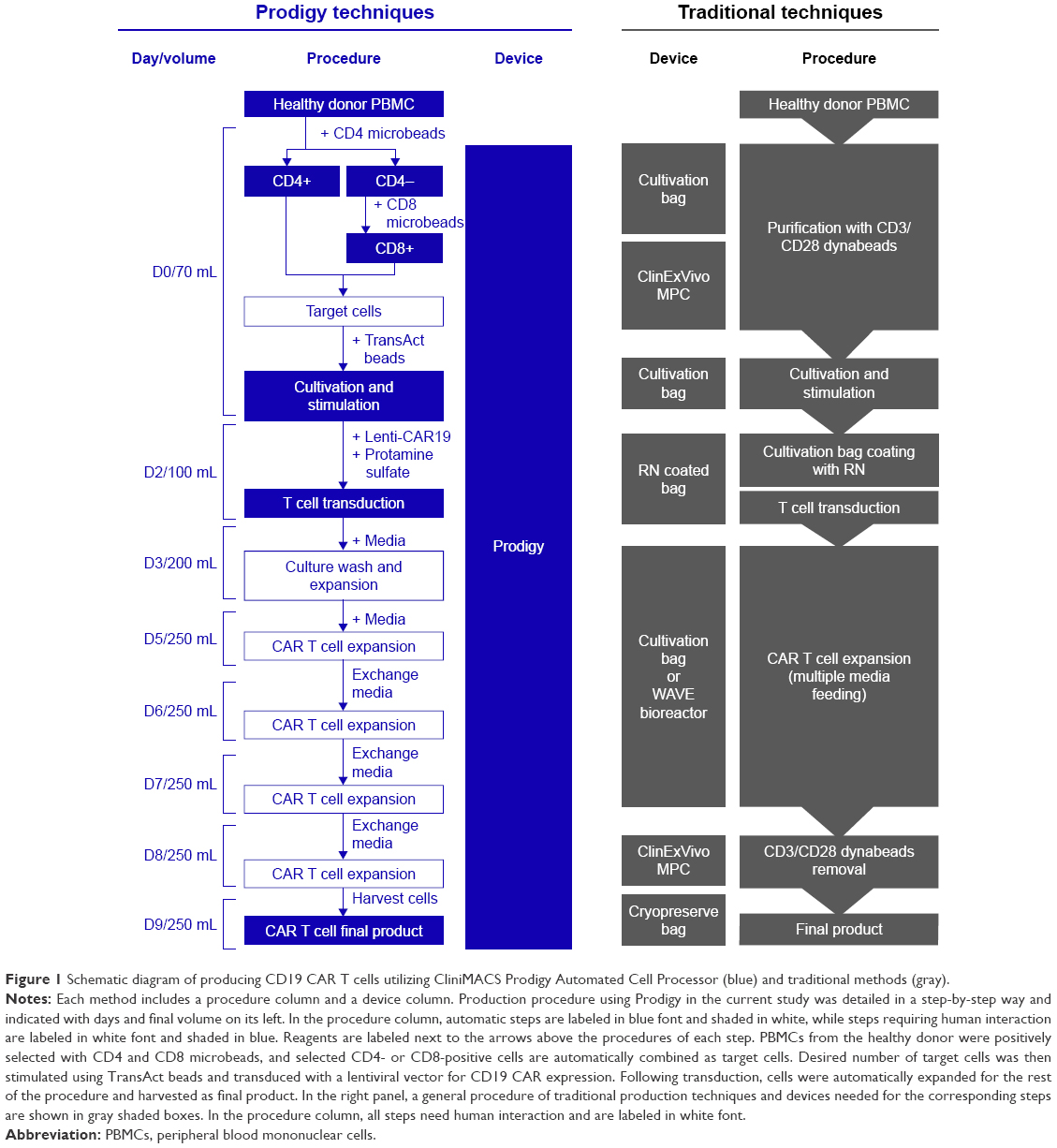 | Figure 1 Schematic diagram of producing CD19 CAR T cells utilizing CliniMACS Prodigy Automated Cell Processor (blue) and traditional methods (gray). |
CliniMACS Prodigy System, hereafter referred to as Prodigy, is a self-contained, closed, and largely automated device that obviates the need for human involvement in numerous steps, reducing human resource utilization and the risk of cell contamination (Figure 1). Prodigy has recently been shown to successfully produce CAR T-cell products at different facilities, demonstrating its potential feasibility in decentralizing CAR T-cell production.27–29 However, the immunophenotypes, activation status, or cytokine profile of the Prodigy-produced CAR T-cell products have not been fully characterized. In this study, we utilized Prodigy for the production of clinical grade CAR T cells from a healthy donor’s (HD’s) lymphocytes at lower cost than traditional methods. We produced CAR T cells in as short as 8 days and characterized the immunophenotypes, activation status, and anti-tumor function of CD19 CAR T cells produced in the GMP facilities at the University of Colorado Anschutz Medical Campus. This research demonstrates the feasibility of successful decentralized CAR T-cell production using Prodigy for Phase I clinical trials.
Materials and methods
Clinical grade CD19 CAR lentiviral vector
Clinical grade lentiviral vector encoding a CD19 CAR applied in this study was provided by Lentigen Technology, Inc. (Gaithersburg, MD, USA). The DNA encoding this receptor was cloned into the lentiviral vector backbone. The CAR construct contains a single-chain variable fragment (scFv) derived from CD19 antibody (clone FMC63) and specifically binds to CD19, a transmembrane (TM) domain derived from TNF superfamily member 19 (TNFSF19), a costimulatory domain 4-1BB and a signaling domain TCR-ζ (Figure 2). The clinical grade lentiviral vector was produced at Lentigen Technology Inc. (Gaithersburg, MD, USA) following GMP. CD19 CAR lentiviral vector thus produced was cryopreserved and stored at −80°C prior to use.
 | Figure 2 Schematic diagram of CD19 CAR. |
Production of clinical grade CD19 CAR T cells using Prodigy automated cell processor
Clinical grade CD19 CAR T cells from the HD were produced at University of Colorado using Prodigy (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s instructions. Briefly, the frequency of HD CD3+ T cells in leukapheresis product was determined by flow cytometry. Leukapheresis cells containing desired number of T cells were diluted in CliniMACS PBS/EDTA buffer with 0.5% human serum albumin (Miltenyi Biotec) and transferred into Prodigy Tubing Set TS520 for the selection of T cells by CD4 and CD8 Microbeads (Miltenyi Biotec) sequentially on day 0. About 1×108 post-enrichment cells combined for CD4+ and CD8+ T cells were transferred into a transfer bag and attached to the TS520 by sterile welding, cultivated in 70 mL TexMACS™ GMP Medium (Miltenyi Biotec) containing 200 IU/mL premium grade human IL-2 Improved Sequence (Miltenyi Biotec) and MACS GMP T cell TransAct beads (Miltenyi Biotec) with 5% CO2 at 37°C for 2 days. On day 2, 10 mL of 2.1×109 pfu/mL Lenti-CAR19 (Lentigen Technology) was transferred into a transfer bag with protamine sulfate (USP grade) at a final concentration of 10 μg/mL in 30 mL total volume and attached to Prodigy via sterile welding to start transduction in a final volume of 100 mL. Two media supplements of 100 and 50 mL including IL-2 were added on day 3 and day 5 to wash off the TransAct beads and expand the culture to 200 and 250 mL sequentially. Later, three media exchanges were preceded over the following 3 days to promote CAR T-cell proliferation by removing half the media without disrupting the cells and adding in the same volume of new media with IL-2 (Figure 1). CAR T cells were sampled daily until the end of production for the examination of number of cells and viability by Live/Dead™ viability stain (Thermo Fisher Scientific, Waltham, MA, USA), and on days 0, 5, and 8 for the analyses of cell composition and transduction efficiency by flow cytometry (described as below).
CD19 CAR expression and purity during production
Viability was analyzed using Fixable Viability Dye eFluor®-APC-EF780 (Affymetrix eBioscience, Santa Clara, CA, USA). T-cell purity and T helper and killer subpopulations were determined by flow cytometry analysis using human CD19, CD14, CD45, CD3, CD4, and CD8 antibodies (BD Biosciences, San Jose, CA, USA). T cells were gated using a hierarchy gating strategy as single cells/Live (VIDYE-)/Lymphocytes/CD14− CD19−/CD3+ CD45+.30 CD19 CAR expression was determined using recombinant human CD19 Fc chimera (R&D Systems, Inc., Minneapolis, MN, USA) and goat anti-human IgG Fc-γ (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). Samples were analyzed by flow cytometry using a CLIA certified BD Canto™ flow cytometer with a minimum number of 100,000 cells per sample analyzed and FlowJo Software (FlowJo LLC, Ashland, OR, USA).
Cell lines and cell culture
Raji CCL-86 lymphoma cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in complete media (RPMI-1640 containing L-glutamine and sodium bicarbonate, 10% heat inactivated FBS, and 10 mM Pen/Strep) in a 37°C incubator with 5% CO2. After two passages, frozen aliquots were preserved in FBS containing 10% dimethyl sulfoxide (DMSO) and stored in liquid nitrogen. Two cell lines, MOLM-13 and MDS-L, were included as negative target cells. MOLM-13 cell line was purchased from ATCC and cultured in complete media. MDS-L is a cell line derived from a non-leukemic phase of a patient with myelodysplastic syndrome (MDS) obtained from Dr Daniel T Starczynowski of Cincinnati Children’s hospital as a gift and cultured in complete media containing 10 ng/mL human IL-3 (PeproTech Inc., Rocky Hill, NJ, USA).31 Stocks of MOLM-13 and MDS-L cells were cryopreserved in freezing media containing 45% RPMI-1640, 45% FBS, and 10% DMSO prior to using.
Phenotype and activation status
CD19 CAR T cells produced from peripheral blood mononuclear cells (PBMCs) of an HD and untransduced PBMCs from three different HDs were plated at 1×106 cells per well in a 96-well plate. The plate was spun at 500× g for 5 minutes, and cells were incubated in flow cytometry blocking buffer (1× PBS containing 10% human serum and 10% mouse serum) for 10 minutes at room temperature. Cells were washed with flow cytometry wash buffer (1× PBS containing 2% FBS) and incubated with the following antibodies for 1 hour at 4°C: CD66 (B1.1/CD66), CD3 (UCHT1), CD4 (SK3), CD8 (SK1), and CD25 (2A3) from BD Biosciences, and LAG-3 (11C3C65), PD-1 (EH122H7), and TIM-3 (F382E2) from Biolegend (San Diego, CA, USA). After washing, cells were fixed and permeabilized with Transcription Factor Phospho Buffer Set (BD Biosciences) according to the manufacturer’s instructions. After washing, cells were then stained intracellularly with the following antibodies for 1 hour at 4°C: CTLA-4 (I4D3) from BD Biosciences, FOXP3 (150D) and Tbet (4B10) from Biolegend, and EOMES (WD1928) from Thermo Fisher Scientific. Samples were analyzed by flow cytometry on a BD LSRFortessa X-20 instrument with a minimum number of 50,000 cells per sample analyzed and FlowJo Software (FlowJo LLC).
Cytokine production
CD19 CAR T cells were quick-thawed in a 37°C water bath, washed in complete media, counted, and resuspended in complete media. A total of 7.5×105 CD19 CAR T cells were plated in a 96-well round bottom plate with ±2.5×105 Raji cells and incubated for 18 hours in a 37°C incubator with 5% CO2. The supernatants were harvested after spinning the plate at 500× g for 10 minutes and stored at −80°C. A multiplex cytokine array (V-PLEX; MesoScale Discovery, Rockville, MA, USA) was used to measure cytokines in the supernatants according to the manufacturer’s instructions. Briefly, supernatants were thawed, spun at 2,000× g for 3 minutes, and diluted 1:1 in assay diluent to measure IL-10, IL-12p40, IL-13, IL-1β, IL-4, and IL-6 and diluted 1:100 to measure IL-2, IL-8, IFN-γ, and TNF-α. Pre-coated V-PLEX plates were washed using an automated plate washer (BioTek ELX5012), 50 μL of calibrators or diluted supernatants were added, and plates were incubated for 2 hours at room temperature on a Compact Digital Microplate shaker (Thermo Fisher Scientific) at 600 rpm. Plates were washed, and 25 μL of diluted detection antibodies was added and incubated for 2 hours at room temperature. After washing, 2× Read Buffer (MesoScale Discovery) was added, and the plates were immediately read on a MesoQuickPlex SQ120 electrochemiluminescence plate reader (MSD).
Cytotoxic activity
Raji, MDS-L, and MOLM13 target cells were labeled with Cell Trace Violet (Thermo Fisher Scientific) according to the manufacturer’s instructions. About 2.5×105 Raji target cells were co-cultured with 1.25×105, 2.5×105, 5×105, or 7.5×105 CD19 CAR T cells or untransduced matched HD T cells for 18 hours in a 37°C incubator with 5% CO2. For antigen specificity assays, 2.5×105 MDS-L and MOLM13 cells were incubated with 7.5×105 CD19 CAR T cells or cultured alone. After 18 hours, plates were spun at 500× g for 5 minutes, supernatants were removed for cytokine measurements as described above, and cells were stained with Zombie Green Fixable Viability Kit (Biolegend) according to the manufacturer’s instructions. After washing, cells were stained with CD19 (HIB19; Biolegend) and analyzed by flow cytometry on a BD LSRFortessa X-20 instrument and FlowJo Software (FlowJo, LLC).
Statistical analyses
All statistical analyses in this study were performed using GraphPad Prism 7 software (GraphPad Software, San Diego, CA, USA).
Results
Production of clinical grade CD19 CAR T cells using Prodigy
To characterize clinical grade CAR T cells produced in Prodigy, we produced clinical grade CD19 CAR T cells from an HD leukapheresis product by transducing the cells with a lentiviral vector encoding a CAR protein targeting CD19 after T-cell selection and stimulation (Figures 1 and 2). The production procedure was completed in Prodigy and decreased on labor interaction compared to traditional methods (Figure 1). Viability, cell count, T-cell purity, subpopulation, and CAR expression of T cells were analyzed by flow cytometry with a hierarchy gating strategy throughout the production procedure (Figure 3A). About 4.0×109 HD lymphocytes (40% of total PBMCs) were transferred into Prodigy for T-cell enrichment and 9.0×108 (9% of total PBMCs) enriched cells were harvested (Figure 3B). The selection of T cells resulted in the enrichment of T-cell compartment from 9% in pre-selection PBMCs to 82% in post-selection target cells (Figure 3A and B), similar to that achieved by manual enrichment using human CD3 Microbeads and LS columns (Miltenyi Biotec, unpublished data). Additionally, lymphocytes, CD4+ T cells, and CD8+ T cells were also enriched from 41%, 4%, and 4% in PBMCs to 89%, 45%, and 34% in target cells, respectively (Figure 3B). About 1.0×108 target cells were processed for transduction (Figure 3C). As expected, the number of total live cells increased by 16-fold over the production process of manufacturing from 1 ×108 target cells on day 0 to 1.6×109 total cells on day 8 (Figure 3D). Total number of T cells expanded 20-fold from 8.2×107 on day 0 to 1.6×109 T cells on day 8 (Figure 3C). During this time, the frequency of CD4+ and CD8+ T cells in total live cells changed from 45% and 34% to 22% (3.4×108 cells) and 74% (1.2×109 cells), respectively (Figure 3B and C), showing higher expansion in CD8+ than CD4+ T cells. Overall, the viability of total cells in Prodigy was maintained above 80% throughout the procedure, with a slight drop to 72% on day 3 (1 day after transduction) that was recovered by day 4 (Figure 3D). The percentage of CD19 CAR expression in T cells was 65% in both CD4+ and CD8+ subpopulations on day 5, and slightly dropped to 60% in CD4+ and 50% in CD8+ T cells on day 8, showing similar expression frequencies in the two subpopulations (Figure 3E). CD8+ CAR T cells showed higher expansion rate than CD4+ CAR T cells, with 1.8×108 CD8+ CAR T cells and 1.2×108 CD4+ CAR T cells detected on day 5; this difference further increased by day 8 with 5.4×108 CD8+ CAR T cells and 2.7×108 CD4+ CAR T cells (Figure 3E).
 | Figure 3 Examination of CD19 CAR T cells produced by CliniMACS Prodigy. |
Phenotypic characterization of HD-derived CD19 CAR T cells produced utilizing Prodigy
We compared the immunophenotype and activation status of untransduced HD PBMCs and the final Prodigy-generated CD19 CAR T cells. T lymphocytes were analyzed by cell surface and intracellular flow cytometry for the frequency of FOXP3+ regulatory T cells (Tregs) and by intracellular flow cytometry for the expression of transcription factors that correlate with effector function (Figure 4). T cells and Tregs in CD4+ and CD8+ subpopulations from HD PBMCs and CAR T cells were analyzed using a hierarchy gating strategy (Figure 4A). The frequency of CD4+ Tregs decreased in the CD19 CAR T-cell product compared to untransduced PBMCs from an average of 1.5% to 0.1% of total lymphocytes or from 6.4% to 0.7% of total CD4+ T cells. The frequency of CD8+ Tregs was very low in all samples measured (Figure 4B). In the CD4+ and CD8+ T-cell populations (non-Tregs), there was a significant increase in the frequency of CD19 CAR T cells expressing high levels of T-box transcription factor (Tbet) (67% of the CD4+ T cells and 87% of the CD8+ T cells) compared to untransduced HD T cells (16% of the CD4+ T cells and 39% of the CD8 T cells) (Figure 4C). Although a portion of both CD4+ and CD8+ T cells from untransduced HD T cells expressed high levels of the transcription factor Eomesodermin (EOMES), fewer CAR T cells expressed high levels of EOMES alone or in combination with high levels of Tbet, although the difference was not statistically significant.
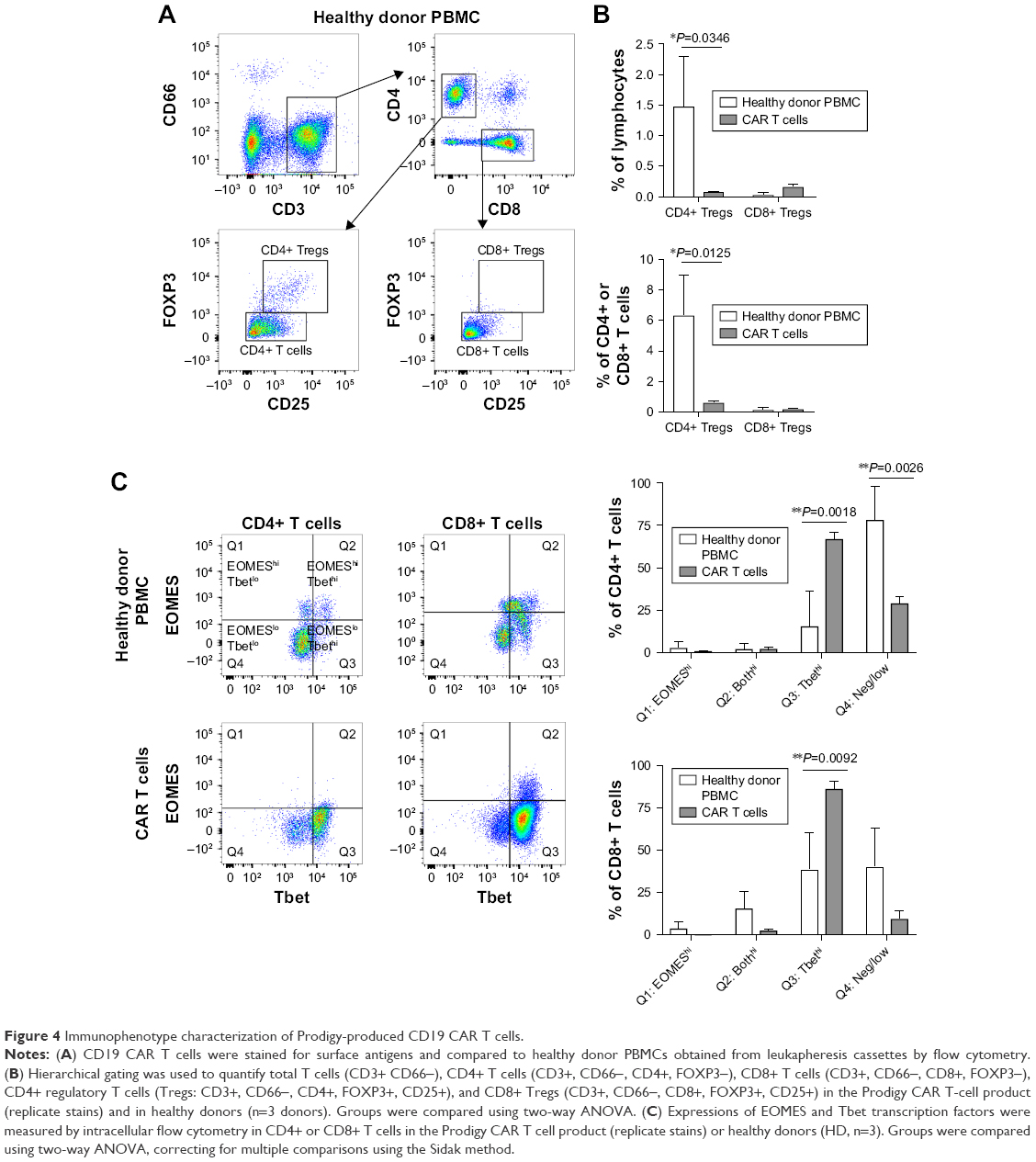 | Figure 4 Immunophenotype characterization of Prodigy-produced CD19 CAR T cells. |
Activation status of Prodigy-generated CD19 CAR T cells
To further examine the activation of CD19 CAR T cells, the surface expression of activation and inhibitory markers were determined by flow cytometry. Compared to untransduced HD PBMCs, CD25 (high-affinity IL-2 receptor), cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), and T-cell immunoglobulin and mucin domain 3 (Tim-3) were increased on CD19 CAR T cells shown by histograms (Figure 5A). The mean fluorescence intensity (MFI) of CTLA-4 was increased 2.3-fold on both CD4+ and CD8+ subsets; the MFI of Tim-3 was increased 7.5- and 5.6-fold on CD4+ and CD8+ T cells, respectively; and the MFI of CD25 was increased 1.3- and 1.7-fold on CD4+ and CD8+ T cells, respectively. The MFI of CTLA-4 and Tim-3 were significantly increased on both the CD4+ and CD8+ T-cell subsets of CD19 CAR T cells compared to HD PBMCs, whereas the MFI of CD25 was only significantly increased on the CD8+ T-cell subset (Figure 5B). In contrast, expression levels of lymphocyte activation gene-3 (LAG-3) and programmed cell death-1 (PD-1) were similarly low on CD19 CAR T cells and HD PBMCs, as shown by both histograms (Figure 5A) and statistical comparisons of MFIs (Figure 5B).
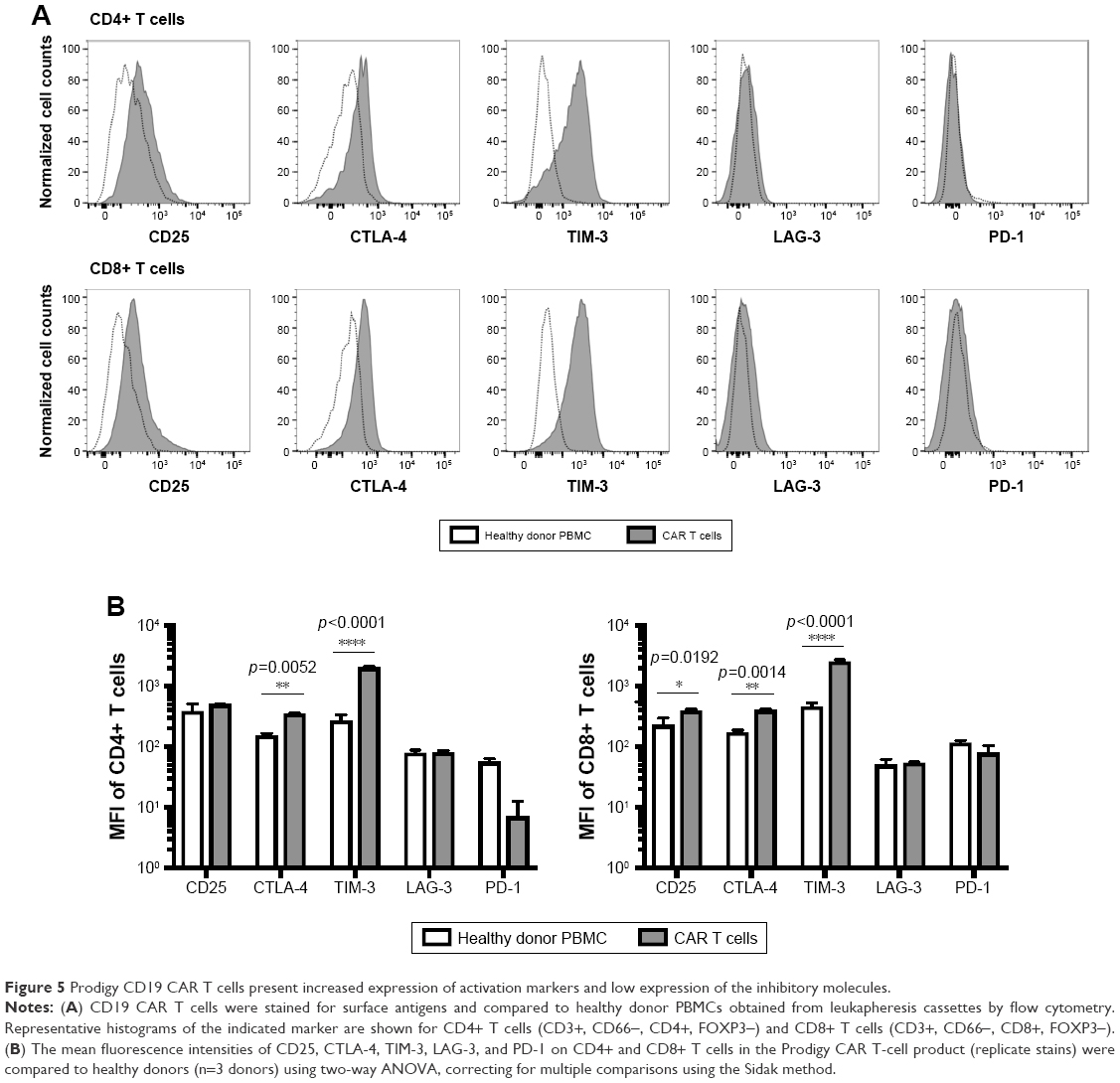 | Figure 5 Prodigy CD19 CAR T cells present increased expression of activation markers and low expression of the inhibitory molecules. |
Production of cytokines by Prodigy-generated CD19 CAR T cells
To further characterize the effector function of CD19 CAR T cells produced with Prodigy, we measured multiple cytokines in cell culture supernatants of CAR T cells stimulated with Raji leukemia cells expressing the cognate antigen, CD19, after 18 hours (Figure 6). Consistent with the increased frequency of CD4+ CAR T cells expressing Tbet transcription factor, stimulated CD19 CAR T cells produced significant amounts of Th1 cytokine, IFN-γ. Importantly for survival and differentiation, they also produced high levels of IL-2 after stimulation. There were smaller increases in the Th1 cytokine, TNF-α, the Th2 cytokines, IL-4 and IL-13, and the pro-inflammatory cytokines, IL-1β, IL-6, and IL-8, after stimulation. However, these increases were not statistically significant after correcting for multiple comparisons, indicating a skewing toward IFN-γ responses and Th1 effector function in CD19 CAR T cells. The anti-inflammatory cytokine IL-10 was produced at low levels by the Raji leukemia cells, as demonstrated by its presence in wells only containing Raji cells.
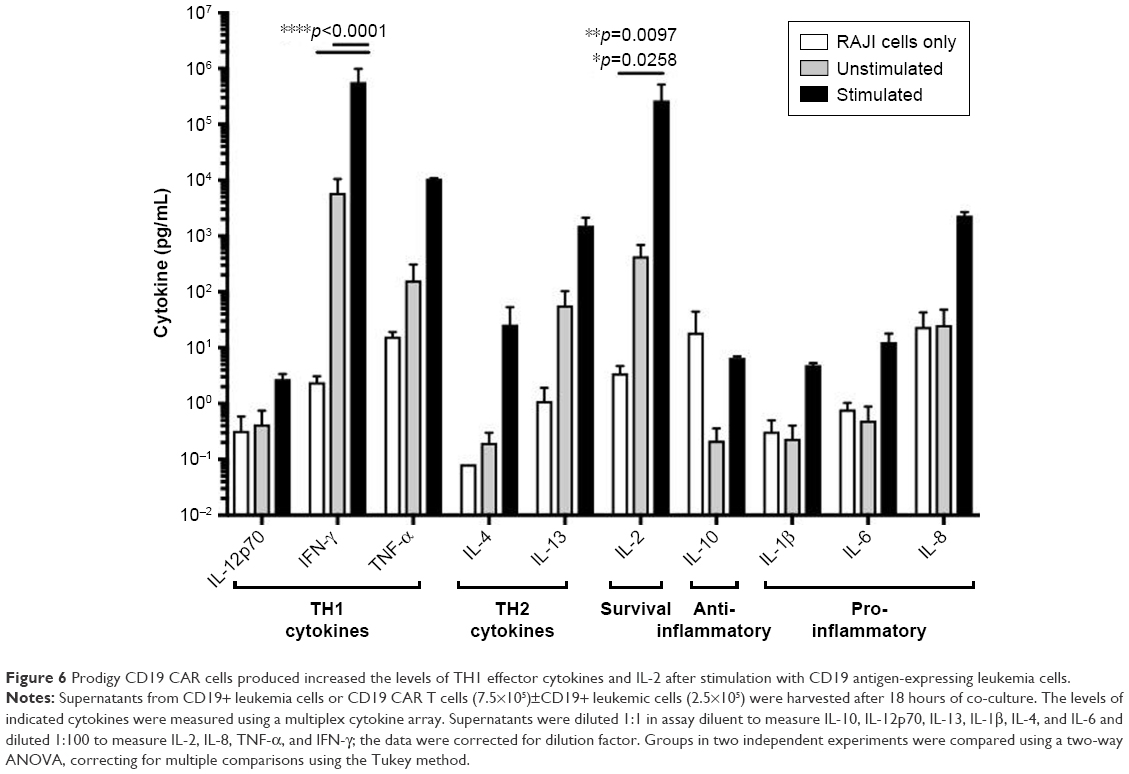 | Figure 6 Prodigy CD19 CAR cells produced increased the levels of TH1 effector cytokines and IL-2 after stimulation with CD19 antigen-expressing leukemia cells. |
Antigen specificity of Prodigy-generated CD19 CAR T cells
We next performed an in vitro killing assay to determine the anti-tumor activity and specificity of CD19 CAR T cells produced at our facility. CD19+ Raji cells (targets) were labeled with CellTrace Violet and co-cultured with CD19 CAR T cells (effectors) or untransduced T cells from HD PBMCs at increasing effector-to-target ratios. After 18 hours, a large portion of the Raji cells incubated with CD19 CAR T cells, but not untransduced T cells, were non-viable as detected by staining with Zombie Green viability dye (Figure 7A). The frequency of non-viable Raji cells after co-culture with CD19 CAR T cells was ~22% at a 1:2 effector-to-target ratio and increased to ~60% at a 3:1 effector-to-target ratio (Figure 7A). To determine the specificity of the anti-tumor function of CD19 CAR T cells, a similar killing assay was performed using two negative control cell lines as targets, MDS-L and MOLM13, that lack CD19 expression (Figure 7B). The frequency of non-viable MDS-L and MOLM-13 cells was similar in the presence and absence of CD19 CAR T cells, indicating that cells lacking CD19 are not effectively killed by CD19 CAR T cells (Figure 7C). To further examine if the death of CD19+ Raji cells was due to specific anti-tumor functions of the transduced CD19 CAR T cells, we measured cytokines in the supernatants from the killing assays. Consistent with the killing assay, IFN-γ, TNF-α, and IL-2 were significantly increased after co-culture with Raji cells, but not MDS-L or MOLM13 cells, suggesting the specific activation and targeting of CD19+ cells by CD19 CAR T cells (Figure 7D).
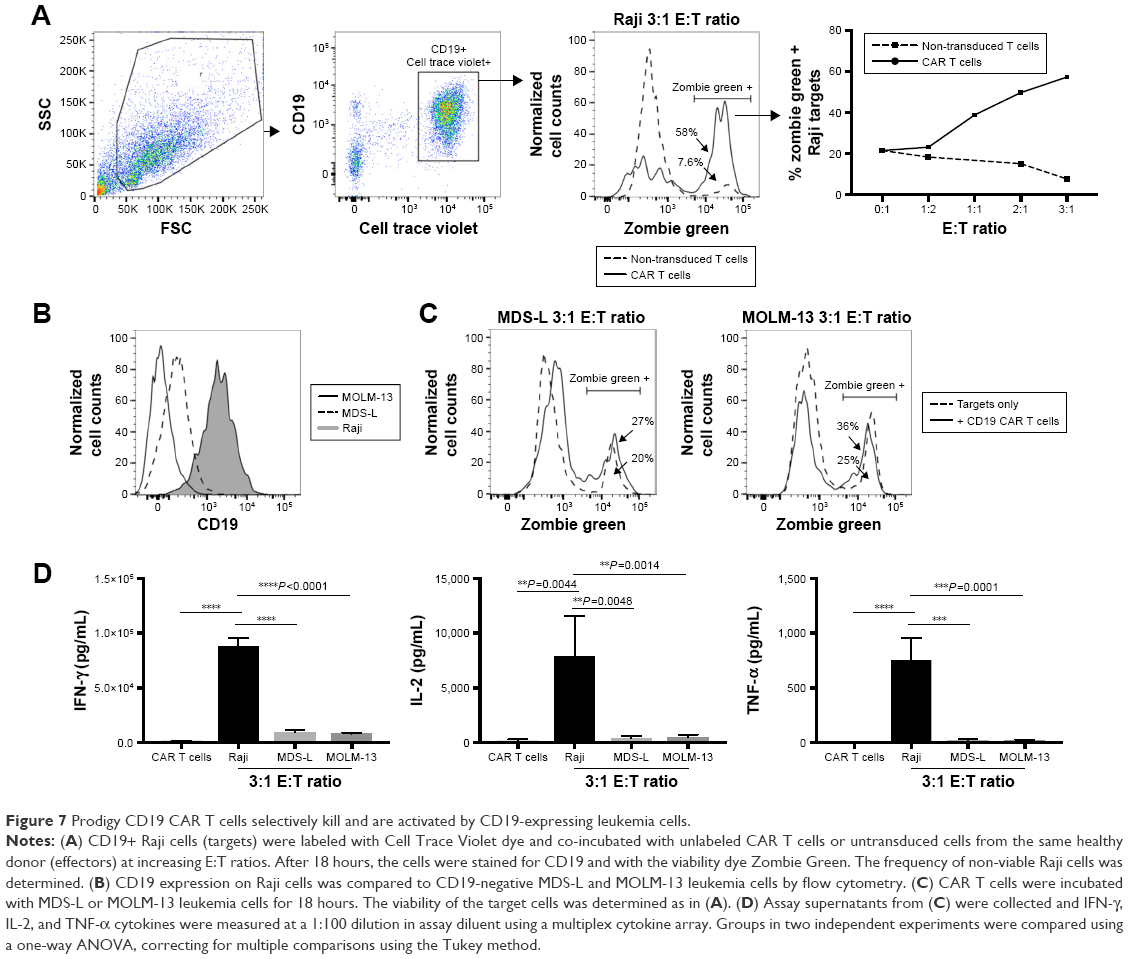 | Figure 7 Prodigy CD19 CAR T cells selectively kill and are activated by CD19-expressing leukemia cells. |
Discussion
Widespread implementation of CAR T-cell therapies has been largely prevented by the burdens of cost, reliance on production expertise, as well as long production time using traditional methods. Development of manufacturing methods that are both cost-efficient and time-efficient therefore has emerged as a big challenge in broadening this largely personalized therapy.23 In the present study, we demonstrate successful manufacturing of CD19 CAR T cells for clinical application by utilizing the fully automated and closed system, CliniMACS Prodigy, at the University of Colorado. The production procedure using this system has cut down both the cost to $25,000.00 including reagents and labor and the production time to 8 days. These are significant improvements compared to the traditional method that requires at least 13 days for manufacturing at a cost of $475,000.00 and $373,000.00 for the current two commercially available CAR T-cell products.26
Prodigy-produced CD19 CAR T cells in the current study achieved significant expansion fold and high transduction efficiency with stable viability that meet requirements for clinical application. Over the manufacturing procedure, viability of the cells has been shown to be consistently above 80% except a temporary drop on day 3 due to transduction. This is consistent with the results from other institutes.26–28 Total T cells expand 20 times and achieved 1.6×109 total cells at the end of production on day 8 with CAR expressing T cells at 1.5×108. The observed fold expansion is significantly higher than traditional methods and similar to a previously published result using Prodigy and HD T cells, indicating a stable advantage of high expansion rate in a short period of the system.26,27 The percentage of T cells expressing the CD19 CAR construct achieved 65% on day 5 and dropped slightly on day 8, indicating that the maximal transduction efficiency was achieved at an earlier stage of manufacturing. Frequency of CD19 CAR expression was similar in general between CD4+ and CD8+ T-cell subpopulations on the examined production days. However, the number of CD8+ CAR T cells was dramatically higher than CD4+ CAR T cells on day 5, and this difference further increased on day 8, indicating an advantage of survival or proliferation of CD8+ CAR T cells in the current CAR T-cell product. Such higher number of CD8+ than CD4+ CAR T cells was also observed by other researchers using a CAR vector composing 4-1BB cositmulatory domain.29 Previous research has shown that 4-1BB molecule preferably promotes proliferation of CD8+ T cells over CD4+ compartment.32 The higher number of CD8+ than CD4+ CAR T cells in current CAR T-cell product might be at least partially contributed to the 4-1BB costimulatory domain incorporated in the current CD19 CAR lentiviral vector.
Currently, second generation of CAR constructs with one costimulatory molecule of either 4-1BB or CD28 intracellular domain is the most commonly used design of CAR in both preclinical and clinical research. Limited research has been done on comparison of phenotype and activation status in CAR T cell incorporating the two different costimulatory molecules. Both CD28 and 4-1BB costimulation induce IL-2 production in T cells, enhance survival of activated T cells, and support cytotoxic activity of T cells.33–40 Depending on how the CAR is designed, the engineered CAR T cell takes on the immunophenotype and activation status of the costimulatory molecule with which it is engineered. Variations in such characters of CAR T cells may play important roles in their therapeutic results.41 Therefore, we further characterized the immunophenotype, activation status, antigen specificity, and cytokine release profile of CD19 CAR T cells produced in Prodigy transduced with a lentiviral vector encoding a CD19-specific CAR with a 4-1BB intracellular signaling motif.
Treg percentage was decreased in CD19 CAR T cells compared to untransduced HD PBMCs, demonstrating a lower immune suppressive status after the multi-step procedure in Prodigy. This result is also consistent with a previous study on CD19/20 dual CAR T cell production using Prodigy, suggesting a stable low immune suppressive status in Prodigy procedure.29 The increased expression of Tbet in the CD19 CAR T-cell product compared to untransduced HD PBMCs indicates a skewing in differentiation toward the Th1 (CD4+) and Tc1 (CD8+) lineages, which corresponded with its increased production of IFN-γ upon stimulation with CD19+ Raji cells.42–44 High expression of Tbet in T cells lacking concomitant high expression of Eomes also indicates a differentiation toward effector memory cells (TEM).45–49 This result is also consistent with a previous observation of higher TEM in Prodigy-produced CD20 CAR T cells costimulated by 4-1BB.28 It would be interesting to investigate in future if Prodigy-produced CAR T cells also hold the other phenotypes contributed by the specific costimulatory molecules.41 Increased Tbet expression in T cells has been shown to facilitate an increased potential to mediate effective anti-tumor responses by inducing CXCR3 in T cells and increasing the number of tumor-infiltrating T cells.50 These results indicate that CD19 CAR T cells produced in the Prodigy might be effective in eliminating leukemia cells in patients.
Multiple activation markers were also increased on Prodigy-generated CD19 CAR T cells without exhaustion phenotype compared to untransduced T cells from HD PBMCs.51 CD25 and CTLA-4 are both commonly upregulated in the setting of antigen stimulation. Increased CD25 indicated IL-2-induced maintenance of activation and proliferation of CAR T cells during the production. It was surprising that the increase of CD25 in CAR T cells was not significant in CD4+ subpopulation, but a trend at 1.3-fold was observed, and it would be interesting to investigate in additional samples from our facility in the future. CTLA-4 is upregulated in both CD4+ and CD8+ subpopulations, indicating maintenance of CAR T-cell homeostasis.52 CTLA-4 is not expressed in rested T cells and peak expression occurs ~48 hours after T-cell activation, returning to basal levels by 96 hours.53–55 Tim-3 augments T-cell receptor-dependent signaling pathways. Tim-3 elevated expression in both CD4+ and CD8+ subpopulations correlated with the Tbet regulated differentiation toward Th1 cells, Tc1 cells, and maybe also CD8+ TEM as well as the elevated expression of IFN-γ and IL-2.56–58 Lag-3 is an inhibitory receptor that outcompetes CD4 for binding to MHC class II and is important for maintaining T-cell homeostasis.59–62 PD-1 is an important immune checkpoint that promotes apoptosis in T cells. The increased expression of CD25, CTLA-4, and Tim-3 without increases in LAG-3 and PD-1 in Prodigy-generated CD19 CAR T cells compared to untransduced T cells suggests that CD19 CAR T cells are activated but not exhausted. It would be important to further compare the activation and exhaustion status between Prodigy-produced CAR T cells and untransduced TransAct-stimulated PBMCs to specifically examine the role of Prodigy in effecting the activation/exhaustion status of the cell products.
The Prodigy-produced CD19 CAR T cells functioned in a CD19-dependent manner and produced a broad panel of cytokines. Ten different cytokines for Th1 and Th2 function, cell proliferation, pro-inflammatory, and anti-inflammatory were examined in the current study. All the examined cytokines were increased at various levels in CD19 CAR T cells after stimulation with CD19+ Raji cells, except IL-10 with a high background level from Raji cell alone. Notably, Th1 cytokine IFN-γ and proliferation cytokine IL-2 were both significantly elevated after stimulation with Raji cells expressing the CD19 target antigen or compared to the Raji cells alone. Robust increases in the production of Th1 cytokine and IL-2 with minimal elevations in Th2 cytokines correlated with the differentiation toward Th1 and indicate highly activated anti-tumor function.63–65 Additionally, pro-inflammatory cytokines IL-1β, IL-8, and IL-6, the major player of cytokine release syndrome, and anti-inflammatory cytokine IL-10 are not much elevated in the produced CAR T cells following tumor target stimulation, indicating a low inflammatory status in the coculture of CD19 CAR T cells and tumor target Raji cells.66
The cytotoxicity of CD19 CAR T cells was proportional to the effector target ratio in the assay, indicating a dose-related anti-tumor function of the CD19 CAR T cells that depended on the expression of CD19 by the target cell. High killing efficacies were detected at serial effector target ratio. 58% of Raji cells were killed at 3:1 effector target ratio, which is higher than other reported Prodigy-produced CAR T cells.27,29 It would be interesting to further examine copy number of CAR in this product to investigate if that affects the killing potency. The killing potency was also shown specific against CD19+ Raji cells as no effect on viability of CD19- cells were observed after being cocultured with CAR T cells. Additionally, significant increases in the production of the effector cytokines TNF-α, IFN-γ and IL-2 were observed after incubating CAR T cells with CD19-positive Raji cells, but not the negative control cell lines MDS-L or MOLM-13.
Together, we demonstrate the feasibility and efficiency of using a closed automated system CliniMACS Prodigy in manufacturing CD19 CAR T cells for clinical use with significantly decreased cost and labor in as short as 8 days. Importantly, we further demonstrate the ability of the system to produce sufficient quantities of cells for therapeutic doses, with a minimal starting cell number. Additionally, detailed characterization of immunophenotype and activation status showed that these CAR T cells presented lower immune suppression, higher activation status, differentiation toward Th1, Tc1 and TEM, and no detectable exhaustion, indicating their potential long-term high efficacy against target tumor cells. Furthermore, in vitro assays of cell function by a broad panel of cytokine production and killing reflect the high activity and specific anti-tumor function of CD19 CAR T cells. These data demonstrate the feasibility of decentralized manufacturing of CAR T cell using Prodigy for Phase I clinical trials at university hospitals. Comparisons of the current research data to Prodigy-produced CAR T cells from other institutes suggested consistency in cell number expansion, transduction efficiency, decreased immune suppression, and increased differentiation toward TEM.27–29 However, due to differences in the manufacturing protocols including viral vectors, cytokines, manufacture time, and volume, more conclusive comparisons for evaluating the stability of Prodigy is warranted. Further studies with additional CAR T-cell products from both HDs and patients will be necessary to further validate the stability as well as the clinical feasibility of CAR T cell production using Prodigy System.
Ethical approval
The study protocol was approved by the University of Colorado Institutional Review Board, and all subjects gave written informed consent in accordance with the Declaration of Helsinki, Protocol #06-0720.
Acknowledgment
The authors thank Lentigen Technology Inc. for providing clinical grade CD19 CAR lentiviral vector and Gates Center for Regenerative Medicine for support in manufacturing CD19 CAR T cells.
Disclosure
The authors report no conflicts of interest in this work.
References
Davila ML, Brentjens R, Wang X, Rivière I, Sadelain M. How do CARs work? Oncoimmunology. 2012;1(9):1577–1583. | ||
Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123(17):2625–2635. | ||
Shalabi H, Angiolillo A, Fry TJ. Beyond CD19: opportunities for future development of targeted immunotherapy in pediatric relapsed-refractory acute leukemia. Front Pediatr. 2015;3:80. | ||
Hartmann J, Schüβler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9(9):1183–1197. | ||
Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050–6056. | ||
Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. | ||
Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. | ||
Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. | ||
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. | ||
Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. | ||
Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531–2544. | ||
Wang CM, Wu ZQ, Wang Y, et al. Autologous T Cells Expressing CD30 Chimeric Antigen Receptors for Relapsed or Refractory Hodgkin Lymphoma: An Open-Label Phase I Trial. Clin Cancer Res. 2017;23(5):1156–1166. | ||
Ramos CA, Ballard B, Zhang H, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest. 2017;127(9):3462–3471. | ||
Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–3148. | ||
Budde L, Song JY, Kim Y, et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood. 2017;130:811. | ||
Cummins KD, Gill S. Anti-CD123 chimeric antigen receptor T-cells (CART): an evolving treatment strategy for hematological malignancies, and a potential ace-in-the-hole against antigen-negative relapse. Leuk & Lymphoma. 2018;59(7):1539–1553. | ||
Kenderian SS, Ruella M, Shestova O, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–1647. | ||
Ritchie DS, Neeson PJ, Khot A, et al. Persistence and Efficacy of Second Generation CAR T Cell Against the LeY Antigen in Acute Myeloid Leukemia. Molecular Therapy. 2013;21(11):2122–2129. | ||
Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–2060. | ||
Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126(8):983–992. | ||
Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. | ||
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. | ||
Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Molecular Therapy – Oncolytics. 2016;3:16015. | ||
Levine BL, Miskin J, Wonnacott K, Keir C. Global Manufacturing of CAR T Cell Therapy. Mol Ther Methods Clin Dev. 2017;4(17):92–101. | ||
Sabatino M, Hu J, Sommariva M, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–528. | ||
Tumaini B, Lee DW, Lin T, et al. Simplified process for the production of anti-CD19-CAR-engineered Tcells. Cytotherapy. 2013;15(11):1406–1415. | ||
Mock U, Nickolay L, Philip B, et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy. 2016;18(8):1002–1011. | ||
Lock D, Mockel-Tenbrinck N, Drechsel K, et al. Automated Manufacturing of Potent CD20-Directed Chimeric Antigen Receptor T Cells for Clinical Use. Hum Gene Ther. 2017;28(10):914–925. | ||
Zhu F, Shah N, Xu H, et al. Closed-system manufacturing of CD19 and dual-targeted CD20/19 chimeric antigen receptor T cells using the CliniMACS Prodigy device at an academic medical center. Cytotherapy. 2018;20(3):394–406. | ||
Groen B, van der Wijk AE, van den Berg PP, et al. Immunological Adaptations to Pregnancy in Women with Type 1 Diabetes. Sci Rep. 2015;5:13618. | ||
Nakamura S, Ohnishi K, Yoshida H, et al. Retrovirus-mediated gene transfer of granulocyte colony-stimulating factor receptor (G-CSFR) cDNA into MDS cells and induction of their differentiation by G-CSF. Cytokines Cell Mol Ther. 2000;6(2):61–70. | ||
Shuford WW, Klussman K, Tritchler DD, et al. 4-1BB Costimulatory Signals Preferentially Induce CD8+ T Cell Proliferation and Lead to the Amplification In Vivo of Cytotoxic T Cell Responses. J Exp Med. 1997;186(1):47–55. | ||
Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3(12):939–951. | ||
Ville S, Poirier N, Blancho G, Vanhove B. Co-Stimulatory Blockade of the CD28/CD80-86/CTLA-4 Balance in Transplantation: Impact on Memory T Cells? Front Immunol. 2015;6:411. | ||
Debenedette MA, Wen T, Bachmann MF, et al. Analysis of 4-1BB ligand (4-1BBL)-deficient mice and of mice lacking both 4-1BBL and CD28 reveals a role for 4-1BBL in skin allograft rejection and in the cytotoxic T cell response to influenza virus. J Immunol. 1999;163(9):4833–4841. | ||
Boise LH, Yinn AJ, Noel PJ, et al. CD28 Costimulation Can Promote T Cell Survival by Enhancing the Expression of Bcl-xL. Immunity. 1995;3(1):87–98. | ||
Schwarz H, Blanco FJ, von Kempis J, Valbracht J, Lotz M. ILA, a member of the human nerve growth factor/tumor necrosis factor receptor family, regulates T-lymphocyte proliferation and survival. Blood. 1996;87(7):2839–2845. | ||
Azuma M, Cayabyab M, Buck D, Phillips JH, Lanier LL. CD28 interaction with B7 costimulates primary allogeneic proliferative responses and cytotoxicity mediated by small, resting T lymphocytes. J Exp Med. 1992;175(2):353–360. | ||
Shuford WW, Klussman K, Tritchler DD, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. 1997;186(1):47–55. | ||
Zhou Z, Pollok KE, Kim KK, Kim YJ, Kwon BS. Functional analysis of T-cell antigen 4-1BB in activated intestinal intra-epithelial T lymphocytes. Immunol Lett. 1994;41(2–3):177–184. | ||
Kawalekar OU, O’Connor RS, Fraietta JA, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44(2):380–390. | ||
Szabo SJ, Kim ST, Costa GL, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100(6):655–669. | ||
Szabo SJ, Sullivan BM, Stemmann C, et al. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295(5553):338–342. | ||
Shinohara ML, Jansson M, Hwang ES, et al. T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc Natl Acad Sci U S A. 2005;102(47):17101–17106. | ||
Intlekofer AM, Takemoto N, Wherry EJ, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6(12):1236–1244. | ||
Knox JJ, Cosma GL, Betts MR, Mclane LM. Corrigendum: Characterization of T-bet and Eomes in Peripheral Human Immune Cells. Front Immunol. 2016;7:337. | ||
Mclane LM, Banerjee PP, Cosma GL, et al. Differential localization of T-bet and Eomes in CD8 T cell memory populations. J Immunol. 2013;190(7):3207–3215. | ||
Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 2013;13(11):777–789. | ||
Marshall HD, Chandele A, Jung YW, et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity. 2011;35(4):633–646. | ||
Zhu Y, Ju S, Chen E, et al. T-bet and eomesodermin are required for T cell-mediated antitumor immune responses. J Immunol. 2010;185(6):3174–3183. | ||
Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. Epub 2017 Oct 5. | ||
McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. 1999;77(1):1–10. | ||
Brunet JF, Denizot F, Luciani MF, et al. A new member of the immunoglobulin superfamily – CTLA-4. Nature. 1987;328(6127):267–270. | ||
Linsley PS, Greene JL, Tan P, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176(6):1595–1604. | ||
Freeman GJ, Lombard DB, Gimmi CD, et al. CTLA-4 and CD28 mRNA are coexpressed in most T cells after activation. Expression of CTLA-4 and CD28 mRNA does not correlate with the pattern of lymphokine production. J Immunol. 1992;149(12):3795–3801. | ||
Hastings WD, Anderson DE, Kassam N, et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009;39(9):2492–2501. | ||
Sabins NC, Chornoguz O, Leander K, et al. TIM-3 Engagement Promotes Effector Memory T Cell Differentiation of Human Antigen-Specific CD8 T Cells by Activating mTORC1. J Immunol. 2017;199(12):4091–4102. | ||
Lee J, Su EW, Zhu C, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. 2011;31(19):3963–3974. | ||
Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004;172(9):5450–5455. | ||
Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol. 2003;33(4):970–979. | ||
Xu F, Liu J, Liu D, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014;74(13):3418–3428. | ||
Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. 1995;25(9):2718–2721. | ||
Ma C, Cheung AF, Chodon T, et al. Multifunctional T-cell analyses to study response and progression in adoptive cell transfer immunotherapy. Cancer Discov. 2013;3(4):418–429. | ||
Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112(5):1557–1569. | ||
Romagnani S. T-cell subsets (Th1 versus Th2). Ann Allergy Asthma Immunol. 2000;85(1):9–18. | ||
Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.