Back to Archived Journals » Transplant Research and Risk Management » Volume 13
Changes in the Composition and Function of the Human Salivary Microbiome After Heart Transplantation: A Pilot Study
Authors Joyce H, Taylor MR, Moffat A ![]() , Hong M, Isaac D, Fine N, Greenway SC
, Hong M, Isaac D, Fine N, Greenway SC
Received 8 July 2021
Accepted for publication 27 August 2021
Published 1 September 2021 Volume 2021:13 Pages 15—21
DOI https://doi.org/10.2147/TRRM.S328467
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Qing Yi
Heidi Joyce,1 Michael R Taylor,1 Andrew Moffat,1 Mia Hong,1 Debra Isaac,2 Nowell Fine,2 Steven C Greenway1– 3
1Department of Pediatrics and Alberta Children’s Hospital Research Institute, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada; 2Department of Cardiac Sciences and Libin Cardiovascular Institute, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada; 3Department of Biochemistry & Molecular Biology, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada
Correspondence: Steven C Greenway
Section of Cardiology, Alberta Children’s Hospital, 28 Oki Drive NW, Calgary, AB, T3B 6A8, Canada
Tel +1-403-955-5049
Fax +1-403-955-7621
Email [email protected]
Introduction: Interactions between our commensal microbes and immune system are well recognized but the impact of immunosuppression on this relationship is less well characterized, particularly outside of the gastrointestinal tract. In this pilot case-control study, we examined microbial composition and inferred function in the saliva of patients after heart transplantation.
Methods: Saliva samples were collected from 26 healthy adolescent and adult heart transplant patients and 27 healthy non-transplant controls. Bacterial DNA was isolated and the V3 and V4 regions of the 16S rRNA gene were sequenced. Measures of bacterial diversity and inferred function were calculated using the software packages DADA2, Phyloseq, QIIME, PICRUSt and STAMP.
Results: Transplant patients were on average 51.6 ± 18.2 years of age and 8.6 ± 5.3 years post-transplant. Both alpha (p = 0.0009) and beta (p = 0.001) diversity differed significantly between the groups, and there were statistically significant changes (p < 0.01) in 101 individual functional pathways.
Discussion: We conclude that the effects of chronic immunosuppression on bacterial composition and function in the oropharynx appear relatively subtle with no obvious ill effects on patient health. Full interpretation is limited by our lack of knowledge for many of the bacterial functional pathways.
Keywords: microbiome, saliva, transplant, heart, immune suppression
Plain Language Summary
Interactions between the microbes that exist within us and the immune system are not well characterized, particularly outside of the intestinal tract. We studied microbial composition and inferred function in the saliva of patients after heart transplantation compared to healthy non-transplant controls. Bacterial diversity differed significantly between the groups and there were statistically significant changes in multiple individual functional pathways. We conclude that immune suppression has broad but subtle effects on the oral microbiome.
Introduction
To improve human health it is increasingly recognized that it is important to understand the relationship between the body and our commensal microbes.1,2 There are many diverse microorganisms within the human microbiome3 which has multiple important functions and works to maintain homeostasis within the human host.4–8 The oral cavity provides primary immune defense, acting to prevent pathogens from entering the host. The oral microbiome9–11 may play an important role in maintaining the health of an individual and alterations, such as are seen in periodontitis, are associated with microbial migration through the bloodstream and increased risk of cardiovascular and non-cardiovascular diseases.12–16
After cellular or solid organ transplantation, patients must take immunosuppressive drugs (ISDs) to prevent immune-mediated injury (ie, rejection) of the donated organ or cells. As the immune system and microbiome are closely intertwined, their relationship is necessarily altered in the presence of systemic immunosuppression.17–21 These changes may have important implications for patient health post-transplant; thus, the microbiome may represent a modifiable environmental variable that can be manipulated to improve outcomes for patients after transplantation. While projects such as the Human Microbiome Project4 have advanced our understanding of the diversity and variability of niche microbiomes in immune competent humans, the microbiome after transplantation remains relatively understudied. This proof-of-concept project aimed to characterize the oral microbiome of adult heart transplant patients both in terms of bacterial composition and function and to provide insight into the effect of chronic immunosuppression on the salivary microbiome.
Materials and Methods
This case-control pilot study was approved by the Conjoint Health Research Ethics Board at the University of Calgary (study ID REB13-0576). This study was conducted in accordance with the Declaration of Helsinki. Adolescent and adult heart transplant (HT) patients were recruited at the Alberta Children’s Hospital and Foothills Medical Centre in Calgary, Alberta, Canada between October and December 2015 and included in the study if they were healthy outpatients. All organs were donated voluntarily with written informed consent and transplantation was conducted in accordance with the Declaration of Istanbul. All patients provided informed written consent. For participants <18 years of age, a parent or legal guardian provided consent. Information was collected from the patient medical chart regarding age, sex, date of transplant, current immunosuppression and relevant recent medical events. Patients with recent exposure to antibiotics, presence of significant comorbidities, recent infection, hospitalization, surgery or rejection events were excluded from the analysis. Control samples were collected between October and December 2016 from healthy, non-transplant student volunteers with no preexisting medical or dental conditions at the University of Calgary. After providing informed written consent, all participants were asked to provide a saliva sample. Samples were collected using the OMNIgene (OM-501) collection kit (DNA Genotek, Canada). Saliva samples were stored at room temperature in the original collection tube until processed. DNA was isolated from all saliva samples using the Genomic DNA MiniPrep Kit (DNA Genotek, Canada) following the protocol provided by the manufacturer. Following isolation, DNA concentration was determined using a Qubit fluorometer (ThermoFisher Scientific, USA) and then stored at −20°C. Purified DNA was used for PCR amplification of the bacterial 16S rRNA gene V3/V4 regions.22 Libraries were constructed and sequenced on the Illumina MiSeq platform in the Nicole Perkins Microbial Communities Core Laboratory at the University of Calgary.
Illumina-sequenced, paired-end FASTQ files were trimmed to remove primers and barcodes using Cutadapt.23 DADA2 was used to produce an OTU (operational taxonomic unit) table with taxonomy assigned through SILVA or de novo clustering.24 Downstream analysis of the final OTU table was done in R using Phyloseq (version 1.16.2).25 Functional analysis of the bacterial populations was performed using QIIME (Quantitative Insights Into Microbial Ecology)26 and PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States).27 STAMP (STatistical Analysis of Metagenomics Profiles)28 was used to analyze the metabolic profiles of patient samples compared to healthy controls using the output produced through PICRUSt. Functional predictions of KEGG (Kyoto Encyclopedia of Genes and Genomes) Orthologs (KOs) were made using the corrected OTU table as input.29 Identified KOs were also collapsed to KEGG pathways to allow for biological interpretation of higher-level systemic functions.
Patient data are presented as mean ± standard deviation with a two-sided Welch’s t-test used to determine differences between the control and HT groups. A Kruskal–Wallis test and a PERMANOVA test were used to test for significant differences between the groups for alpha and beta diversity, respectively. STAMP was used for statistical analysis of microbial function in conjunction with a Benjamini-Hochberg false discovery rate procedure to correct for multiple comparisons. The significance level for all tests was set at p < 0.05.
Results
A total of 53 participants were recruited for this study. Of the 53 participants, 26 were HT patients and 27 were healthy, non-transplant controls. The non-transplant controls had no self-reported medical conditions, and all HT patients were stable outpatients at the time of sample collection with no recent (<1 month) rejection events or changes in immunosuppression. For the 25 HT patients for whom clinical data was available (Table 1), 36% were female and 64% were male. Average age was 51.6 ± 18.2 years and ranged from 14 to 74 years. Patients were 8.6 ± 5.3 years post-transplant (range 0.75–20 years). For all 25 patients, 19 (76%) were on tacrolimus, 20 (80%) were on mycophenolate mofetil, 6 (24%) were on sirolimus, and 5 (21%) were on prednisone. Most patients (64%) were on dual therapy with tacrolimus and mycophenolate mofetil. For the 21 patients where data was available, 9 (43%) had Diabetes Mellitus type II. None of the patients had a history of recent infection or exposure to antibiotics.
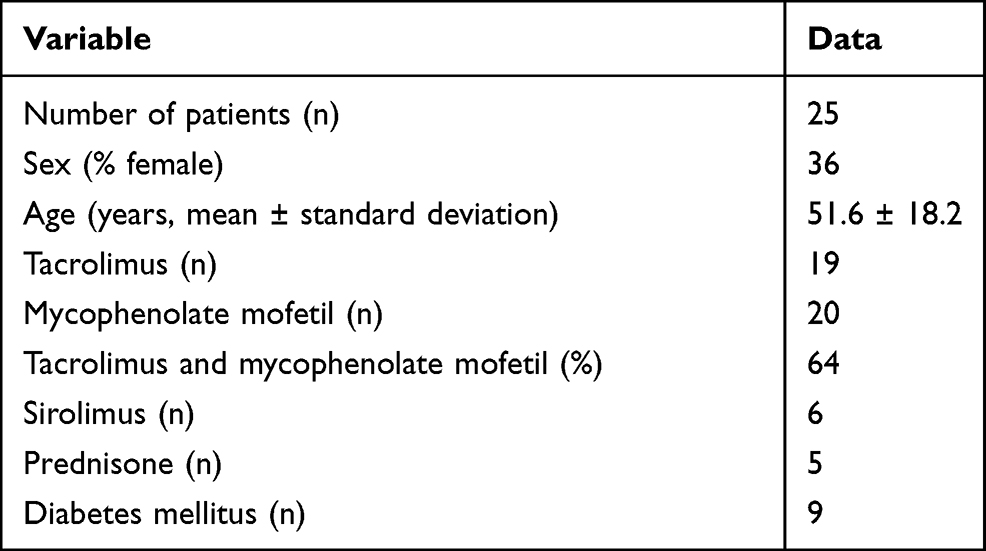 |
Table 1 Demographic Characteristics of Studied Heart Transplant Patients |
Bacterial composition was examined at the class level for all samples. Taxonomic distribution of the top 20 sequences for both control and HT patients is shown in Figure 1. The class Bacilli predominated for most samples with some individual samples from both groups showing relatively larger populations of Bacteroidia and Betaproteobacteria. Fusobacteria and Gammaproteobacteria were well represented in most samples. Alpha diversity (Shannon diversity index), referring to diversity within a sample, was highly significantly different (p = 0.00088) between the groups with reduced diversity in the transplant patient group (Figure 2). Beta diversity was significantly different between the control and transplant patient groups (p = 0.001), as determined using the Bray–Curtis dissimilarity index and visualized with a non-metric multidimensional scaling (NMDS) plot (Figure 3).
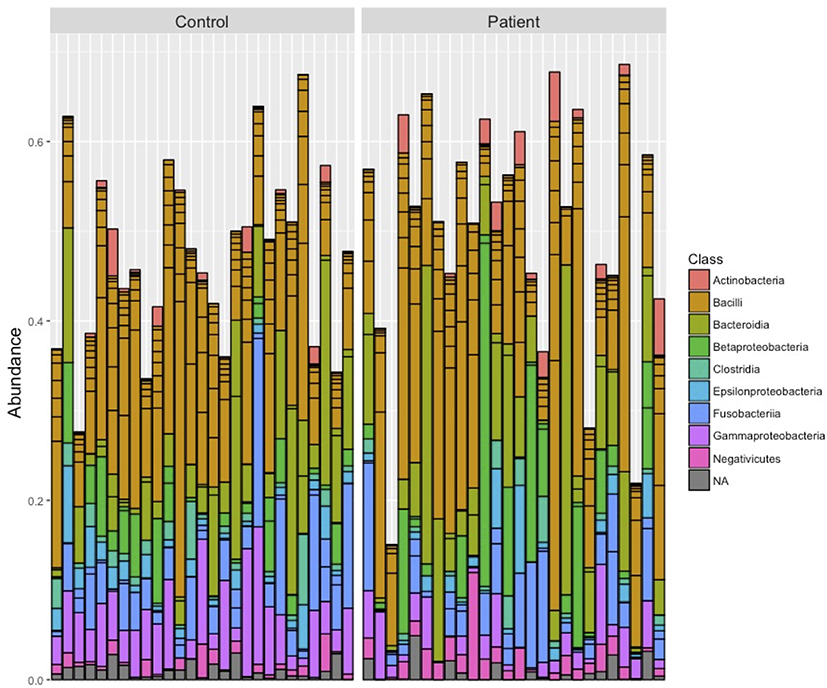 |
Figure 1 Salivary bacterial composition at the class level. The taxonomic distribution of the top 20 sequences are shown for each individual saliva sample in the control and patient groups. |
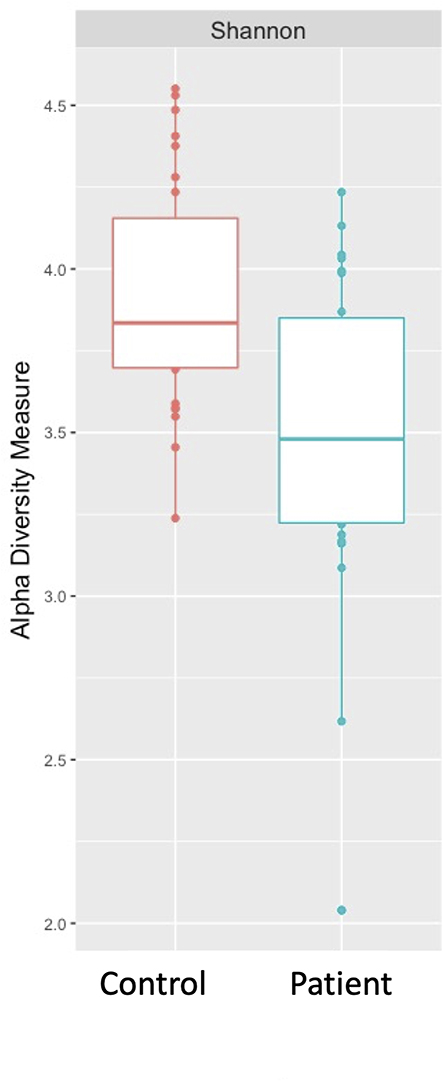 |
Figure 2 Alpha (Shannon) diversity of the saliva microbiome. Intra-sample diversity differed significantly between the control and heart transplant patient groups (p = 0.00088) with decreased diversity in the transplant samples. |
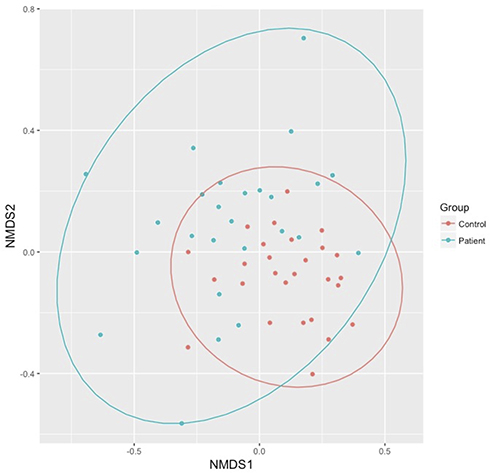 |
Figure 3 Beta (Bray–Curtis) diversity of the saliva microbiome. Microbial composition differed significantly between the control and heart transplant patient groups (p = 0.001). The 95% confidence intervals for both groups are indicated by the colored circles. |
In assessing the impact of immunosuppression on bacterial function, the KEGG pathways of metabolism (p = 0.61), cellular processes and signaling (p = 0.92) and human diseases (p = 0.78) were not found to be significantly different between the groups. KOs were examined and 101 were found to be significantly different between HT patients and controls. A heat map showing the distribution of these differences between patients and controls is shown in Figure 4.
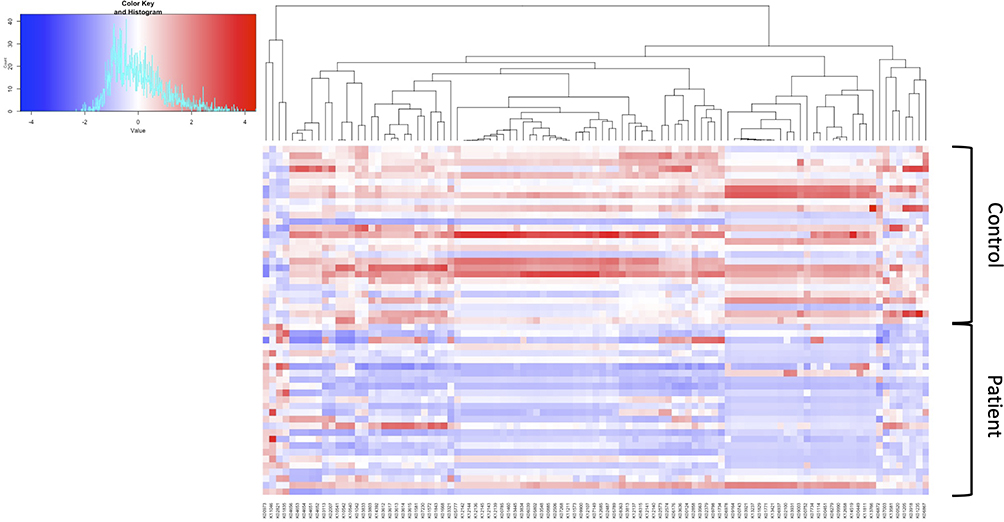 |
Figure 4 Heatmap summarizing the 101 significant KEGG ontology (KO) differences between the control and heart transplant patient groups. |
Discussion
This project aimed to characterize changes in the oral microbiome of patients on immunosuppressive drugs compared to non-transplant controls and test the hypothesis that immunosuppression alters the salivary microbiome. Our focus, specifically the oral microbiome, was chosen due to its association with systemic microbial migration and cardiovascular and non-cardiovascular diseases. Furthermore, analysis of the oral microbiome remains relatively understudied although changes have been identified in chronic diseases including cancer.10,30 Assessment of the saliva microbiota could also potentially provide insight into the immunosuppressive state of an individual, similar to that seen with viruses in the blood,31,32 or provide an alternative biological material other than stool or blood to more readily assess changes in the microbiota post-transplant for an individual patient. Overall, we found definite but relatively subtle changes in bacterial composition and function in stable transplant patients. Our study adds to the limited literature studying the oral microbiome in transplant patients.33
Based on the relative bacterial abundances and relative composition found in HT patients compared to healthy controls, the groups appear to be qualitatively similar as determined using DADA2 and Phyloseq. There is a predominance of the class Bacilli in addition to Fusobacteria and Gammaproteobacteria, consistent with previous findings in the human oral microbiome.13,34 However, there are some samples, amongst both the cases and controls that have a predominance of Bacteroidia or Betaproteobacteria. These population differences are of uncertain significance and were not associated with an obvious phenotype. Alpha diversity looks at both richness (how many OTUs are present) and evenness of distribution within a sample. The alpha diversity of HT patients was found to be significantly reduced in comparison to controls suggesting that immunosuppression does result in an overall loss of bacterial diversity. Beta diversity was also examined, using the Bray–Curtis dissimilarity index, to look at relative differences between samples. Again, a significant difference was found between the two groups suggesting that bacterial composition was affected by immunosuppression. These observations suggest that there may be alterations in bacterial composition in the oral microbiome after transplantation. These changes are supported by findings in the gut microbiome where transplantation consistently leads to a loss of microbial diversity and an increase in the phylum Proteobacteria.12,35,36 Interestingly, Diaz et al in a similarly structured study looking at the saliva microbiome in 20 renal and heart transplant patients did not identify a significant change in alpha diversity but did find immunosuppression to be associated with a significant change in beta diversity.33
It is interesting to note that the functional pathways examined were not significantly affected by immunosuppression even though there were significant changes in microbial composition. However, while there were no significant changes to selected pathways, there were significant changes at lower levels. There were 101 KOs that were found to significantly differ between HT patients and controls. Of the 101 KOs identified, 17 were involved in the ko0110 metabolic pathway. The remaining pathways identified consisted of 1–7 KOs, many with unknown functions. Many KOs were found to be significantly altered but, with no overall difference in microbiome function, the biological significance of these multiple lower-level changes is unknown at this time.
Our findings suggest that there are important alterations to the salivary microbiome after transplantation, but our study does have limitations. Participant demographics differed between the groups with the control group consisting of healthy university students while the HT patients were, on average, older. However, the age difference between the two groups may not be a serious issue since, once established, the microbiome is relatively stable throughout adulthood.37 Additionally, medical history of the control participants was self-reported. Ideally, age-matched or spousal controls would be recruited, and their microbiome used for comparison which would also help to better control for environmental exposures. Over 100 KOs were differentially expressed between HT patients and controls but, since there are still many uncharacterized KOs and pathways, it is unknown what biological impact this may have. One possible approach to assess biological impact may be to explore differences in the salivary metabolome and measure the differences in end-products of bacterial metabolism within the salivary microbiome. While differences can be identified between the two study populations, it is not possible to understand what these differences may mean clinically until more is known about the underlying biological pathways and functions. Finally, in future studies, sample size should be increased in order to make more robust inferences about microbial populations and allow for potential analyses between patient subgroups (eg, effect of different immunosuppressive drugs and impact of non-cardiac comorbidities such as diabetes or chronic kidney disease). Finally, analyses of viruses and eukaryotes present in the saliva need to be studied in order to obtain a complete picture of the oral microbiome and the effects of immunosuppression.
Conclusions
In this small pilot study, we did see significant changes in the salivary microbiota after transplantation. There was a loss of richness in the transplant samples and bacterial composition differed between the groups with both alpha (p = 0.0009) and beta (p = 0.001) diversity differing significantly between the groups. There were also statistically significant changes (p < 0.01) in 101 individual functional pathways which potentially indicate subtle functional differences but the clinical significance of these findings remains unknown at this time.
Data Sharing Statement
Data available on request.
Acknowledgments
The authors acknowledge the kind assistance of Dr. Karen Poon in the Nicole Perkins Microbial Communities Core Lab and Dr. Kevin Rioux in the Snyder Institute for Chronic Diseases both at the University of Calgary.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
SCG received support from the Alberta Children’s Hospital Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Obeng N, Bansept F, Sieber M, Traulsen A, Schulenburg H. Evolution of microbiota-host associations: the microbe’s perspective. Trends Microbiol. 2021;29(9):779–787. doi:10.1016/j.tim.2021.02.005
2. Zmora N, Zeevi D, Korem T, Segal E, Elinav E. Taking it personally: personalized utilization of the human microbiome in health and disease. Cell Host Microbe. 2016;19(1):12–20. doi:10.1016/j.chom.2015.12.016
3. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14(8):e1002533. doi:10.1371/journal.pbio.1002533
4. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–810. doi:10.1038/nature06244
5. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157(1):121–141.
6. Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570(7762):462–467. doi:10.1038/s41586-019-1291-3
7. Daliri EB, Ofosu FK, Chelliah R, Lee BH, Oh DH. Challenges and Perspective in integrated multi-omics in gut microbiota studies. Biomolecules. 2021;11(2):300. doi:10.3390/biom11020300
8. Manor O, Borenstein E. Systematic characterization and analysis of the taxonomic drivers of functional shifts in the human microbiome. Cell Host Microbe. 2017;21(2):254–267. doi:10.1016/j.chom.2016.12.014
9. Martellacci L, Quaranta G, Fancello G, et al. Characterizing peri-implant and sub-gingival microbiota through culturomics. first isolation of some species in the oral cavity. a pilot study. Pathogens. 2020;9(5):5. doi:10.3390/pathogens9050365
10. Patini R. Oral microbiota: discovering and facing the new associations with systemic diseases. pathogens. 2020;9(4):4. doi:10.3390/pathogens9040313
11. Martellacci L, Quaranta G, Patini R, et al. Review of Metagenomics and culturomics of the peri-implant microbiome: current evidence and future perspectives. Materials. 2019;12:18. doi:10.3390/ma12183010
12. Baghai Arassi M, Zeller G, Karcher N, Zimmermann M, Toenshoff B. The gut microbiome in solid organ transplantation. Pediatr Transplant. 2020;24(7):e13866. doi:10.1111/petr.13866
13. Dewhirst FE, Chen T, Izard J, et al. The human oral microbiome. J Bacteriol. 2010;192(19):5002–5017. doi:10.1128/JB.00542-10
14. Lee JR, Magruder M, Zhang L, et al. Gut microbiota dysbiosis and diarrhea in kidney transplant recipients. Am J Transplant. 2019;19(2):488–500. doi:10.1111/ajt.14974
15. Chhibber-Goel J, Singhal V, Bhowmik D, et al. Linkages between oral commensal bacteria and atherosclerotic plaques in coronary artery disease patients. NPJ Biofilms Microbiomes. 2016;2(1):7. doi:10.1038/s41522-016-0009-7
16. Pietiäinen M, Liljestrand JM, Kopra E, Pussinen PJ. Mediators between oral dysbiosis and cardiovascular diseases. Eur J Oral Sci. 2018;126(Suppl 1):26–36. doi:10.1111/eos.12423
17. Gibson CM, Childs-Kean LM, Naziruddin Z, Howell CK. The alteration of the gut microbiome by immunosuppressive agents used in solid organ transplantation. Transpl Infect Dis. 2021;23(1):e13397. doi:10.1111/tid.13397
18. Alegre ML, Mannon RB, Mannon PJ. The microbiota, the immune system and the allograft. Am J Transplant. 2014;14(6):1236–1248. doi:10.1111/ajt.12760
19. Rahim HTM, Hirota SA, Greenway SC. Microbiome alterations following solid organ transplantation: consequences, solutions and prevention. Transplant Res Risk Management. 2018;10:1–11. doi:10.2147/TRRM.S143063
20. Flannigan KL, Rajbar T, Moffat A, et al. Changes in composition of the gut bacterial microbiome after fecal microbiota transplantation for recurrent clostridium difficile infection in a pediatric heart transplant patient. Front Cardiovasc Med. 2017;4(17):17. doi:10.3389/fcvm.2017.00017
21. Zhang Z, Liu L, Tang H, et al. Immunosuppressive effect of the gut microbiome altered by high-dose tacrolimus in mice. Am J Transplant. 2018;1:5847.
22. Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads. Appl Environ Microbiol. 2011;77(11):3846–3852. doi:10.1128/AEM.02772-10
23. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–12.
24. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi:10.1038/nmeth.3869
25. McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi:10.1371/journal.pone.0061217
26. Gregory Caporaso J, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nature Publishing Group. 2010;5:7.
27. Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi:10.1038/nbt.2676
28. Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. (1367-4811 (Electronic)). Bioinformatics. 2014;30(21):3123
29. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–d361. doi:10.1093/nar/gkw1092
30. Mohammed H, Varoni EM, Cochis A, et al. Oral Dysbiosis in Pancreatic Cancer and Liver Cirrhosis: a Review of the Literature. Biomedicines. 2018;6(4):115. doi:10.3390/biomedicines6040115
31. Pradier A, Masouridi-Levrat S, Bosshard C, et al. Torque Teno Virus as a Potential Biomarker for Complications and Survival After Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol. 2020;11:998. doi:10.3389/fimmu.2020.00998
32. Rezahosseini O, Drabe CH, Sørensen SS, et al. Torque-Teno virus viral load as a potential endogenous marker of immune function in solid organ transplantation. Transplant Rev. 2019;33(3):137–144. doi:10.1016/j.trre.2019.03.004
33. Diaz PI, Hong BY, Frias-Lopez J, et al. Transplantation-associated long-term immunosuppression promotes oral colonization by potentially opportunistic pathogens without impacting other members of the salivary bacteriome. Clin Vaccine Immunol. 2013;20(6):920–930. doi:10.1128/CVI.00734-12
34. Verma D, Garg PK, Dubey AK. Insights into the human oral microbiome. Arch Microbiol. 2018;200(4):525–540. doi:10.1007/s00203-018-1505-3
35. Lee JR, Muthukumar T, Dadhania D, et al. Gut microbial community structure and complications after kidney transplantation: a pilot study. Transplantation. 2014;98(7):697–705.
36. Sun L-Y, Yang Y-S, Qu W, et al. Gut microbiota of liver transplantation recipients. Sci Rep. 2017;7(1):3762. doi:10.1038/s41598-017-03476-4
37. Rodríguez JM, Murphy K, Stanton C, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis. 2015;26:26050.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.