Back to Journals » Drug Design, Development and Therapy » Volume 8
Changes in surface expression of N-methyl-D-aspartate receptors in the striatum in a rat model of Parkinson's disease
Authors Gan J, Qi C, Mao L, Liu Z
Received 15 July 2013
Accepted for publication 3 October 2013
Published 17 January 2014 Volume 2014:8 Pages 165—173
DOI https://doi.org/10.2147/DDDT.S51559
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Jing Gan,1 Chen Qi,1 Li-Min Mao,2 Zhenguo Liu1
1Department of Neurology, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, People's Republic of China; 2Department of Basic Medical Science, University of Missouri-Kansas City School of Medicine, Kansas City, MO, USA
Background: N-methyl-D-aspartate (NMDA) receptors play a central role in glutamatergic synaptic transmission in the mammalian brain and are linked to the pathophysiology and symptomatology of Parkinson's disease (PD). However, changes in NMDA receptor expression in distinct subcellular compartments in PD have not been elucidated. In this study, we investigated changes in subcellular expression of NMDA receptors in striatal neurons in a rodent PD model.
Methods: Intracranial injection of the neurotoxin 6-hydroxydopamine (6-OHDA) was selectively lesioned into the nigrostriatal dopaminergic pathway in adult Sprague Dawley rats, which is a common rat model of PD. A surface receptor crosslinking assay was conducted to examine the response of individual NMDA receptor subunits to dopamine depletion in isolated and confined surface and intracellular compartments of striatal neurons.
Results: In PD rats where 6-OHDA was selectively lesioned, surface expression of NMDA receptor GluN1 subunits as detected by surface protein crosslinking assays was increased in the striatum. In contrast, intracellular levels of GluN1 were decreased in the lesioned region. The NMDA receptor GluN2B subunit was elevated in its abundance in the surface pool of the lesioned striatum, while intracellular GluN2B levels were not altered. GluN2A subunits in both surface and intracellular fractions remained stable. In addition, total cellular levels of striatal GluN1 and GluN2A were not changed in lesioned tissue, while total GluN2B proteins showed an increase.
Conclusion: These results demonstrate the differential sensitivity of principal NMDA receptor subunits to dopamine depletion. GluN1 and GluN2B expression in the distinct surface compartment underwent upregulation in striatal neurons after selective lesions of the dopaminergic pathway by 6-OHDA.
Keywords: glutamate, excitatory amino acid, NMDA, GluN, dopamine, 6-hydroxydopamine, caudate putamen, nucleus accumbens
Introduction
L-glutamate is an important neurotransmitter in the mammalian brain. This transmitter interacts with a family of ionotropic and metabotropic glutamate receptors to regulate a variety of cellular and synaptic activities.1 N-methyl-d-aspartate (NMDA) receptors are a major subtype of ionotropic glutamate receptors. These receptors are assembled into functional ion channels by composing obligatory GluN1 (formerly known as NR1) subunits and modulatory GluN2 subunits, mainly GluN2A (NR2A) and GluN2B (NR2B).2,3 NMDA receptors are distributed in broad areas of brain, including the striatum. In this key structure of the basal ganglia, NMDA receptors are enriched in the medium spiny projection neurons and interneurons, in parallel with massive glutamatergic afferents from multiple forebrain sites, such as cerebral cortex, hippocampus, amygdala, and thalamus.4 As such, these receptors are actively involved in the modulation of striatal neuronal activities.
In addition to the role of NMDA receptors in the regulation of normal striatal activities, malfunction of the receptor is linked to the pathogenesis of various neurological and neuropsychiatric disorders. Central among neurological disorders is Parkinson’s disease (PD). This disease is a progressive neurodegenerative illness that results from dopamine deficiency in the striatum. In addition to dopamine, the glutamatergic corticostriatal pathway is suggested to play a significant role in the pathophysiology and symptomatology of PD.5 Early studies with ligand binding assays demonstrated reductions in NMDA binding sites in the striatum after dopamine denervation.6,7 In an animal model of PD, Lai et al8 found enhancement of GluN1 messenger RNA (mRNA) expression with no change in GluN2B mRNA levels in the striatum; others have reported elevated GluN2B mRNA expression.9,10 One study showed that GluN1 and GluN2B proteins were decreased in membrane fractions of the rat lesioned striatum relative to the unlesioned striatum, while GluN2A abundance was unchanged.11 However, to date, there is still no general consensus about the changes in the NMDA receptor subunits in the striatum after dopamine denervation6–10,12–14 and changes in expression of NMDA receptors in distinct subcellular compartments (eg, surface versus intracellular pools) in response to dopamine depletion has not been fully elucidated in the striatum.11
In this study, we investigated changes in NMDA receptor expression in the striatum in response to dopamine lesions. We used the most common rat model of PD, in which 6-hydroxydopamine (6-OHDA) was injected to achieve the unilateral lesion of the nigrostriatal dopamine pathway. A surface receptor crosslinking assay was conducted to examine the response of individual NMDA receptor subunits to dopamine depletion in isolated and confined surface and intracellular compartments of striatal neurons.
Methods
Animals
Adult male Sprague Dawley rats weighing 200–250 g were housed in standard Plexiglas® (Evonik Specialty Chemicals Co. Ltd. Shanghai, People’s Republic of China) cages with free access to food and water. Environmental conditions were strictly controlled to a light/dark cycle of 12 hours, a temperature of 22°C, and humidity at 44%. The number of animals used was the minimum required for statistical analysis. All protocols involving the animals were approved by the Institutional Review Board of Xinhua Hospital and were performed according to the guidelines of the US National Institutes of Health for the care and use of laboratory animals.
6-OHDA lesions and rotation screening
Rats were anesthetized with ketamine (100 mg/kg) by intraperitoneal (ip) injection. The rat head was mounted in a stereotaxic frame (Narishige Group, Tokyo, Japan). The skull was exposed and a burr hole was drilled to introduce a syringe for injection of 6-OHDA (Sigma-Aldrich, St Louis, MO, USA) solution containing 4 μg 6-OHDA/μL in 0.9% saline with 0.02% ascorbic acid (pH 5.0). The 6-OHDA solution was freshly prepared, kept on ice, and protected from exposure to light in order to minimize the degradation of the toxin. Each animal received an injection of 6-OHDA into two points: the coordinates for medial forebrain bundle (MFB) lesions were: anteroposterior, −3.7 mm; mediolateral, +1.7 mm; and dorsoventral, −7.8 mm. Right ventral tegmental area lesions were: anteroposterior, −4.4 mm; mediolateral, +1.2 mm; and dorsoventral, −7.8 mm. The tooth bar was set to −2.4 mm.15–17 A total volume of 4 μL 6-OHDA was injected at a flow rate of 1μL/minute. After injection, the needle was kept in position for additional 10 minutes to prevent backflow of injected drugs. After the injection, the skin was sutured and the animals were removed from the stereotaxic instrument and placed on a heating pad for 30 minutes before returning to the animal housing facility. As controls, sham lesions were made by injecting a vehicle solution.
Rat rotation screening was performed in order to validate the 6-OHDA lesion.16,17 Twenty-one days after surgery, the lesioned rats were injected with apomorphine (Sigma-Aldrich) at a dose of 0.25 mg/kg (ip) and were then placed in a stainless steel cylindrical bowl. The number of contralateral rotations was counted for a 30-minute period beginning 5 minutes after administration of apomorphine. The rats exhibiting a vigorous rotational response to apomorphine and turning with an average of more than seven turns per minute were justified as successfully lesioned animals and selected for further study.
Western blot analysis
Western blot was performed as described previously.18 Rats were anesthetized with pentobarbital (50 mg/kg, ip) and decapitated. Brains were immediately removed and transferred to a plastic plate cooled on ice to dissect the both sides of striatum. Tissues were homogenized by sonication in radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris, pH 7.5, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1 mM EDTA (ethylenediaminetetraacetic acid), 1% Nonidet P-40 (octylphenoxypolyethoxyethanol), 2 mM PMSF (phenylmethylsulfonyl fluoride), and protease inhibitors (Roche Applied Science, Indianapolis, IN, USA). The homogenate was centrifuged at 1,000 g for 10 minutes at 4°C. The pellet containing mainly nuclei and large debris was discarded. The supernatant was used and protein concentration was determined using a Pierce bicinchoninic acid (BCA) assay kit (Thermo Fisher Scientific, Rockford, IL, USA). Samples were boiled 5 minutes in Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein were loaded on 4%–15% Tris-HCl gradient gels (Bio-Rad Laboratories). After electrophoresis, proteins were transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories). The membranes were blocked in blocking buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20 and 5% w/v nonfat dry milk for 1 hour at room temperature. The blots were incubated overnight at 4°C in a primary rabbit antibody against GluN1 (1:1,000; EMD Millipore, Billerica, MA, USA), GluN2A (1:1,000; EMD Millipore), or GluN2B (1:1,000; EMD Millipore), or a primary mouse antibody against α-actinin (1:1,000; EMD Millipore), or a mouse monoclonal β-actin antibody (1:5,000; Sigma-Aldrich), or tyrosine hydroxylase (TH) (1:5,000; Sigma-Aldrich). The membranes were subsequently washed with TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) and followed by 1-hour incubation in an horseradish peroxidase-conjugated goat secondary antibody against rabbit or mouse IgG (1:2,000; Cell Signaling Technology, Danvers, MA, USA) at room temperature. Bound antibodies were visualized using the enhanced chemiluminescence detection system (EMD Millipore) and analyzed quantitatively using the Image Lab Software (Bio-Rad Laboratories).
Surface receptor crosslinking assays
Assays were performed as described previously.18–20 Briefly, assays were carried out with a membrane-impermeable crosslinking reagent bis(sulfosuccinimidyl) suberate (BS3) (Pierce; Thermo Fisher Scientific, Rockford, IL, USA), which only crosslinks proteins on the surface membrane of intact cells. Rats were anesthetized with pentobarbital (50 mg/kg, ip) and were decapitated. Brains were removed and sliced. The striatum was removed to an Eppendorf tube containing ice-cold phosphate buffered solution. BS3 was added to a final concentration of 2 mM and incubated with gentle agitation for 30 minutes at 4°C. The crosslinking reaction was terminated by quenching with 20 mM glycine (10 minutes, 4°C). The tissue was then washed four times in cold artificial cerebrospinal fluid (10 minutes each). Samples were homogenized and analyzed directly by sodium dodecyl sulfate polyacrylamide gel electrophoresis (4%–15% Tris-HCl gel; Bio-Rad Laboratories). To determine that BS3 does not have access to intracellular proteins, we use α-actinin protein based on selective immunodetection of the monomeric band but no crosslinked band for this protein. α-Actinin is an intracellular protein. Using the same crosslinked and non-crosslinked samples, verify that probing with antibodies (α-actinin) to intracellular proteins yields only a single band at the predicted molecular weight. This confirms that BS3 did not have access to the intracellular compartment during incubation of brain slices.18,20,21
Statistics
The experimental data are presented as means ± standard error of the mean. One-way analysis of variance followed by least significant difference post hoc comparison tests was employed to analyze data among different groups. Student’s t-test was used to observe differences between the lesioned side and the unlesioned side. Significance was set at P<0.05.
Results
The depletion of TH in the striatum after 6-OHDA lesions
We first examined abundance of TH, a marker for dopaminergic neurons, in the striatum to validate the lesion of dopamine neurons with 6-OHDA. As shown in Figure 1, TH protein levels in the striatum remained stable in the lesioned side as compared with the unlesioned side in a sham group of rats (92.60%±5.15% of the TH levels in the unlesioned side, P>0.05). In contrast, in 6-OHDA–lesioned rats, the striatal TH abundance in the lesioned side was reduced to 2.52%±0.26% of that in the unlesioned striatum. This confirms the denervation of the nigrostriatal dopaminergic pathway in the injected side.
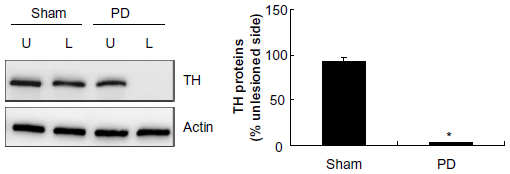 | Figure 1 Tyrosine hydroxylase levels in the striatum. |
Surface and intracellular expression of NMDA receptors
BS3 selectively crosslinks cell surface receptors, forming high molecular weight aggregates, while intracellular receptors are not accessible to this membrane-impermeable reagent. The BS3-linked high molecular weight surface receptors and the BS3-unlinked intracellular receptor monomers can be readily separated by gel electrophoresis and visualized through the following immunoblots.16–18 Indeed, as shown in Figure 2A, a high molecular weight band (BS3-linked) was observed for all subunits (GluN1, GluN2A, and GluN2B) in BS3-treated striatal tissue. No such band was seen in control tissue. In addition, a single band (ie, BS3-unlinked band) was seen for each individual subunit, which corresponds to the predicted molecular weight of GluN1 (~120 kDa), GluN2A (~180 kDa), and GluN2B (~180 kDa) monomers. For an intracellular protein α-actinin, no crosslinking effect of BS3 was seen (Figure 2B). These results are consistent with those reported previously16 and validate our BS3 crosslinking assays in detecting surface versus intracellular receptor expression in striatal neurons.
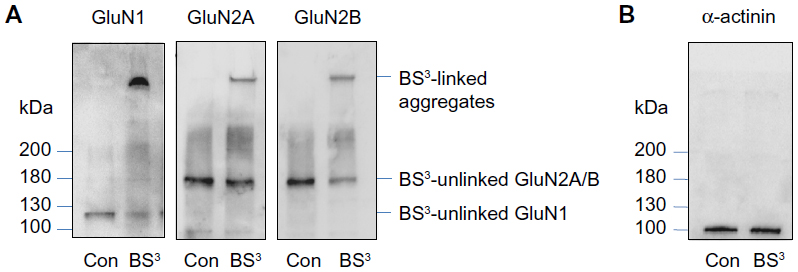 | Figure 2 BS3 crosslinking measures surface and intracellular pools of NMDA receptors. |
Changes in cellular expression of NMDA receptors in the striatum after 6-OHDA lesions
We next examined changes in NMDA receptor expression in the striatum after dopamine depletion. We first investigated changes in total cellular levels of GluN1, GluN2A, and GluN2B proteins. No significant change in GluN1 levels in the striatum was seen in the 6-OHDA–lesioned side as compared with the unlesioned side (Figure 3A). Similar results were found for GluN2A (Figure 3B). In contrast to GluN1 and GluN2A levels, the GluN2B protein level was significantly higher in the lesioned side than in the unlesioned side in PD rats. GluN2B levels in the 6-OHDA–lesioned side reached 126.13%±5.41% of the sham animal unlesioned side (P<0.05; Figure 3C). Thus, dopamine lesions by 6-OHDA increased GluN2B expression in the striatum, while GluN1 and GluN2A expression remained stable.
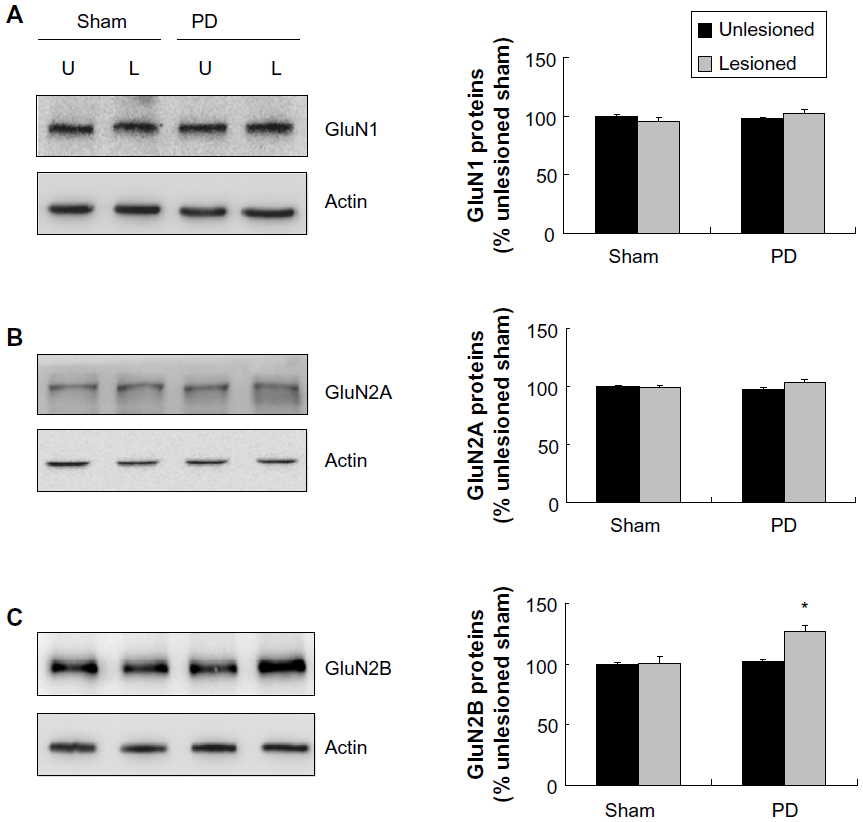 | Figure 3 Effect of 6-OHDA on total GluN1/GluN2A/GluN2B proteins of rat striatum. |
Changes in surface and intracellular expression of NMDA receptors in the striatum after 6-OHDA lesions
We next examined whether 6-OHDA alters receptor expression in confined subcellular pools of striatal neurons. In assays analyzing GluN1, we found that GluN1 in surface fractions was increased in the 6-OHDA–lesioned side relative to the unlesioned side in PD rats and to both lesioned and unlesioned sides in sham rats (157.06%±7.99% of surface GluN1 in the sham unlesioned side, P<0.05; Figure 4A and B). In contrast, intracellular GluN1 levels in lesioned side of PD rats were decreased to 76.49%±1.96% of levels in the sham unlesioned side (P<0.05; Figure 4A and C). Due to an increase in surface pools and a decrease in intracellular pools, the surface to intracellular ratio of GluN1 expression was significantly higher in the 6-OHDA–lesioned side than in other sides (Figure 4D). These data suggest a redistribution of GluN1 (from intracellular to surface locations) in striatal neurons after dopamine lesion by 6-OHDA.
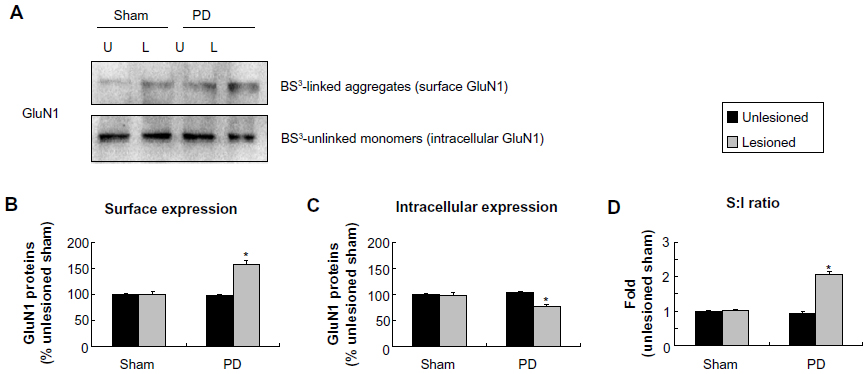 | Figure 4 Effect of 6-OHDA lesions on GluN1 receptor expression in surface and intracellular pools of rat striatum. |
Unlike subcellular expression of GluN1, subcellular expression of GluN2A was insensitive to 6-OHDA lesion. No significant change in this subunit in either surface or intracellular fractions was seen in PD and sham rats (Figure 5A–C). As a result, the surface-to-intracellular ratio of GluN2A expression was not altered (Figure 5D).
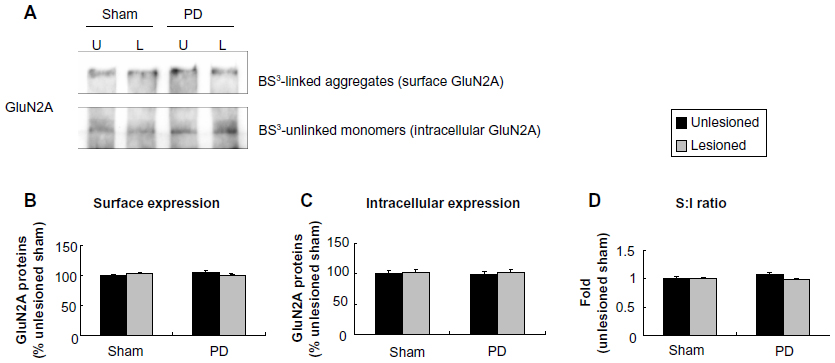 | Figure 5 Effect of 6-OHDA lesions on GluN2A receptor expression in surface and intracellular pools of rat striatum. |
A significant increase in surface expression of GluN2B occurred in the lesioned striatum (172.47%±5.43% of GluN2B surface expression in the sham unlesioned side, P<0.05; Figure 6A and B). However, intracellular expression of GluN2B was unchanged (Figure 6C). The surface-to-intracellular ratio of GluN2B expression was elevated primarily due to an increase in surface expression of the subunit (Figure 6D). These results demonstrate that GluN2B in surface membranes is subject to upregulation in response to 6-OHDA lesion.
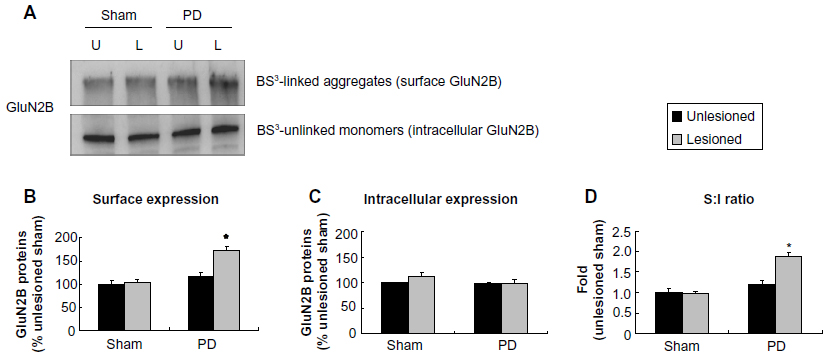 | Figure 6 Effect of 6-OHDA lesions on GluN2B receptor expression in surface and intracellular pools of rat striatum. |
Finally, we investigated the effect of 6-OHDA on expression of an intracellular protein α-actinin. Protein levels of α-actinin were not significantly altered in the lesioned side compared with the unlesioned side in both PD and sham rats (Figure 7).
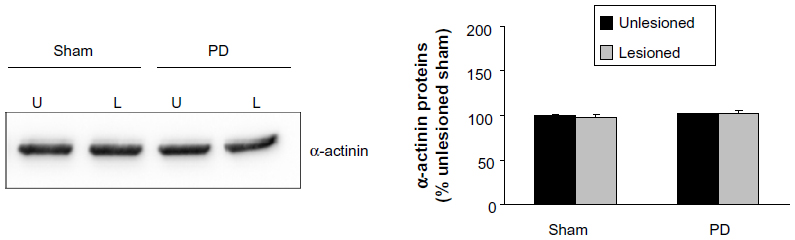 | Figure 7 α-Actinin expressions. |
Discussion
In this study, we investigated changes in NMDA receptor expression in striatal neurons in response to dopamine depletion in a rat PD model. We focused on the receptor expression in specific pools of receptors, ie, surface versus intracellular compartments, with a surface receptor crosslinking assay. Our result verified that the BS3 crosslinking method allowed discrimination between surface proteins and intracellular components in NMDA subunits. We found that NMDA receptors underwent drastic subunit-selective changes. While GluN2A expression in both surface and intracellular pools remained unchanged, GluN1 and GluN2B were upregulated in their abundances in the surface pool of striatal neurons in 6-OHDA–lesioned rats. Intracellular expression of GluN1 was decreased in the lesioned striatum. To our knowledge, this study is the first to measure surface-expressed NMDA receptor subunits in a PD animal model with a crosslinking method. These data reveal the response of major individual NMDA receptor subunits in confined subcellular compartments of striatal neurons to dopamine depletion. Selective lesions of the nigraostriatal dopaminergic pathway may upregulate surface expression of GluN1/GluN2B receptors in the striatum.
A major finding in this study is the change in GluN1 expression in response to dopamine lesions. This change was characterized by an increase in abundance in the surface pool and a proportional decrease in the intracellular pool, while total GluN1 protein levels were not changed. This unique pattern of changes implies a redistribution of GluN1 proteins from intracellular to surface locations. This may result from an increase in GluN1 exocytosis or a decrease in GluN1 internalization or both. Another important finding is that GluN2B, like GluN1, was another subunit sensitive to dopamine lesions. 6-OHDA lesions caused a significant increase in surface expression of GluN2B as well as in the total level of GluN2B. 6-OHDA lesions caused a significant increase in surface expression of GluN2B as well as in the total level of GluN2B, while intracellular GluN2B expression remained the same. This suggested that the increase of new GluN2B receptors preferentially expressed on the cell surface. It is well documented that in the intracellular pool, the endoplasmic reticulum is involved in synthesis, assembly, and secretion of NMDA receptors.11,18 One likely possibility is that overall increase of new GluN2B protein synthesis could account for the NR2B increase after dopamine denervation, and enhancement of trafficking or delivery of NR2B from the intracellular pool to the surface membrane is likely concurrence. Given that surface expression of both GluN1 and GluN2B were increased in parallel while GluN2A was not altered, GluN1/GluN2B are believed to be a sensitive subtype subjected to the modulation by dopamine depletion. An early report by Dunah et al11 showed insignificant changes in total striatal GluN1, GluN2A, and GluN2B proteins after 6-OHDA lesions. However, GluN1 and GluN2B levels in striatal synaptosomal membranes were reduced.11 One reason for the discrepancy as compared with our results may be the different survival durations employed in the two studies (21 days in our study and 14 days in the study by Dunah et al11). The different survival durations after 6-OHDA lesion may lead to the different degrees of nigrostiratal damage. Evidence showed that the loss of dopaminergic terminals increased linearly, over the days after 6-OHDA injection, peaking at the third week.22 The different degrees of nigrostriatal damage is a possible reason for the differences between the two studies. Another reason may be related to different methods for detecting surface receptors used in the two studies.
The striatum receives both dopaminergic input from substantia nigra pars compacta and glutamatergic input from the cerebral cortex and thalamus. In PD, the loss of dopaminergic innervation elicits secondary alterations in glutamate receptors, especially NMDA receptors. It is current opinion that after 6-OHDA–lesion, glutamate is at high concentrations and leads to the upregulation of total glutamate receptors. Then, altered corticostriatal transmission and aberrant activation of postsynaptic signal transduction pathways (eg, calcium/calmodulin-dependent protein kinase II) lead to persistent maladaptive changes in the regulation of late-activated genes, including alteration in membrane receptors.23 The increased GluN1/GluN2B surface expression may be a response to altered excitatory transmission. Also, CaMKII autophosphorylation is an important regulatory mechanism involved in subcellular distribution and anchoring to the membrane.24 Upregulation of NMDA receptors may increase the intracellular Ca2+ and subsequent increase in second messengers, which causes enhanced metabolic stress on mitochondria, excessive oxidative phosphorylation, increased production of reactive oxygen species, and final neurodegeneration. Thus, NMDA receptor-mediated excitotoxicity has been suggested to be one of the possible causes of the neuronal degeneration.25–27 According to our experimental results, GluN1 and GluN2B may be more potentially involved in the development of PD. Some research has shown that the striatal projection neurons are enriched in NMDA receptors composed of GluN1/GluN2B subunits.25,28 GluN1/GluN2B receptors contributed to the cell death pathway, and GluN2A-containing NMDA receptors mediated the cell survival signaling pathway.10,29 Pharmacological studies have also demonstrated that the use of selective GluN1/GluN2B NMDA antagonists in the therapeutics of a PD animal model was supported. Reduction in expression of GluN1 or blocking expression of GluN2B could ameliorate the motor symptoms in the 6-OHDA–lesioned rat.30–32 The application of the gene silencing targeting the GluN2B subunit could effectively ameliorate the Parkinsonian motor symptoms, reduce GluN2B protein expression, and attenuate the dopaminergic cell loss in the striatum and substantia nigra regions.10 However, a randomized, double-blind, placebo-controlled study conducted in PD patients did not support the benefit of GluN2B antagonist.33 Although administration dose and time of GluN2B antagonist were discussible in this study, it takes a long time to move to clinical trials from animal studies.
In conclusion, NMDA receptors are key modulators of striatal functions with upregulation of GluN1 and GluN2B surface compartments in striatal neurons after 6-OHDA–lesion. Our results demonstrated that GluN1/GluN2B receptors were important mechanisms of PD and provides support for this possible useful therapeutic target.
Acknowledgments
The study was supported by the Projects of National Science Foundation of China (No 81071025 and 81171203) and Projects of the Shanghai Committee of Science and Technology, China (No 11nm0503300, No 11410708900, and No 12XD1403800).
Disclosure
The authors report no conflicts of interest in this work.
References
Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62(3):405–496. | |
Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51(1):7–61. | |
Kew JN, Kemp JA. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl). 2005;179(1):4–29. | |
Standaert DG, Testa CM, Young AB, Penney JB. Organization of N-methyl-D-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J Comp Neurol. 1994;343(1):1–16. | |
Loftis JM, Janowsky A. The N-methyl-D-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol Ther. 2003;97(1):55–85. | |
Tarazi FI, Campbell A, Yeghiayan SK, Baldessarini RJ. Localization of ionotropic glutamate receptors in caudate-putamen and nucleus accumbens septi of rat brain: comparison of NMDA, AMPA, and kainate receptors. Synapse. 1998;30(2):227–235. | |
Zavitsanou K, Mitsacos A, Giompres P, Kouvelas ED. Changes in [3H]AMPA and [3H]kainate binding in rat caudate-putamen and nucleus accumbens after 6-hydroxydopamine lesions of the medial forebrain bundle: an autoradiographic study. Brain Res. 1996;731(1–2):132–140. | |
Lai SK, Tse YC, Yang MS, Wong CK, Chan YS, Yung KK. Gene expression of glutamate receptors GluR1 and NR1 is differentially modulated in striatal neurons in rats after 6-hydroxydopamine lesion. Neurochem Int. 2003;43(7):639–653. | |
Nandhu MS, Paul J, Kuruvila KP, Abraham PM, Antony S, Paulose CS. Glutamate and NMDA receptors activation leads to cerebellar dysfunction and impaired motor coordination in unilateral 6-hydroxydopamine lesioned Parkinson’s rat: functional recovery with bone marrow cells, serotonin and GABA. Mol Cell Biochem. 2011;353(1–2):47–57. | |
Ng OT, Chen LW, Chan YS, Yung KK. Small interfering RNA specific for N-methyl-D-aspartate receptor 2B offers neuroprotection to dopamine neurons through activation of MAP kinase. Neurosignals. 2013;21(1–2):42–54. | |
Dunah AW, Wang YH, Yasuda RP, et al. Alterations in subunit expression, composition, and phosphorylation of striatal N-methyl-D-aspartate glutamate receptors in a rat 6-hydroxydopamine model of Parkinson’s disease. Mol Pharmacol. 2000;57(2):342–352. | |
Gardoni F, Picconi B, Ghiglieri V, et al. A critical int eraction between NR2B and MAGUK in L-DOPA induced dyskinesia. J Neurosci. 2006;26(11):2914–2922. | |
Kong M, Ba M, Song L, Liu Z. Comparative effects of acute or chronic administration of levodopa to 6-OHDA–lesioned rats on the expression and phosphorylation of N-methyl-D-aspartate receptor NR1 subunits in the striatum. Neurochem Res. 2009;34(8):1513–1521. | |
Fiorentini C, Rizzetti MC, Busi C, et al. Loss of synaptic D1 dopamine/N-methyl-D-aspartate glutamate receptor complexes in L-DOPA-induced dyskinesia in the rat. Mol Pharmacol. 2006;69(3):805–812. | |
Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6th ed. London, UK: Academic Press/Elsevier; 2007. | |
Ye M, Wang XJ, Zhang YH, et al. Transplantation of bone marrow stromal cells containing the neurturin gene in rat model of Parkinson’s disease. Brain Res. 2007;1142:206–216. | |
Wang XJ, Liu WG, Zhang YH, Lu GQ, Chen SD. Effect of transplantation of c17.2 cells transfected with interleukin-10 gene on intracerebral immune response in rat model of Parkinson’s disease. Neurosci Lett. 2007;423(2):95–99. | |
Mao LM, Wang W, Chu XP, et al. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci. 2009;12(5):602–610. | |
Boudreau AC, Milovanovic M, Conrad KL, Nelson C, Ferrario CR, Wolf ME. A protein cross-linking assay for measuring cell surface expression of glutamate receptor subunits in the rodent brain after in vivo treatments. Curr Protoc Neurosci. 2012;Chapter 5:Unit 5.30.1– Unit 5.3019. | |
Carino C, Fibuch EE, Mao LM, Wang JQ. Dynamic loss of surface-expressed AMPA receptors in mouse cortical and striatal neurons during anesthesia. J Neurosci Res. 2012;90(1):315–323. | |
Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25(40):9144–9151. | |
Blandini F, Armentero MT, Martignoni E. The 6-hydroxydopamine model: news from the past. Parkinsonism Relat Disord. 2008;14 Suppl 2:S124–S129. | |
Simola N, Morelli M, Carta AR. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox Res. 2007;11(3–4):151–167. | |
Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369(6477):233–235. | |
Truong L, Allbutt HN, Coster MJ, Kassiou M, Henderson JM. Behavioural effects of a selective NMDA NR1A/2B receptor antagonist in rats with unilateral 6-OHDA+parafascicular lesions. Brain Res Bull. 2009;78(2–3):91–96. | |
Rao VL, Bowen KK, Dempsey RJ. Transient focal cerebral ischemia down-regulates glutamate transporters GLT-1 and EAAC1 expression in rat brain. Neurochem Res. 2001;26(5):497–502. | |
Nandhu MS, Paul J, Kuruvilla KP, Malat A, Romeo C, Paulose CS. Enhanced glutamate, IP3 and cAMP activity in the cerebral cortex of unilateral 6-hydroxydopamine induced Parkinson’s rats: effect of 5-HT, GABA and bone marrow cell supplementation. J Biomed Sci. 2011;18:5. | |
Löschmann PA, De Groote C, Smith L, et al. Antiparkinsonian activity of Ro 25-6981, a NR2B subunit specific NMDA receptor antagonist, in animal models of Parkinson’s disease. Exp Neurol. 2004;187(1):86–93. | |
Liu Y, Wong TP, Aarts M, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27(11):2846–2857. | |
Lai SK, Ng TK, Lau WK, et al. Selective knockdown of gene expression of N-methyl-D-aspartate receptor one ameliorates Parkinsonian motor symptom in 6-hydroxydopamine-lesioned rats. Neurochem Int. 2004;45(1):11–22. | |
Sze SC, Wong CK, Yung KK. Modulation of the gene expression of N-methyl-D-aspartate receptor NR2B subunit in the rat neostriatum by a single dose of specific antisense oligodeoxynucleotide. Neurochem Int. 2001;39(4):319–327. | |
Johnson KA, Conn PJ, Niswender CM. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol Disord Drug Targets. 2009;8(6):475–491. | |
Addy C, Assaid C, Hreniuk D, et al. Single-dose administration of MK-0657, an NR2B-selective NMDA antagonist, does not result in clinically meaningful improvement in motor function in patients with moderate Parkinson’s disease. J Clin Pharmacol. 2009;49(7):856–864. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at