Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Characteristics and potential role of M2 macrophages in COPD
Authors He S ![]() , Xie L, Lu J, Sun S
, Xie L, Lu J, Sun S
Received 24 July 2017
Accepted for publication 12 September 2017
Published 17 October 2017 Volume 2017:12 Pages 3029—3039
DOI https://doi.org/10.2147/COPD.S147144
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chunxue Bai
Shengyang He, Lihua Xie, Junjuan Lu, Shenghua Sun
Department of Respiratory Medicine, The Third Xiangya Hospital of Central South University, Changsha, Hunan, People’s Republic of China
Background: COPD is a multi-pathogenesis disease mainly caused by smoking. A further understanding of the mechanism of smoking-related COPD might contribute to preventions and treatments of this disease in the early stages. This study was designed to identify the characteristics of M2 macrophages in COPD for a better understanding about their potential role.
Materials and methods: COPD models were built in the C57BL/6 mouse by cigarette smoke (CS) exposure combined with intraperitoneal injection of cigarette smoke extract (CSE). The modeling efficiency was evaluated by lung function and hematoxylin and eosin (H&E) staining. The number of different macrophage phenotypes was detected by immunohistochemical staining (IHS) of CD206, CD86 and CD68 on the lung tissue paraffin section. The RAW264.7 cells were polarized toward the M2 phenotype by interleukin IL-4 and confirmed by a flow cytometer. The gene expression levels of TGF-βRII, Smad2, Smad3 and Smad7 in CSE-treated M2 macrophages were detected by real-time reverse transcription polymerase chain reaction (RT-PCR). The expression levels of TGF-β/Smad pathway-related makers (TGF-βRII, p-Smad2, p-Smad3, Smad7 and TGF-β) in alveolar M2 macrophages were detected by two consecutive paraffin section IHS.
Results: The COPD model is well established, which is confirmed by the lung function test and lung H&E staining. The whole number of macrophages and the ratio of M2/M1 phenotype are both increased (p<0.05). The level of CD206+ cells in IL-4-stimulated RAW264.7 cells is up to 93.4%, which is confirmed by a flow cytometer. The gene expression of TGF-βRII, Smad2, Smad3 and Smad7 are all enhanced (p<0.05) in CES-treated M2 macrophages, which is detected by RT-PCR. The protein levels of TGF-β/Smad pathway-related markers are all increased in alveolar M2 macrophages of the model group.
Conclusion: This study found an increased deposition of alveolar M2 macrophages in the mouse COPD model and an increased expression level of TGF-β/Smad pathway in M2 macrophages, both in vitro and in vivo, induced by CSE and/or CS exposure, indicating that M2 macrophages might contribute to COPD through changing of phenotype and TGF-β/Smad pathway.
Keywords: COPD, M2 macrophage, TGF-β/Smad pathway
Introduction
By the end of 2016, COPD had officially become the third leading cause of mortality worldwide, imposing a heavy financial burden to every country. COPD, primarily caused by smoking, is generally defined as a common preventable and treatable disease characterized by persistent irreversible airflow limitation.1,2 Pathologically speaking, COPD is featured by four main characteristics: emphysema, small airway remodeling, chronic bronchitis and pulmonary hypertension.3 We could conclusively describe all the pathological features as two main parts: emphysema and small airway disease.
Many studies have demonstrated a strong link between the pathological changes and the disordered immune system, especially the monocyte–macrophage system. Macrophages, a type of white blood cell, participate in the dissolving and development of inflammation and the repairing and remodeling of tissue by their functions of phagocytosis, antigen presentation and immunoregulation.4 Many researchers have depicted two main activated macrophages, corresponding to T helper 1 (Th1)/T helper 2 (Th2) immune response. One is the classically activated macrophages (related to Th1 immune response), which could produce a great amount of inflammatory cytokines such as TNF-α, IL-6 and IL-12, defending the host from exogenous microbes and tumor cells and are activated by cytokinesis such as interferon INF-γ and LPS. The other one is the alternatively activated macrophages (connected with Th2 immune response), which are responsible for the dissolvement of inflammation, tissue repairing and remodeling; they could be induced by Th2 cytokines such as IL-4 and IL-13.5 Further, through the alternative activation, macrophages could not only be activated into the M2 phenotype, but also M2-like macrophages.6 Although these two subtypes of M2 macrophages have many overlapping functions due to highly expressed functional surface molecules like CD206 and CD163 on both of them,7 there still exist some differences between them. The M2 macrophages are mainly stimulated by IL-4 and IL-13, while the M2-like phenotype is primarily activated by immunocomplexes, glucocorticoids and TGF-β, as well as IL-10, and so on. Another study found 12/15-lipoxygenase might also contribute to the production of M2-like macrophages.8 Moreover, with the increasingly developed understanding of M2 macrophages, they could even be further divided into three subtypes: M2a, M2b, and M2c.7 Studies have confirmed an enhanced polarization level of M2 phenotype in the lungs of smokers and even higher in COPD patients, indicating a crucial role of M2 macrophages in the pathogenesis of COPD induced by smoking.9
Small airway remodeling is the most important part of small airway disease, resulting from the accumulation of inflammatory secretions and the thickened and distorted airway wall (caused by airway-connected tissue repairing).10 Researchers found that an essential change in small airway remodeling is the disordered components of airway extracellular matrix (ECM; such as the deposition of collagen, fibronectin and glycosaminoglycan).11 TGF-β is considered to be the key to the stability of ECM, because it could regulate the balance of synthesis and decomposition of many component-related proteases in ECM dynamically. In its downstream molecular pathway, the TGF-β signal is received by TGF-βRII, which in turn activates TGF-βRI, resulting in the phosphorylation of the Smad2 and Smad3, which in turn could interact with the transporter Smad4, becoming a new complex. This complex could enter the nucleus and initiate gene transcription. Smad7 in turn can affect this pathway by inhibiting the phosphorylation of the Smad2 and Smad3.12
According to all the studies mentioned earlier, we believe that M2 macrophages and TGF-β/Smad pathway are both indispensable to the pathogenesis of COPD. To further enhance our understanding of M2 macrophages in COPD, we observed the number and ratio of the phenotype of macrophages in the mouse COPD model and then detected the expression level of TGF-β/Smad pathway of M2 macrophages affected by cigarette smoke (CS) exposure and cigarette smoke extract (CSE) in vivo and in vitro. We then made some reasonable speculations about the role of M2 macrophages in the pathogenesis of COPD based on all the results we obtained in this study.
Materials and methods
Animal model
A total of 20 6-week-old mice (weight: 21.1±1.92 g), bought from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China), were enrolled in this study and divided into two groups: the control group (n=10) and the model group (n=10; CS exposure combined with intraperitoneal injection of CSE). The breeding environment and details of the modeling method had been previously described.13 In short, the whole modeling period was 28 days. Briefly, the model group mice were put to CS exposure for two cycles per day except on days 1, 12 and 23. The control group was maintained in fresh air with no special treatment. On days 1, 12 and 23, mice in the control group were given an intraperitoneal injection of phosphate-buffered saline (PBS; 0.3 mL/20 g), and meanwhile, mice in the model group were given an intraperitoneal injection of 100% CSE solution (0.3 mL/20 g; preparation method is described in the following) in the same way. On day 29, all the mice underwent lung function measurement using a small animal spirometer. The study was approved by the institutional review board of Central South University and conformed to the guiding principles for research involving animals and human beings.14
Preparation of CSE
The preparation method of CSE was also described previously.13 Briefly, each non-filter Furong cigarette (tar: 13 mg, nicotine: 1.0 mg, carbon monoxide: 14 mg/cigarette; China Tobacco Hunan Industrial Co. Ltd., Changsha, China) was burned, and all the smoke was dissolved in 4 mL PBS as the 100% CSE. The solution was utilized for CSE intraperitoneal injection and cell culture before being filtrated via a 0.22-μm pore filter (Thermo Fisher Scientific, Waltham, MA, USA) to remove particles and bacteria. The solution was prepared freshly for each utilization.
Lung function measurement
Lung function was measured by a small animal spirometer (PLY3211 system; Buxco Electronics, Sharon, CT, USA), following the protocol described previously by our group.13 In brief, each mouse was weighed first and anesthetized by intraperitoneal injection of 10% chloral hydrate (3 mL/kg) before being tracheostomized. The trachea was cannulated, and the cannula was connected to a computer-controlled small animal spirometer. Airway resistance (Raw), lung dynamic compliance (Cdyn), peak expiratory flow (PEF) and inspiratory time/expiratory time (Ti/Te) were measured and noted.
Cell culture
The RAW264.7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA), being seeded in six-well plates with Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, Carlsbad, CA, USA) containing 10% (v/v) heat-inactivated fetal bovine serum (FBS; Life Technologies) and 100 U/mL penicillin G and 100 mg/mL streptomycin (HyClone Laboratories, Inc., Logan, UT, USA). The complete medium was free of mycoplasma. The cells were incubated at 37°C in a 5% CO2 incubator.
M2 macrophage polarization
We polarized M2 macrophages by stimulating RAW264.7 cells with IL-4 (20 ng/mL) as described in another study.15 Then, we prepared the IL-4 medium by adding 20 ng/mL of recombinant murine IL-4 (PeproTech, Rocky Hill, NJ, USA) to the complete medium described earlier. After being stimulated by IL-4 for 24 h, the RAW264.7 cells were thought to have become M2 macrophages. The polarization efficiency was tested by flow cytometry.
Flow cytometry
To confirm the percentage of M2 macrophages in the IL-4-stimulated RAW264.7 cells, we analyzed the positive rate of CD206, a widely utilized surface marker of M2 macrophages,16 by flow cytometry. Corresponding fluorescence label-conjugated isotype controls were utilized in this experiment. Briefly, all the IL-4-stimulated RAW264.7 cells were harvested 24 h later and washed by flow buffer (PBS supplemented with 1% [v/v] FBS and 0.05% NaN3) once in room temperature and once sequentially in ice-cold flow buffer. Then, CD206-specific antibody (BD Pharmingen, San Jose, CA, USA; diluted 1:40) was used to analyze the expression of CD206 via a BD Cyan flow cytometer using CellQuest software (BD Biosciences, San Jose, CA, USA). Further analysis was performed using FloJo software (Tree Star Inc., Ashland, OR, USA).
Hematoxylin and eosin (H&E) staining of lung tissue
We chose the lower left lobes to be the sample. The lung tissue was inflated by 4% paraformaldehyde at a constant pressure of 25 cmH2O and then fixed with 4% paraformaldehyde for 24 h. The fixed lung tissue was embedded by paraffin (Sigma-Aldrich Co., St Louis, MO, USA) and sectioned into 4-μm sections for further staining with H&E (Sigma-Aldrich Co.). We collected the data of the mean linear intercept (MLI), mean alveolar septal thickness (MAST) and destructive index (DI) to evaluate the pathological change in emphysema of the model as previously described in another study.17
Immunohistochemical staining (IHS)
In the first part, we ran the IHS to assess the expression levels of CD68, CD86 and CD206 (representing macrophages, M1 macrophage and M2 macrophage, respectively) in lung tissue by utilizing the specific antibodies of CD68 (diluted 1:100; Proteintech, Chicago, IL, USA), CD86 (Santa Cruz Biotechnology Inc., Dallas, TX, USA; diluted 1:200) and CD206 (diluted 1:100; Proteintech).18 In the second part, we detected the expression level of key factors of TGF-β/Smad (TGF-β: diluted 1:200, Abcam, Shanghai, China; TGF-βRII: diluted 1:100, Abcam; p-smad2: diluted 1:100, Abcam; p-smad3: diluted 1:100, Abcam and smad7: diluted 1:100, R&D Systems, Inc., Minneapolis, MN, USA) pathway in M2 macrophages of mouse lung tissue. Since the diameter of a cell is more than the thickness of two continuous sections, these two continuous sections could almost represent the information from the same cell layer. Thus, we utilized the two continuous section-contrasted staining method (putting two consecutive lung paraffin sections as A and B on the same slide and running IHS of CD206 on section A and target proteins [TGF-β, TGF-βRII, p-Smad2, p-Smad3, Smad7] on section B to detect the expression level of the target proteins in CD206 located cells) to detect the expression of the target proteins in M2 macrophages. Briefly, 4 μm-thick paraffin-embedded lung tissue slice was deparaffinized and rehydrated. Antigen was retrieved using sodium citrate at 95°C for 10 min (Tris/EDTA, 95°C, 20 min for p-Smad2). To block endogenous peroxidase activity, these sections were incubated with 3% H2O2 for 30 min at room temperature. Then, the lung tissues were incubated overnight at 4°C with all the first antibodies. The samples were then washed carefully by PBS and incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies for 15 min at 37°C. Then, the target proteins were visualized by diaminobenzidine (DAB) (Sigma-Aldrich Co.) and counterstained with hematoxylin. The tissues were photographed by a microscope (Olympus Corporation, Tokyo, Japan). In the first part, the number of positive cells were collected. In the second part, we calculated the ratio of double-positive cell/CD206-positive cell (double positive: the CD206+ and the corresponding target protein + in the same cell layer from the two continuous sections) to represent the expression level of the target proteins in M2 macrophages.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
We treated the IL-4-stimulated RAW264.7 cells with CSE (10%, 24 h) and then utilized the TRIzol reagent (TaKaRa, Dalian, Liaoning, China) to isolate the total RNA of them following the manufacturer’s protocol. Then, the total RNA sample (optical density [OD] A260/A280>1.8) was reversely transcribed by PrimeScript™ RT Reagent Kit (TaKaRa). Then, the relative expression level of mRNA was assessed by SYBR Green PCR Master Mix (Bio-Rad, Laboratories Inc., Hercules, CA, USA) and quantified by two-step quantitative real-time polymerase chain reaction (PCR; Applied Biosystems, Carlsbad, CA, USA). β-actin was used as an internal control, and ΔCT method was used to calculate the expression of mRNA. The primers for quantitative polymerase chain reaction (qPCR) analysis are shown in Table 1.
 | Table 1 Information of primers for PCR used in this study |
Statistical analysis
All the data were analyzed by SPSS 22.0 statistical software (IBM Corporation, Armonk, NY, USA) and are presented as mean ± standard error of the mean (SEM). The differences between groups were examined using Student’s unpaired t-test. And we used the Pearson’s correlation coefficient between groups to analyze the correlation. p-value <0.05 was considered as statistically significant.
Results
Evaluation of mouse COPD model
The changes in lung function of the model group mice were similar to those of human COPD patients whose Cdyn, PEF and Ti/Te were all decreased (p<0.05; Figure 1B–D) compared to the normal ones, and meanwhile, Raw was increased (p<0.05; Figure 1A). The pathological changes were mainly described as enlarged alveolar space, thinner alveolar septum and destroyed alveolar wall. Quantitatively speaking, the MIL, DI and MAST were all decreased compared to the control group (p<0.05; Figure 2A–C).
 | Figure 1 Comparison of lung function in the control group and model group according to (A) Raw (airway resistance), (B) Cdyn (lung dynamic compliance), (C) PEF (peak expiratory flow) and (D) Ti/Te (inspiratory time/expiratory time). |
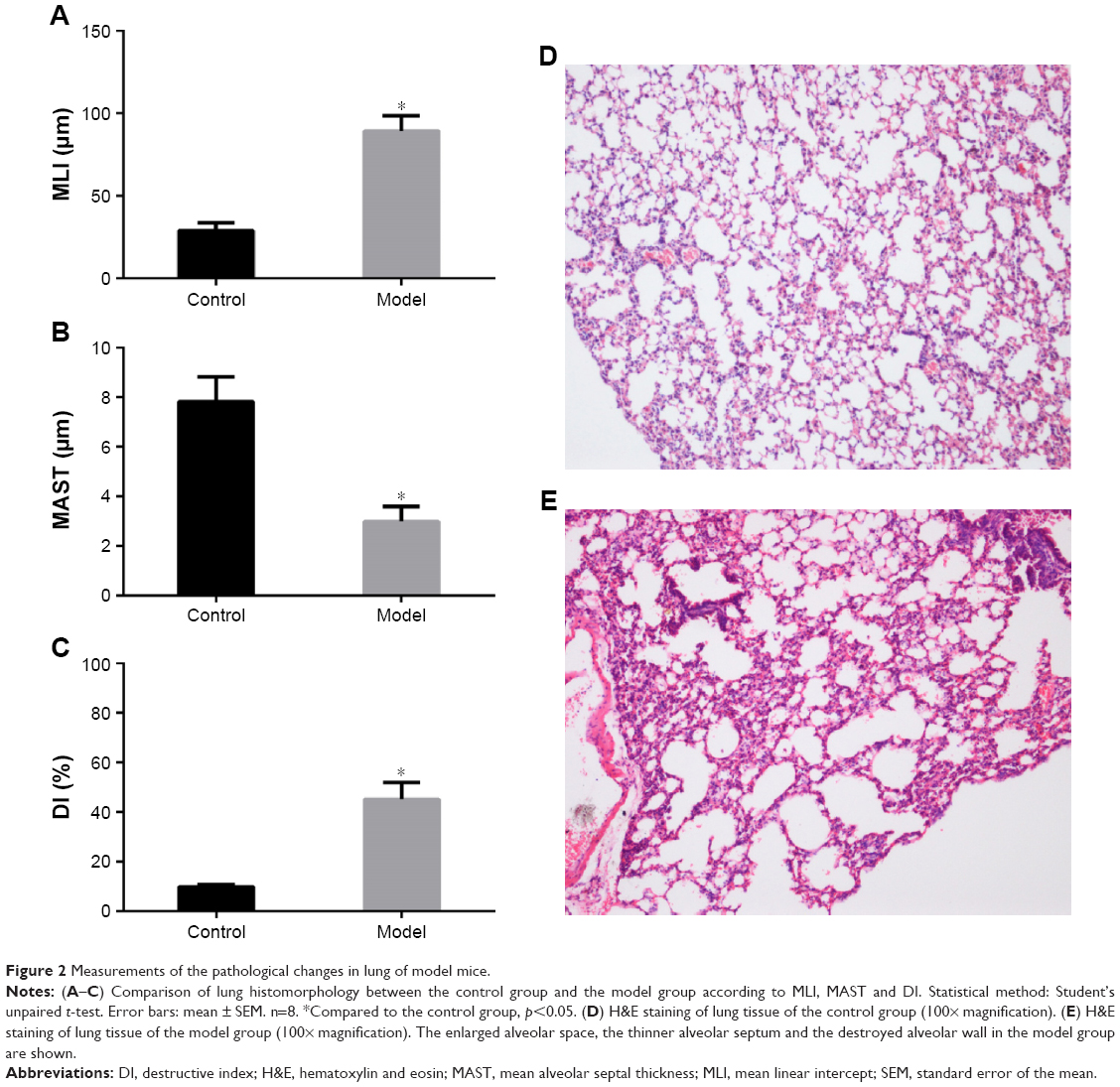 | Figure 2 Measurements of the pathological changes in lung of model mice. |
Macrophages were M2 polarized in mouse COPD model
In the first IHS part, we found an enhanced number of macrophages (p<0.05; Figures 3A and 4A and B) and an increased ratio of M2/M1, indicating a tendency that showed a M2-directed polarization of macrophages in the lung of model group mice (p<0.05; Figures 3B and 4C–F).
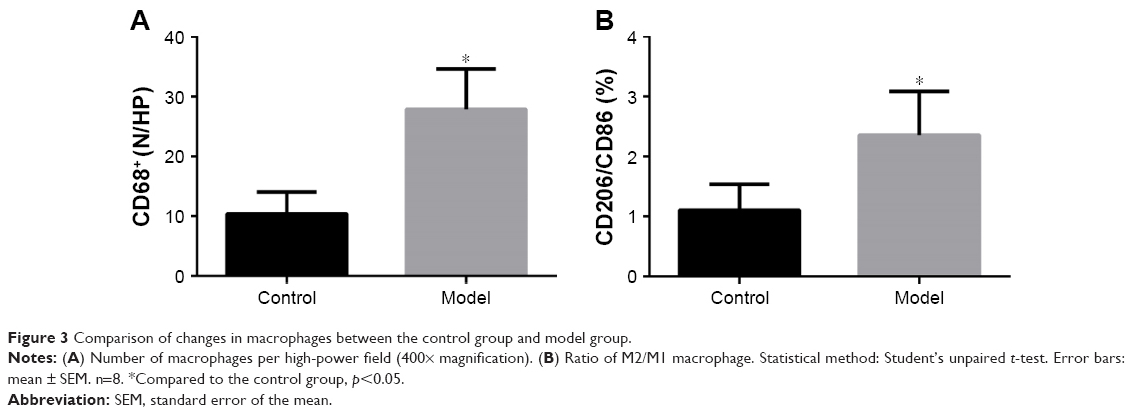 | Figure 3 Comparison of changes in macrophages between the control group and model group. |
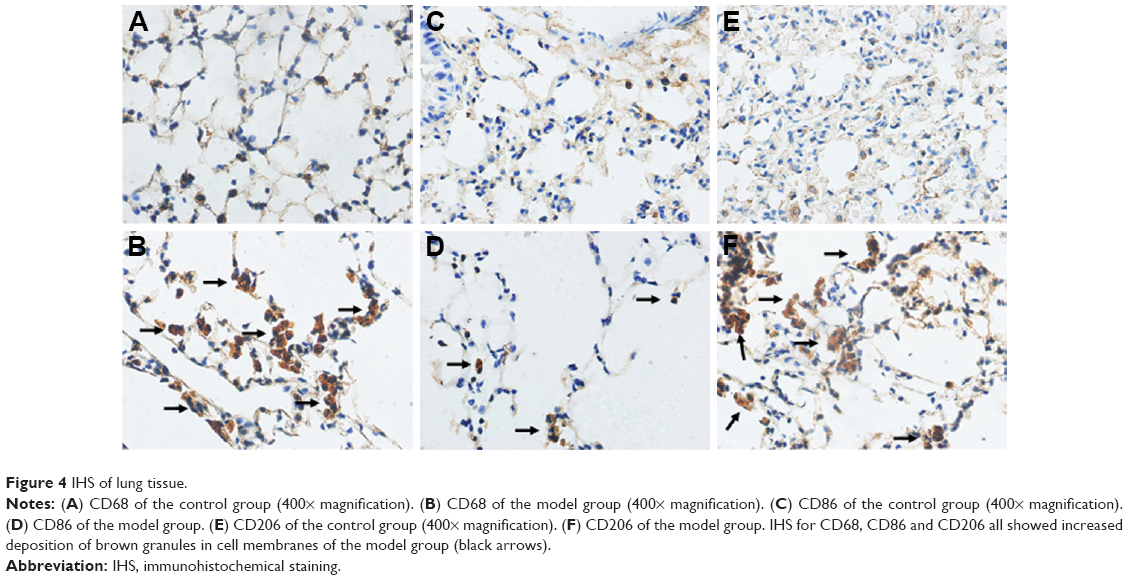 | Figure 4 IHS of lung tissue. |
Correlation analysis for lung function and the ratio of M2/M1 macrophages in lung tissue among the control and model groups
We found, by Pearson analysis, that the ratio of M2/M1 in lung tissue tended to have a positive correlation with Raw of lung function (Figure 5A). However, the other criteria of lung function such as Cdyn, PEF and Te/Ti were all negatively correlated with the ratio of M2/M1 macrophages (Figure 5B–D).
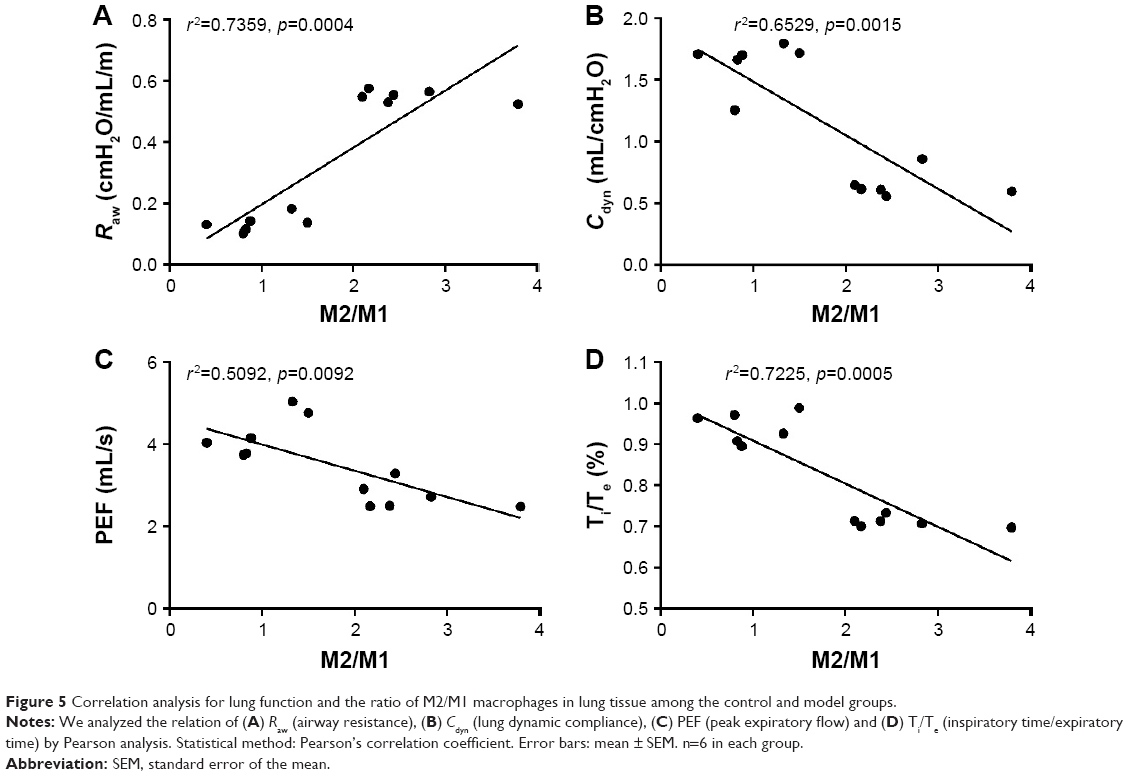 | Figure 5 Correlation analysis for lung function and the ratio of M2/M1 macrophages in lung tissue among the control and model groups. |
Polarization efficiency of IL-4-stimulated RAW264.7 cells
The RAW264.7 cells were detected for the surface marker of CD206 after IL-4 stimulation (20 ng/mL, 24 h) by flow cytometry. The positive rate of CD206+ cells was up to 93.4% (Figure 6), which is sufficient for the following experiments.
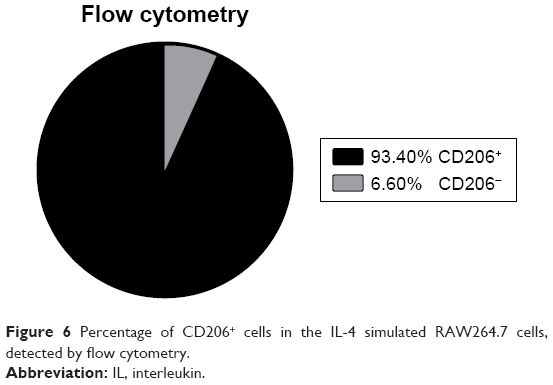 | Figure 6 Percentage of CD206+ cells in the IL-4 simulated RAW264.7 cells, detected by flow cytometry. |
Gene expression change of TGF-β/Smad pathway in CSE-treated M2 macrophage
The ratio of OD A260/A280 of the total RNA we isolated was between 1.8 and 2.1. Analyzed by RT-PCR, the gene expression values of TGF-βRII, Smad2, Smad3 and Smad7 were all increased compared to the control group (p<0.05; Figure 7).
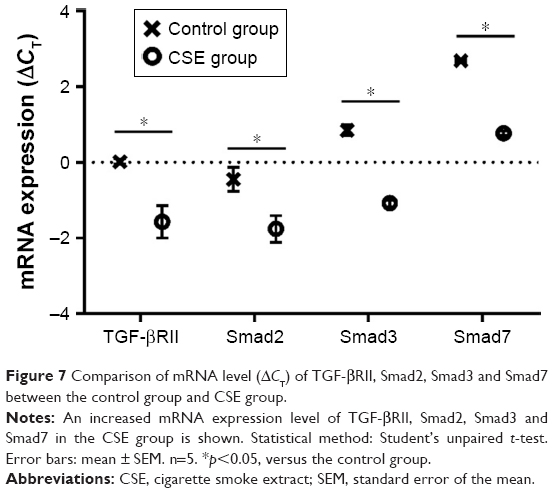 | Figure 7 Comparison of mRNA level (ΔCT) of TGF-βRII, Smad2, Smad3 and Smad7 between the control group and CSE group. |
Protein expression change of TGF-β/Smad pathway in M2 macrophages in lungs of mouse COPD model
We ran the two continuous section-contrasted staining in immunohistochemical way to detect the expression levels of TGF-β, TGF-βRII, p-Smad2, p-Smad3 and Smad7. We found, in the model group, that the ratio of all double-positive/CD206-positive cells was higher than those of the corresponding control group, which means that the expression levels of all target proteins in M2 macrophages were increased compared to the those of the control group (p<0.05; Figures 8 and 9).
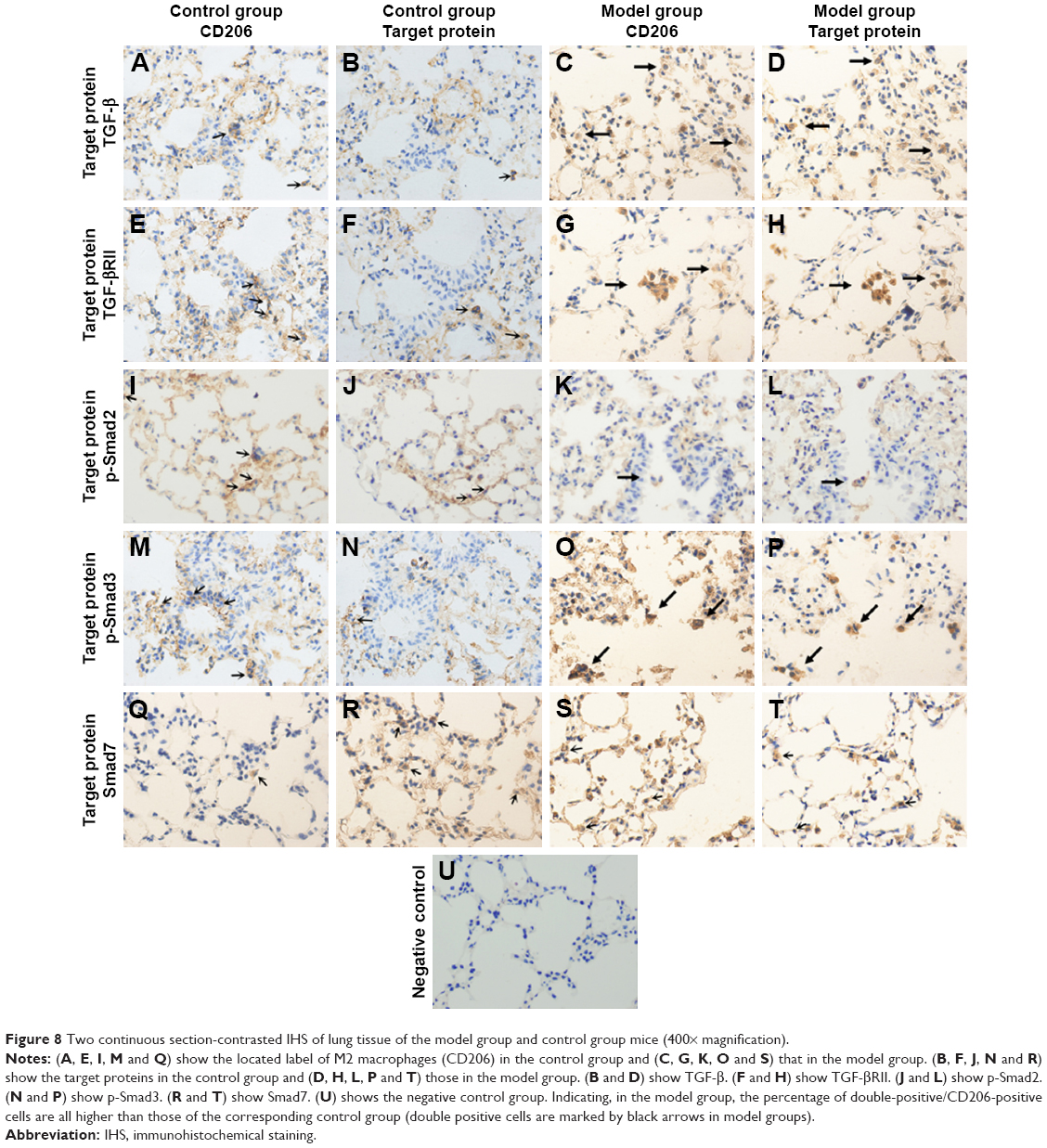 | Figure 8 Two continuous section-contrasted IHS of lung tissue of the model group and control group mice (400× magnification). |
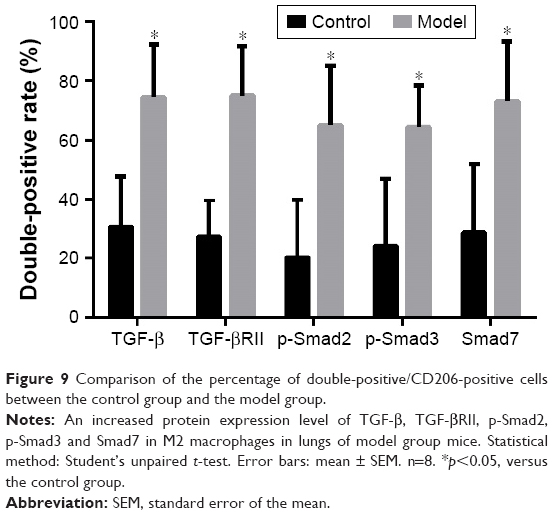 | Figure 9 Comparison of the percentage of double-positive/CD206-positive cells between the control group and the model group. |
Discussion
In the present study, we successfully built the mouse COPD model and confirmed a M2-directed polarization tendency of alveolar macrophages. Moreover, we initially found that the expression level of TGF-β/Smad pathway in M2 macrophages is aberrantly activated both in vitro and in vivo and TGF-β from M2 macrophages themselves might accelerate this activation process in an autocrine manner.
The COPD model-building method of CS exposure combined with CES intraperitoneal injection was first reported by our group, and the changes were similar with those of human smokers and COPD patients pathologically and physiopathologically.13 The long-term smoking-induced mouse COPD model is the most classical method to build COPD model, and the method of CES intraperitoneal injection alone was also reported previously.19 This new method is more efficient compared to the classical ones.
Generally speaking, macrophages could be divided into different subgroups according to their surface markers and functions as is mentioned earlier.5 Briefly, the M2 phenotype, induced by Th2-related cytokines, presenting a feature of anti-inflammation, is also considered to be related with antiparasite, tissue repairing and remodeling as well as tumor promotion.20 The alveolar macrophages present a polarization tendency of M2 phenotype, which is identical with the previous studies.9,21
Small airway disease is one of the most crucial pathological change in COPD. As is demonstrated by the other researchers,22 the small airway narrowing results from the remodeling and fibrosis of surrounding tissue of peripheral airway and the destruction of small airway-attached alveolar. These processes are all related to TGF-β/Smad pathway, which is a key factor to tissue fibrosis and remodeling and adjusting of ECM elements.23,24 Research has reported that serum amyloid P presents an anti-fibrotic feature through inhibiting M2 polarization in an Smad3-dependent manner.25 Other research has found that, in insulin-resistant patients, adipose tissue demonstrated increased fibrosis, which might be resulting from M2 macrophage abundance and TGF-β activity.26 There is also research concerned with the cytokine environment of abdominal cavities of peritoneal fibrosis (PF) rats, promoting macrophages to differentiate to M2 phenotype. The function of M2 macrophages in PF might be related to the TGF-β/Smad pathway.27 All this research demonstrated M2-directed and TGF-β/Smad pathway-activated changes to be involved in a tissue-remodeling and fibrosis-related pathogenic environments. When people smoke, the alveolar macrophages are under direct exposure of CS. To observe the characteristics of M2 macrophages in such a pathogenic environment in vitro and vivo, we treated the M2 macrophages with CSE and build mouse COPD model, respectively. We found an obviously enhanced expression level of TGF-β/Smad signaling in M2 macrophages both in vitro and in vivo compared to that in the control group. These results indicate that CS exposure might not only promote M2-directed polarization of macrophages28 but also activate the TGF-β/Smad pathway in them. Smad7 took effect as a pathway antagonist. Our present results were in accordance with the previous studies to a large extent. Although there are many limitations utilizing RAW264.7 cells (mouse leukemic macrophages) as a research object, their functions of macrophage still remain, and many studies utilized RAW264.7 to design M2-related research and obtained ideal results.15,29
Previously, many studies had established a strong relationship between matrix metalloproteinases (MMPs) and COPD, because MMPs are the regulatory factors of elastin and collagen in ECM, which are indispensable to the occurrence of emphysema.30–32 Studies found, under CS exposure, that the expression levels of MMPs were increased in lungs of mouse.9,33,34 Moreover, MMP-9 and MMP-12 had been confirmed to have strong causal relationship with COPD. To the best of our knowledge, macrophages could secrete MMPs themselves,35 and smoking might contribute to a higher level of MMP-9 from alveolar macrophages;36 moreover, it went further from COPD patients’ alveolar macrophages with a higher elastolytic activity.37 Besides the disordered cytokines secretion of macrophages under CS exposure, the functions of antigen presentation and phagocytosis were damaged,38 leading to a defect in inflammation regulation. Nevertheless, all this research did not concern the subgroup of macrophages. Gao et al39 found that smoking might give rise to the Smad7 level, which might in turn cause a higher MMP-9 level, leading to emphysematous changes in Fut8−/− mouse, and they did not mention the sources of Smad7 and TGF-β. However, it is reasonable to speculate, largely, that those aberrantly secreted Smad7 and TGF-β are related to M2 macrophages in COPD. The studies cited above all surmised that, more or less, CS exposure, M2 macrophages and TGF-β/Smad signaling are responsible for COPD respectively, but none of these studies had debated the inner connection of these three items. Based on all the previous studies and what we found in our present study, we could make a hypothesis that smoking might cause a M2-directed polarization tendency of alveolar macrophages with an activated TGF-β/Smad signaling, which might promote the secretion of MMP-9 from these M2 macrophages, contributing to the occurrence of COPD eventually. Therefore, blocking the alternative activation process of macrophages and the activation of TGF-β/Smad in M2 macrophages might be novel ways to prevent and treat COPD in the early stage.
Some limitations of the present study merit consideration. Initially, the original intention of this study was to observe the characteristics of M2 macrophages in COPD both in vitro and in vivo and speculate the probable pathogenic mechanism behind it reasonably. Thus, this study might not be able to demonstrate a complete mechanism of M2 macrophages in COPD but a potential role based on all the data we collected. However, it might still sever as an enlightenment for further research on mechanisms. Moreover, the method of two continuous section-contrasted IHS might, inevitably, like every other experimental method, cause some bias in results because the two continuous tissue paraffin sections could be damaged physically, more or less, during the manufacturing process. While according to our observation, these biases of results are acceptable and have not made any significant difference to the trend of primary outcomes. This novel method of IHS is feasible theoretically and simple to handle, working without advanced experiment instruments such as laser confocal fluorescence microscope. Nevertheless, it could be affected by improper and careless operational processes; therefore, a detailed and canonical protocol might be necessary to make this method more reliable and credible.
Conclusion
We demonstrated that macrophages tend to polarize to the M2 phenotype with an activated TGF-β/Smad signaling under CS and/or CSE exposure both in vitro and in vivo. Initially, we indicated that functions of M2 macrophages in the pathogenesis of COPD might associate with TGF-β/Smad signaling. However, this study was still an observational one. Further studies about the accurate molecular mechanism are necessary to address the complete role of M2 macrophages in the pathogenesis of COPD; yet, intervention of macrophages’ polarization and activation of TGF-β/Smad pathway might be novel directions to prevent and treat COPD in the early stage.
Acknowledgment
This study was supported by grants from the National Natural Science Foundation of China (Youth Science Foundation; No 81500033).
Author contributions
All authors made substantial contributions to conception and design, acquisition of data or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Gnatiuc L, Caramori G. COPD in nonsmokers: the biomass hypothesis – to be or not to be? Eur Respir J. 2014;44(1):8–10. | ||
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. | ||
Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L1–L15. | ||
Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol. 2011;12(3):231–238. | ||
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. | ||
Madsen DH, Leonard D, Masedunskas A, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol. 2013;202(6):951–966. | ||
Boorsma CE, Draijer C, Melgert BN. Macrophage heterogeneity in respiratory diseases. Mediators Inflamm. 2013;2013:769214. | ||
Uderhardt S, Herrmann M, Oskolkova OV, et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity. 2012;36(5):834–846. | ||
Shaykhiev R, Krause A, Salit J, et al. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183(4):2867–2883. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Kranenburg AR, Willems-Widyastuti A, Moori WJ, et al. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. 2006;126(5):725–735. | ||
Eurlings IM, Dentener MA, Cleutjens JP, et al. Similar matrix alterations in alveolar and small airway walls of COPD patients. BMC Pulm Med. 2014;14:90. | ||
Lu J, Xie L, Liu C, Zhang Q, Sun S. PTEN/PI3k/AKT regulates macrophage polarization in emphysematous mice. Scand J Immunol. 2017;85(6):395–405. | ||
World Medical Association, American Physiological Society. Guiding principles for research involving animals and human beings. Am J Physiol Regul Integr Comp Physiol. 2002;283(2):R281–R283. | ||
He Z, Zhang H, Yang C, et al. The interaction between different types of activated RAW 264.7 cells and macrophage inflammatory protein-1 alpha. Radiat Oncol. 2011;6:86. | ||
Song L, Huang Y, Zhao M, et al. A critical role for hemolysin in Vibrio fluvialis-induced IL-1beta secretion mediated by the NLRP3 inflammasome in macrophages. Front Microbiol. 2015;6:510. | ||
He ZH, Chen P, Chen Y, et al. Comparison between cigarette smoke-induced emphysema and cigarette smoke extract-induced emphysema. Tob Induc Dis. 2015;13(1):6. | ||
Hernandez C, Barrachina MD, Cosin-Roger J, et al. Progastrin represses the alternative activation of human macrophages and modulates their influence on colon cancer epithelial cells. PLoS One. 2014;9(6):e98458. | ||
Chen Y, Hanaoka M, Chen P, Droma Y, Voelkel NF, Kubo K. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. Am J Physiol Lung Cell Mol Physiol. 2009;296(4):L648–L656. | ||
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. | ||
Kaku Y, Imaoka H, Morimatsu Y, et al. Overexpression of CD163, CD204 and CD206 on alveolar macrophages in the lungs of patients with severe chronic obstructive pulmonary disease. PLoS One. 2014;9(1):e87400. | ||
Contoli M, Santus P, Papi A. Small airway disease in asthma: pathophysiological and diagnostic considerations. Curr Opin Pulm Med. 2015;21(1):68–73. | ||
Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364(9435):709–721. | ||
Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35(2):83–92. | ||
Murray LA, Chen Q, Kramer MS, et al. TGF-beta driven lung fibrosis is macrophage dependent and blocked by serum amyloid P. Int J Biochem Cell Biol. 2011;43(1):154–162. | ||
Spencer M, Yao-Borengasser A, Unal R, et al. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab. 2010;299(6):E1016–E1027. | ||
Hu W, Jiang Z, Zhang Y, et al. Characterization of infiltrating macrophages in high glucose-induced peritoneal fibrosis in rats. Mol Med Rep. 2012;6(1):93–99. | ||
Yuan F, Fu X, Shi H, Chen G, Dong P, Zhang W. Induction of murine macrophage M2 polarization by cigarette smoke extract via the JAK2/STAT3 pathway. PLoS One. 2014;9(9):e107063. | ||
Liu CY, Xu JY, Shi XY, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. 2013;93(7):844–854. | ||
Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol. 2003;28(1):12–24. | ||
Elkington PT, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax. 2006;61(3):259–266. | ||
Gueders MM, Foidart JM, Noel A, Cataldo DD. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: potential implications in asthma and other lung diseases. Eur J Pharmacol. 2006;533(1–3):133–144. | ||
Stevenson CS, Docx C, Webster R, et al. Comprehensive gene expression profiling of rat lung reveals distinct acute and chronic responses to cigarette smoke inhalation. Am J Physiol Lung Cell Mol Physiol. 2007;293(5):L1183–L1193. | ||
Wright JL, Tai H, Wang R, Wang X, Churg A. Cigarette smoke upregulates pulmonary vascular matrix metalloproteinases via TNF-alpha signaling. Am J Physiol Lung Cell Mol Physiol. 2007;292(1):L125–L133. | ||
Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. | ||
Lim S, Roche N, Oliver BG, Mattos W, Barnes PJ, Chung KF. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10. Am J Respir Crit Care Med. 2000;162(4 pt 1):1355–1360. | ||
Russell RE, Culpitt SV, DeMatos C, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26(5):602–609. | ||
Hodge S, Hodge G, Ahern J, Jersmann H, Holmes M, Reynolds PN. Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2007;37(6):748–755. | ||
Gao C, Maeno T, Ota F, et al. Sensitivity of heterozygous alpha1, 6-fucosyltransferase knock-out mice to cigarette smoke-induced emphysema: implication of aberrant transforming growth factor-beta signaling and matrix metalloproteinase gene expression. J Biol Chem. 2012;287(20):16699–16708. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.