Back to Archived Journals » Biosimilars » Volume 5
Challenging issues in assessing analytical similarity in biosimilar studies
Authors Chow S
Received 6 March 2015
Accepted for publication 2 April 2015
Published 22 May 2015 Volume 2015:5 Pages 33—39
DOI https://doi.org/10.2147/BS.S84141
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Laszlo Endrenyi
Shein-Chung Chow
Biostatistics and Bioinformatics, Duke University School of Medicine, Durham, NC, USA
Abstract: For assessing biosimilarity of biosimilar products, the US Food and Drug Administration (FDA) proposed a stepwise approach for providing totality-of-the-evidence of similarity between a proposed biosimilar product and a US-licensed (reference) product. The stepwise approach starts with an assessment of critical quality attributes (CQAs) that are relevant to clinical outcomes in structural and functional characterization in manufacturing process of the proposed biosimilar product. The FDA suggests that these critical quality–relevant attributes be identified and classified into three tiers depending their criticality or risk ranking. To assist the sponsors, the FDA also suggests some statistical approaches for the assessment of analytical similarity for CQAs from different tiers, namely equivalence test for Tier 1, quality range approach for Tier 2, and descriptive raw data and graphical comparison for Tier 3. In this paper, challenging issues to the FDA's recommended approaches are discussed followed by alternative methods for the assessment of analytical similarity (mainly for CQAs from Tier 1).
Keywords: stepwise approach, critical quality attribute, CQA, equivalence test, quality range approach
Background
Following the passage of the Biologics Price Competition and Innovation Act in 2009, the US Food and Drug Administration (FDA) released three draft guidances on the demonstration of biosimilarity of biosimilar products in February 2012. These guidances are intended not only i) to assist sponsors to demonstrate that a proposed therapeutic protein product is biosimilar to a reference product for the purpose of submitting a marketing application under section 351(k) of the Public Health Service Act but also ii) to describe the FDA’s current thinking on the factors considered to demonstrate that a proposed protein product is highly similar to a reference product, which was licensed under section 351(a) of the Public Health Service Act. In the draft guidance on Scientific Considerations in Demonstrating Biosimilarity to a Reference Product, the FDA introduces the concept of stepwise approach for obtaining totality-of-the-evidence for the regulatory review and approval of biosimilar applications.1
The stepwise approach starts with the assessment of analytical similarity of critical quality attributes (CQAs) that are commonly seen in the structural and functional characterization in manufacturing process of biosimilar products. In practice, there are often a large number of CQAs that may be relevant to clinical outcomes. Thus, it is almost impossible to assess analytical similarity for all these CQAs. As a result, the FDA suggests that the sponsors to identify CQAs are relevant to clinical outcomes and classify them into three tiers depending upon their criticality (or risk ranking), ie, most, mild to moderate, and least relevant to clinical outcomes. To assist the sponsors, the FDA also proposes some statistical approaches for the assessment of analytical similarity for CQAs from different tiers. For example, the FDA recommends equivalence test for CQAs from Tier 1, quality range approach for CQAs from Tier 2, and descriptive raw data and graphical presentation for CQAs from Tier 3.2
The purpose of this paper is not only to provide a close look at these approaches by providing interpretation and/or statistical justification whenever possible but also to discuss some challenging issues to the FDA’s proposed approach (mainly on the equivalence test for Tier 1 CQAs). In addition, recommendations and alternative methods are proposed.
The stepwise approach for demonstrating biosimilarity as suggested by the FDA draft guidance is briefly outlined in the stepwise approach for demonstrating biosimilarity section. The third section, FDA’s approaches for tier analysis, provides brief descriptions of the equivalence test, quality range approach, and the method of descriptive raw data and graphical comparison. Some challenging issues to the FDA’s proposed approaches are discussed in the challenging issues to the FDA’s approaches section. The fifth section, recommendations and alternative methods, provides recommendations and alternative methods for the assessment of analytical similarity in CQAs from different tiers. Some concluding remarks are given in the last section.
Stepwise approach for demonstrating biosimilarity
As defined in the Biologics Price Competition and Innovation Act, a biosimilar product is a product that is highly similar to the reference product notwithstanding minor differences in clinically inactive components and there are no clinically meaningful differences in terms of safety, purity, and potency. Based on the definition of the Biologics Price Competition and Innovation Act, biosimilarity requires that there are no clinically meaningful differences in terms of safety, purity, and potency. Safety could include pharmacokinetics (PK) and pharmacodynamics, safety and tolerability, and immunogenicity studies. Purity includes all CQAs during manufacturing process. Potency is referred to as efficacy studies. In the 2012 FDA draft guidance on scientific considerations, the FDA recommends that a stepwise approach be considered for providing the totality-of-the-evidence to demonstrating biosimilarity of a proposed biosimilar product as compared to a reference product.1
The stepwise approach is briefly summarized by a pyramid illustrated in Figure 1. The stepwise approach starts with analytical studies for structural and functional characterization. The stepwise approach continues with animal studies for toxicity, clinical pharmacology studies such as PK/pharmacodynamics studies, followed by investigations of immunogenicity, and clinical studies for safety/tolerability and efficacy.
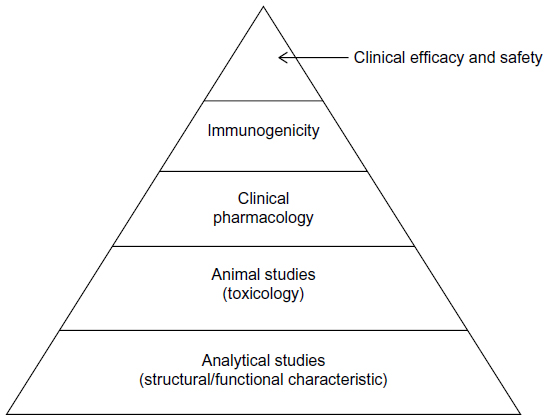 | Figure 1 A stepwise approach to demonstrate biosimilarity. |
The sponsors are encouraged to consult with medical/statistical reviewers of the FDA with the proposed plan or strategy of the stepwise approach for regulatory agreement and acceptance. This is to make sure that the information provided is sufficient to fulfill the FDA’s requirement for providing the totality-of-the-evidence for the demonstration of biosimilarity of the proposed biosimilar product as compared to the reference product. As an example, more specifically, the analytical studies are to assess similarity in CQAs at various stages of the manufacturing process of the biosimilar product as compared to those of the reference product. To assist the sponsors to fulfill the regulatory requirement for providing the totality-of-the-evidence of analytical similarity, the FDA suggests several approaches depending upon the criticality of the identified quality attributes relevant to the clinical outcomes.
FDA’s approaches for tier analysis
Analytical similarity assessment is referred to as the comparisons of functional and structural characterization between a proposed biosimilar product and a reference product in terms of CQAs that are relevant to clinical outcomes. The FDA suggests that the sponsors identify CQAs that are relevant to clinical outcomes and classify them into three tiers depending the criticality or risk ranking (eg, most, mild to moderate, and least) relevant to clinical outcomes. At the same time, the FDA also recommends some statistical approaches for the assessment of analytical similarity for CQAs from different tiers. The FDA recommends an equivalence test for CQAs from Tier 1, quality range approach for CQAs from Tier 2, and descriptive raw data and graphical presentation for CQAs from Tier 3,2 which are briefly outlined in the following subsections.
Equivalence test for Tier 1
For Tier 1, the FDA recommends that an equivalency test be performed for the assessment of analytical similarity. As indicated by the FDA, a potential approach could be a similar approach to bioequivalence testing for generic drug products.3–6 In other words, for a given critical attribute, we may test for equivalence by the following interval (null) hypothesis:
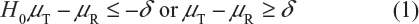
where δ > 0 is the equivalence limit (or similarity margin), and μT and μR are the mean responses of the test (the proposed biosimilar) product and the reference product lots, respectively. Analytical equivalence (similarity) is concluded if the null hypothesis of nonequivalence (nonsimilarity) is rejected. Note that Yu defined inequivalence as when the confidence interval falls entirely outside the equivalence limits.7 Similar to the confidence interval approach for bioequivalence testing under the raw data model, analytical similarity would be accepted for a quality attribute if the (1 − 2α)100% two-sided confidence interval of the mean difference is within (−δ, δ).
Under the null hypothesis (1), the FDA indicates that the equivalence limit (similarity margin), δ, would be a function of the variability of the reference product, denoted by σR. It should be noted that each lot contributes one test value for each attribute being assessed. Thus, σR is the population standard deviation of the lot values of the reference product. Ideally, the reference variability σR should be estimated based on some sampled lots randomly selected from a pool of reference lots for the statistical equivalence test. In practice, it may be a challenge when there is a limited number of available lots. Thus, the FDA suggests that the sponsor provide a plan on how the reference variability σR will be estimated with a justification.
Quality range approach for Tier 2
For Tier 2, the FDA suggests that analytical similarity be performed based on the concept of quality ranges, ie, ±xσ, where σ is the standard deviation of the reference product and x should be appropriately justified. Thus, the quality range of the reference product for a specific quality attribute is defined as 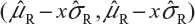 . Analytical similarity would be accepted for the quality attribute if a sufficient percentage of test lot values (eg, 90%) fall within the quality range.
. Analytical similarity would be accepted for the quality attribute if a sufficient percentage of test lot values (eg, 90%) fall within the quality range.
As it can be seen, for a given critical attribute, the quality range is set based on test results of available reference lots. If x = 1.645, we would expect 90% of the test results from reference lots to lie within the quality range. If x is chosen to be 1.96, we would expect that about 95% test results of reference lots will fall within the quality range. As a result, the selection of x could impact the quality range and consequently the percentage of test lot values that will fall within the quality range. Thus, the FDA indicates that the standard deviation multiplier (x) should be appropriately justified.
Raw data and graphical comparison for Tier 3
For CQAs in Tier 3 with lowest risk ranking, the FDA recommends an approach that uses raw data/graphical comparisons. The examination of similarity for CQAs in Tier 3 is by no means less stringent, which is acceptable because they have the least impact on clinical outcomes in the sense that a notable dissimilarity will not affect clinical outcomes.
Challenging issues to the FDA’s approaches
The idea of the FDA’s proposed equivalence test for Tier 1 CQAs comes from the bioequivalence assessment for generic drugs, which contain the same active ingredient(s) as the reference drug product. It may not be appropriate to apply the idea directly to the assessment of biosimilarity of biosimilar products. The FDA’s proposed equivalence test is sensitive to i) the primary assumptions made, ii) the selection of c, and iii) the estimation of σR. In what follows, I will comment on these issues.
Primary assumptions
Basically, the FDA’s proposed equivalence test ignores i) lot-to-lot variability of both the reference product and the proposed biosimilar product, ii) the difference between means, and iii) the inflation/deflation in variability between the reference product and the proposed biosimilar product. Suppose that there are K reference lots that will be used to establish equivalence acceptance criterion (EAC) for equivalence test. The FDA suggests that one sample is randomly selected from each lot. The standard deviation of the reference product σR can be estimated based on the K test results. Let xi, i = 1, 2., … K be the test result of the ith lot. xi, i = 1, 2., … K, is assumed independently and identically distributed with mean and variance 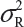 . In other words, we assume that μRI = μRj = μR and
. In other words, we assume that μRI = μRj = μR and 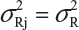 for i ≠ j, i, j = 1, 2, …, K. Thus, the expected value of and
for i ≠ j, i, j = 1, 2, …, K. Thus, the expected value of and 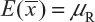 and
and 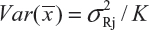 . In practice, it is well recognized that μRi ≠ μRj and
. In practice, it is well recognized that μRi ≠ μRj and 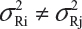 for i ≠ j, where μRi and
for i ≠ j, where μRi and 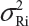 are the mean and variance of the ith lot of the reference product. A similar argument applies to the proposed biosimilar (test) product. As a result, the selection of reference lots for the estimation of σR is critical for the proposed approach.
are the mean and variance of the ith lot of the reference product. A similar argument applies to the proposed biosimilar (test) product. As a result, the selection of reference lots for the estimation of σR is critical for the proposed approach.
In addition, the FDA assumes that the difference in mean responses between the reference product and the proposed biosimilar product is proportional to the variability of the reference product. In other words, Δ = μT − μR (in log scale) ∞ σR The FDA suggests that the power for detecting a clinically meaningful difference be evaluated at σR/8. Thus, under the assumption, the FDA’s proposed equivalence testing is straightforward and easy to implement. However, Chow4 indicated that the FDA’s proposed testing procedure depends upon the selection of the regulatory standard c = 1.5, the anticipated difference Δ = μT − μR, and the compromise between the test size (type I error) and statistical power (type II error) for detecting Δ.5
Justification for the selection of c
The FDA indicates that one potential approach is to assume that the equivalence limit (similarity margin) is proportional to the reference product variability, ie, δ = C*σR. The constant c can be selected as the value that provides adequate power to show equivalence if there is only a small difference in the true mean between the biosimilar and the reference product, when a moderate number of reference product and biosimilar lots are available for testing. The FDA’s recommended approach for the assessment of analytical similarity for a critical attribute is to choose δ = 1.5 σR (ie, c = 1.5) and then to select an appropriate sample size for achieving a desired power in order to establish similarity at the α = 5% level of significance when the true underlying mean difference between the proposed biosimilar and reference product lots is equal to σR/8. The FDA did not provide scientific/statistical justification for the selection of c = 1.5 for EAC. Because the FDA’s proposed equivalence test was motivated from the bioequivalence assessment for generic drug products, the selection of c = 1.5 can be justified by the following steps:
Step 1. We start with 0.8 = δL ≤ μT - μR ≤δU = 1.25, where μT and μR are the reference mean and test mean (in log scale), respectively.
Step 2. For drug products with large variabilities (ie, highly variable drug products), the FDA recommends the scaled average bioequivalence criterion by adjusting the bioequivalence limits for variability of the reference product.8,9 This gives
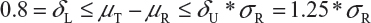
Step 3. The FDA assumes that the difference between means is proportional to σR and allows a mean shift of σR /8 = 0.125, which is the half width of the margin. The worst possible scenario for the shift is that the true mean difference falls on 1.25 *σR. In this case, the FDA expands the margin 0.25 *σR. Thus, the upper margin of EAC becomes
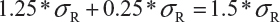
Estimate of σR
The FDA proposed that the equivalence test using available lot values be mainly based on the assumptions that i) there is no lot-to-lot variability within the reference product and the test product and ii) the difference in mean responses is proportional to the variability of reference product. In practice, however, it is recognized that μRi ≠ μRj and 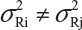 for i ≠ j. The differences between lots and heterogeneity among lots are major challenges to the validity of the FDA’s proposed approaches for both equivalence testing for CQAs in Tier 1 and the concept of quality range CQAs from Tier 2. Under the assumptions that μRi ≠ μRj and
for i ≠ j. The differences between lots and heterogeneity among lots are major challenges to the validity of the FDA’s proposed approaches for both equivalence testing for CQAs in Tier 1 and the concept of quality range CQAs from Tier 2. Under the assumptions that μRi ≠ μRj and 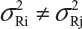 for i ≠ j, it is not clear what are the statistical properties/finite sample performances and corresponding impact on the assessment of analytical similarity and consequently on providing the totality-of-the-evidence to demonstrate similarity.
for i ≠ j, it is not clear what are the statistical properties/finite sample performances and corresponding impact on the assessment of analytical similarity and consequently on providing the totality-of-the-evidence to demonstrate similarity.
Heterogeneity within and between the test and reference products
Let 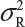 and
and 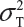 be the variabilities associated with the reference product and the test product, respectively. Also, let nR and nT be the number of lots for analytical similarity assessment for the reference product and the test product, respectively. Thus, we have
be the variabilities associated with the reference product and the test product, respectively. Also, let nR and nT be the number of lots for analytical similarity assessment for the reference product and the test product, respectively. Thus, we have
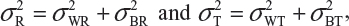
where 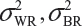 and
and 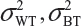 are the within-lot variability and between-lot (lot-to-lot) variability for the reference product and the test product, respectively. In practice, it is very likely that
are the within-lot variability and between-lot (lot-to-lot) variability for the reference product and the test product, respectively. In practice, it is very likely that 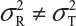 and often
and often 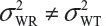 and
and 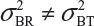 even
even 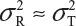 . This has posted a major challenge to the FDA’s proposed approaches for the assessment of analytical similarity for CQAs from both Tier 1 and Tier, especially when there is only one test sample from each lot from the reference product and the test product. The FDA’s proposal ignores lot-to-lot (between lot) variability, ie, when
. This has posted a major challenge to the FDA’s proposed approaches for the assessment of analytical similarity for CQAs from both Tier 1 and Tier, especially when there is only one test sample from each lot from the reference product and the test product. The FDA’s proposal ignores lot-to-lot (between lot) variability, ie, when 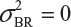 or
or 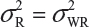 . In other words, sample variance based on xi, i = 1, …, K, from the reference product may underestimate the true
. In other words, sample variance based on xi, i = 1, …, K, from the reference product may underestimate the true 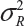 and consequently may not provide a fair and reliable assessment of analytical similarity for a given quality attribute.
and consequently may not provide a fair and reliable assessment of analytical similarity for a given quality attribute.
Matching lots
In practice, it is well recognized that μRi ≠ μRj and 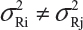 for i ≠ j, where μRi and
for i ≠ j, where μRi and 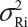 are the mean and variance of the ith lot of the reference product. A similar argument is applied to the proposed biosimilar (test) product. As a result, the selection of reference lots for the estimation of σR is critical for the proposed approach. The selection of reference lots has an impact on the estimation of σR and consequently on the EAC. Suppose there are K reference lots available and n lots will be tested for analytical similarity. The FDA suggests using the remaining K − n lots to establish EAC to avoid selection bias. It sounds a reasonable approach if K >> n. In practice, however, there are few lots available. In this case, the FDA’s proposed approach may not be feasible.
are the mean and variance of the ith lot of the reference product. A similar argument is applied to the proposed biosimilar (test) product. As a result, the selection of reference lots for the estimation of σR is critical for the proposed approach. The selection of reference lots has an impact on the estimation of σR and consequently on the EAC. Suppose there are K reference lots available and n lots will be tested for analytical similarity. The FDA suggests using the remaining K − n lots to establish EAC to avoid selection bias. It sounds a reasonable approach if K >> n. In practice, however, there are few lots available. In this case, the FDA’s proposed approach may not be feasible.
Sample size
In practice, one of the major problems to a biosimilar sponsor is the availability of reference lots for analytical similarity testing. The FDA suggests that an appropriate sample size (the number of lots from the reference product and from the test product) be used for achieving a desired power (say 80%) to establish similarity based on a two-sided test at the 5% level of significance assuming that the mean response of the test product differs from that of the reference product by σR/8.
Furthermore, because sample size is a function of α (type I error), β (type II error or 1 minus power), δ (treatment effect), and σ2 (variability), it is a concern that we may have inflated the type I error rate for achieving a desired power to detect a clinically meaningful effect size (adjusted for variability) with a preselected small sample size (ie, a small number of lots).
Remarks
Different assumptions may lead to different conclusions due to the difference between mean responses of the various lots and the heterogeneity among lots. It should be noted that the difference between the mean responses of the lots may be offset by the heterogeneity across lots in the FDA’s proposed equivalence test. Thus, one of the major criticisms of the FDA’s proposed equivalence test procedure is the validity of the primary assumptions, especially the assumption that the difference in the mean responses between the reference product and the proposed biosimilar product is proportional to the variability of the reference product. In addition, for a given CQA, the FDA only requires that a single sample obtained from a lot be tested. In this case, an independent estimate of the variability associated with the test result of the given lot is not available. Similar comments apply to the quality range approach for CQAs from Tier 2.
Recommendations and alternative methods
Recommendations to current approaches for the assessment of analytical similarity
Suppose that there are K reference lots to establish EAC for the equivalence test for Tier 1 CQAs. The FDA suggests that one sample is randomly selected from each lot. The standard deviation of the reference product σR can be estimated based on the K test results. Let xi, i = 1, 2 …, K, be the test result of the ith lot. xi, i = 1, 2 …, K, are assumed to be independently and identically distributed with mean μR and variance 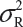 . In other words, we assume that μRi =μRj =μR and
. In other words, we assume that μRi =μRj =μR and 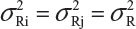 for i ≠ j, i, j = 1, 2 …, K. Thus, the expected value of
for i ≠ j, i, j = 1, 2 …, K. Thus, the expected value of 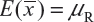 and
and 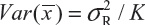 . Under the assumption that μRi ≠ μRj and
. Under the assumption that μRi ≠ μRj and 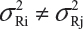 for i ≠ j, where μRi and
for i ≠ j, where μRi and 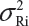 are the mean and variance of the ith lot of the reference product. In this case, we have
are the mean and variance of the ith lot of the reference product. In this case, we have
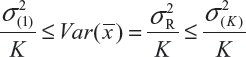
where 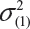 and
and 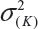 are the smallest and largest within-lot variance among the K lots. Thus, it is recommended that the current approach of equivalence test for analytical similarity be modified as follows:
are the smallest and largest within-lot variance among the K lots. Thus, it is recommended that the current approach of equivalence test for analytical similarity be modified as follows:
Randomly select at least two samples from each lot. The replicates will provide independent estimates of within-lot variability 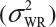 and lot-to-lot variability
and lot-to-lot variability 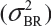 .
. 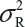 is the sum of
is the sum of 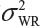 and
and 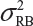 . In the interest of the same total number of tests, the sponsor can test on two samples from each lot among K/2 randomly selected lots.
. In the interest of the same total number of tests, the sponsor can test on two samples from each lot among K/2 randomly selected lots.
For the establishment of EAC, it is then suggested that σ(K) be used in order to take lot-to-lot and within-lot variabilities into consideration.
In case only one sample from each lot is tested, it is suggested that the lower 95% confidence bound be used as σR for the establishment of EAC for equivalence testing of the identified CQAs in Tier 1. In other words, under the FDA’s proposed approach, we will use the following to estimate σR
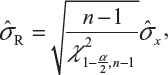
where 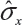 is the sample standard deviation obtained from the n reference lot test values and
is the sample standard deviation obtained from the n reference lot test values and 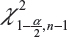 is the
is the 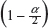 th upper quantile of a chi-square distribution with n − 1 degrees of freedom.
th upper quantile of a chi-square distribution with n − 1 degrees of freedom.
Alternative approaches
Alternatively, we may consider a Bayesian approach with appropriate choices of priors for the mean and standard deviation of the reference product in order to take into consideration the heterogeneity in mean and variability. The Bayesian approach is to obtain a Bayesian creditable interval, which will consider EAC for the assessment of analytical similarity.
Concluding remarks
For identifying CQAs at various stages of the manufacturing process, most sponsors assign CQAs based on the mechanism of action or PK, which are believed to be relevant to clinical outcomes. It is a reasonable assumption that change in mechanism of action or PK of a given quality attribute is predictive of clinical outcomes. However, the primary assumption that there is a well-established relationship between in vitro assays and in vivo testing (ie, in vitro assays and in vivo testing correlation) needs to be validated. Under the validated in vitro assays and in vivo testing correlation relationship, the criticality (or risk ranking) can then be assessed based on the degree of the relationship. In practice, however, most sponsors provide clinical rationales for the assignment of the CQAs without using statistical approach for the establishment of in vitro assays and in vivo testing correlation. The assignment of the CQAs without using a statistical approach is considered subjective and hence is somewhat misleading.
For a given quality attribute, the FDA suggests a simple approach by testing one sample (randomly selected) from each of the lots. Basically, the FDA’s approach ignores lot-to-lot variability for the reference product. In practice, however, lot-to-lot variability inevitably exists even when the manufacturing process has been validated. In other words, we would expect that there are differences in mean and variability from lot-to-lot, ie, μRi ≠ μRj and 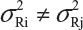 for i ≠ j, i, j = 1,2,,…,K. In this case, it is suggested that the FDA’s approach be modified (eg, performing tests on multiple samples from each lot) in order to account for the within-lot and between-lot (lot-to-lot) variabilities for fair and reliable comparisons.
for i ≠ j, i, j = 1,2,,…,K. In this case, it is suggested that the FDA’s approach be modified (eg, performing tests on multiple samples from each lot) in order to account for the within-lot and between-lot (lot-to-lot) variabilities for fair and reliable comparisons.
For the quality range approach for CQAs in Tier 2, the FDA recommends to use x = 3 by default for 90% of values of test lots contained in the range. It allows approximately one standard deviation of reference for shifting, which may be adjusted based on biologist reviewers’ recommendations. However, some sponsors propose using the concept of tolerance interval in order to ensure that there are a high percentage of test values for the lots from the test products that fall within the quality range. It, however, should be noted that the percentage decreases when the difference in mean between the reference product and the proposed biosimilar product increases. This is also true when σT << σR. Even the tolerance interval is used as the quality range. This problem is commonly encountered mainly because the quality range approach does not take into consideration i) the difference in means between the reference product and the proposed biosimilar product and ii) the heterogeneity among lots within and between products. In practice, it is very likely that a biosimilar product with small variability but a mean response which is away from the reference mean (eg, within the acceptance range of σR/8 per FDA), will fall outside the quality range. In this case, a further evaluation of the data points that fall outside the quality range is necessary to rule out the possibility by chance alone.
The FDA’s current thinking for analytical similarity assessment using a three-tier analysis is encouraging. It provides a direction for statistical methodology development for a valid and reliable assessment toward providing the totality-of-the-evidence for demonstrating biosimilarity. The three-tier approach is currently under tremendous discussion within the pharmaceutical industry and academia. In addition to the challenging issues discussed in the section “Challenging issues to the FDA’s approaches”, there are some issues that remain unsolved and require further research. These issues include, but are not limited to, i) the degree of similarity (ie, how similar is considered highly similar?), ii) multiplicity (ie, is there a need to adjust α for controlling the overall type I error at a prespecified level of significance), iii) acceptance criteria (eg, about what percentage of CQAs in Tier 1 need to pass an equivalence test in order to pass the analytical similarity test for Tier 1?), iv) multiple references (ie, what if there are two reference products such as US-licensed and European Union–approved reference products), and v) credibility toward the totality-of-the-evidence.
Acknowledgments
The author thanks Dr Yi Tsong from the FDA for his review, constructive comments, and discussion, which led to a significant improvement of this paper.
Disclosure
The author reports no conflicts of interest in this work.
References
FDA. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Silver Spring, MD: The United States Food and Drug Administration; 2012. | |
Christl L. Overview of the regulatory pathway and FDA’s guidance for the development and approval of biosimilar products in the US. Presented at: the Oncologic Drugs Advisory Committee meeting; January 7, 2015; Silver Spring, MD. | |
Chow SC, Liu JP. Design and Analysis of Bioavailability and Bioequivalence Studies. 3rd ed. New York: Chapman Hall/CRC Press, Taylor & Francis; 2008. | |
Chow SC. On assessment of analytical similarity in biosimilar studies. Drug Des. 2014;3:119. | |
FDA. Guidance on Bioavailability and Bioequivalence Studies for Orally Administrated Drug Products – General Considerations. Rockville, MD: Center for Drug Evaluation and Research, the US Food and Drug Administration; 2003. | |
Chow SC. Biosimilars: Design and Analysis of Follow-on Biologics. New York: Chapman and Hall/CRC Press, Taylor and Francis; 2013. | |
Yu LX. Bioinequivalence: concept and definition. Presented at: Advisory Committee for Pharmaceutical Science of the Food and Drug Administration; April 13–14, 2004; Rockville, MD. | |
Haidar SH, Davit B, Chen ML, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res. 2008;25:237–241. | |
Tothfalusi L, Endrenyi L, Garcia Areta A. Evaluation of bioequivalence for highly-variable drugs with scaled average bioequivalence. Clin Pharmacokinet. 2009;48:725–743. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.