Back to Journals » International Journal of Nanomedicine » Volume 11
Cerium oxide nanoparticles inhibit the migration and proliferation of gastric cancer by increasing DHX15 expression
Authors Xiao Y, Li J, Wang S, Yong X, Tang B, Jie M, Dong H, Yang X ![]() , Yang S
, Yang S
Received 5 January 2016
Accepted for publication 15 March 2016
Published 15 July 2016 Volume 2016:11 Pages 3023—3034
DOI https://doi.org/10.2147/IJN.S103648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Yu-Feng Xiao,1 Jian-Mei Li,2 Su-Min Wang,1 Xin Yong,1 Bo Tang,1 Meng-Meng Jie,1 Hui Dong,1 Xiao-Chao Yang,2 Shi-Ming Yang1
1Department of Gastroenterology, Xinqiao Hospital, Third Military Medical University, Chongqing, People’s Republic of China; 2School of Biomedical Engineering, Third Military Medical University, Chongqing, People’s Republic of China
Abstract: Gastric cancer is one of the leading causes of tumor-related deaths in the world. Current treatment options do not satisfy doctors and patients, and new therapies are therefore needed. Cerium oxide nanoparticles (CNPs) have been studied as a potential therapeutic approach for treating many diseases. However, their effects on human gastric cancer are currently unknown. Therefore, in this study, we aimed to characterize the effects of CNPs on human gastric cancer cell lines (MKN28 and BGC823). Gastric cancer cells were cocultured with different concentrations of CNPs, and proliferation and migration were measured both in vitro and in vivo. We found that CNPs inhibited the migration of gastric cancer cells when applied at different concentrations, but only a relatively high concentration (10 µg/mL) of CNPs suppressed proliferation. Furthermore, we found that CNPs increased the expression of DHX15 and its downstream signaling pathways. We therefore provide evidence showing that CNPs may be a promising approach to suppress malignant activity of gastric cancer by increasing the expression of DHX15.
Keywords: cerium oxide nanoparticles, gastric cancer, DHX15, p38
Introduction
Gastric cancer is the fifth most common cancer worldwide and the third leading cause of cancer-related deaths.1 Based on the results of a recent study, more than 900,000 new cancer cases and approximately 700,000 cancer deaths are estimated to have occurred in 2012.1,2 These data indicate that gastric cancer is a serious public health problem worldwide, especially in Eastern Asia. Current methods are far from satisfactory for doctors and patients, because the survival rate for gastric cancer remains at 20%–30%3 even when surgery, chemotherapy, and radiotherapy are combined. The invasion and metastasis are the major cause of mortality in gastric cancer patients. It is therefore urgent that effective therapeutic methods for inhibiting the migration and proliferation of gastric cancer are established.
In the past decades, nanomaterials with various kinds of unique characteristics4–7 have shown great potential in solving clinical problems that the traditional biological strategies could not handle.8–10 Particularly, nanomaterials have been well explored in the field of cancer research. For example, iron oxide nanoparticles, gold nanoparticles, and copper-alloyed gold nanoparticles have been used for cancer imaging.11 Gold nanoparticles, polymer nanoparticles, and polymersomes have been widely used as carriers for the delivery of anticancer drugs. Gold-coated FeO nanoroses, gold nanoclusters, and SiO2@GN-serum composites have been directly used as anticancer reagents.11–13 Within these nanomaterials, the cancer inhibition reagents are of great importance as they hold the potential of substituting chemical reagents, which are usually associated with serious side effects. However, the development of cancer is a complicated process involving numerous signaling pathways. A useful nanomaterial for cancer therapy should have the capability to interfere in one or more of these signaling pathways.
It is well known that the oxygen and reactive oxygen species (ROS) are very important in cancer development. Therefore, the nanomaterials that can effectively modulate intracellular oxygen and ROS would be potentially useful in cancer therapy. Recently, it has been reported that cerium oxide nanoparticles (CNPs) could mimic several kinds of enzyme activity, including superoxide oxidase, catalase, oxidase, and phosphatase because of the coexistence of Ce3+ and Ce4+ at the nanoparticle surface.14 Because the function of these enzymes is to regulate the intracellular oxygen and ROS levels, the CNPs might have the ability to interfere in cancer development. This technique has been preliminarily attempted by several studies. For example, CNPs could induce cytotoxicity and oxidative stress in human lung cancer cells.15 CNPs could also inhibit melanoma (518A2) and colorectal adenocarcinoma (HT-29) cell lines.16 However, the effects of CNPs on human gastric cancer cells have not been investigated to date.
In this study, we explored the effect of CNPs on the migration and proliferation of human gastric cancer. We found that CNPs could promote the expression of putative ATP-dependent RNA helicase DEAH (Asp-Glu-Ala-His) box helicase 15 (DHX15), which is one of the DEXD/H box helicase family that plays important roles in the innate immune response against viral infection. The increased expression of DHX15 in turn activates p38 MAPK signal pathway, leading to the inhibition of proliferation and metastasis in gastric cancer in vitro and in vivo.
Materials and methods
Synthesizing the CNPs
CNPs were synthesized using a thermal decomposition method. Cerium nitrate was used as the precursor. The details of the procedure have been previously described.17 The characteristics of the synthesized CNPs were as follows: 1) core size: approximately 3 nm and 2) zeta potential: −18 mV (Supplementary figures; Figure S1).
Cell cultures and stimulation with CNPs
The BGC823 and MKN28 gastric cancer cell lines were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, People’s Republic of China). They were cultured in Dulbecco’s Modified Eagle’s Medium (high glucose) (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA). Both gastric cancer cell lines were cocultured with various concentrations of CNPs (0, 0.01, 0.1, 0.5, 1, and 10 μg/mL) for 3 days. All experimental procedures were approved by the Institutional Review Board of the Third Military Medical University.
Cell migration and proliferation assays
Transwell (Millipore, Billerica, MA, USA) assays were used to measure migration. After the cells were cocultured for 72 hours with various concentrations of CNPs, 1×104 BGC823 and MKN28 cells were seeded into the upper chamber in serum-free culture medium. Medium supplemented with 10% serum was added to the lower chamber (the medium in the lower well also contained the appropriate concentration of CNPs). After 24 hours of incubation, the cells were fixed and stained using 0.05% crystal violet. The number of cells that migrated from the upper chamber to the lower chamber was counted using a light microscope. Cells were counted in five random views in each experiment, and the assays were carried out three times.
A Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) was used for the proliferation assay. A total of 2×103 BGC823 or MKN28 cells were plated in 96-well plates with different concentrations of CNPs. At previously determined time points, the numbers of cells were measured at an absorbance of 450 nm. Each data point is represented as the mean ± standard deviation (n=3).
Gene expression microarray analysis
MKN28 cells were cocultured with CNPs at different concentrations (0, 0.01, and 10 μg/mL). A 1 mL volume of Triol (TaKaRa, Kusatsu, Japan) was then added. The mixtures were sent to the Genminix Biological Company (Shanghai, People’s Republic of China) for microarray analysis. Affymetrix GeneChip Human transcriptome assays 2.0 (Affymetrix, Santa Clara, CA, USA) were used for the gene expression microarray analyses. The Genminix Biological Company performed the bioinformatics analysis. Microarray data were uploaded to the Gene Expression Omnibus.
Western blot analysis
Glyceraldehyde 3-phosphate dehydrogenase antibodies were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). DHX15 antibodies were obtained from Abcam (Cambridge, UK). p38, p65, phosphor-p38, and phosphor-p65 antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). An ECL chemiluminescence kit was purchased from Thermo (Fisher Scientific). Assays for each gene expression were examined three times, using three different batches of cell samples (biological replicates). The protein bands in the Western blots were quantified using densitometry analysis in Quantity One software Quantity One v4.4.0.36 (Bio-Rad, Hercules, CA, USA), and then the data were analyzed by using Prism 6.0 software (GraphPad, La Jolla, CA, USA). Each data point represents the mean ± standard deviation (n=3).
Real-time polymerase chain reaction assays
Messenger RNA (mRNA)-specific primers were designed by the Shanghai Biotechnology Company (Shanghai, People’s Republic of China). Total RNA was extracted using the TRIZOL method (Thermo Fisher Scientific) according to the manufacturer’s instructions. cDNA was synthesized using a PrimeScript RT Reagent Kit (TaKaRa), and quantitative real-time polymerase chain reaction (RT-PCR) was performed using SYBR Premix Ex Taq (TaKaRa). Assays for each gene expression were examined three times, using three different batches of cell samples, and the data were analyzed using Prism 6.0 software (GraphPad).
Immunofluorescence to analyze protein localization
Cells were cocultured with different concentrations (0.01 and 10 μg/mL) of CNPs for 3 days and then seeded on coverslips and grown to 80% confluence. The cells were washed once with phosphate-buffered saline (PBS), and 4% paraformaldehyde was used to fix the cells for 10 minutes at room temperature. The cells were solubilized in 0.5% Triton-X100 in PBS for 15 minutes. Then, the cells were incubated in 5% bovine serum albumin for 30 minutes at room temperature followed by an overnight incubation with primary anti-DHX15 (Abcam), anti-p38 (CST), or anti-p65 (CST) antibodies at 4°C. The secondary antibodies that were used were Alexa Fluor 647 goat-anti rabbit IgG, which was applied at a 1:100 dilution for 1 hour. 4′,6-Diamidino-2-phenylindole (DAPI) was used to stain the cell nuclei.
Transmission electron microscopy
Cells were cocultured with different concentrations of CNPs (0, 0.01, and 10 μg/mL) and then fixed using 2.5% glutaraldehyde and 1% osmic acid. The samples were dehydrated in a series of ethanol washes (50%, 70%, 90%, and 100%). The samples were then embedded and stained. Transmission electron microscopy (TEM) was used to locate the CNPs in these samples.
Flow cytometry to analyze apoptosis
MKN28 and BGC823 cells were cocultured with different concentrations of CNPs. The cells were then harvested to analyze apoptosis using flow cytometry. The effects of different concentrations of CNPs on the cells were detected using flow cytometry according to the instructions in the operation manual for the Fluorescein isothiocyanate (FITC)–Annexin V/Dead Cell Apoptosis Kit (with FITC annexin V and propidium iodide (PI) for flow cytometry) (Thermo Fisher Scientific). Each data point represents the mean ± standard deviation (n=3).
Flow cytometry to analyze ROS
MKN28 and BGC823 cells were cocultured with different concentrations (0, 0.01, and 10 μg/mL) of CNPs. The cells were harvested after they reached 80% confluence. The ROS levels in the MKN28 and BGC823 cells were detected using flow cytometry, according to the operation manual for MitoSOX™ Red Mitochondrial Superoxide Indicator (Thermo Fisher Scientific). Each data point represents the mean ± standard deviation (n=3).
Immunofluorescence to analyze ROS
Cells were seeded on coverslips and cocultured with different concentrations of CNPs (0, 0.01, and 10 μg/mL). Then, the cells were washed once with PBS and then cocultured with MitoSOX™ Red Mitochondrial Superoxide Indicator (Thermo Fisher Scientific) for 30 minutes at 37°C. PBS was then used to wash the cells, and 4% paraformaldehyde was used to fix the cells for 10 minutes at room temperature. DAPI was used to stain the cell nuclei. The ROS levels in the MKN28 and BGC823 cell cultures were detected using immunofluorescence.
Gastric cancer xenograft assays
This study was performed in accordance with the National Institutes of Health (NIH) Animal Use Guidelines and a protocol that was approved by the Southern Illinois University (SIU) Animal Care Committee. All experimental procedures were approved by the Institutional Review Board of the Third Military Medical University. Each group contained five nude mice. The nude mice were kept in the SPF (specific-pathogen free) environment. The animals were housed with standard chow and water ad libitum, and sustained on a 12 h: 12 h light and dark cycle at a temperature of 21~25°C. For the gastric cancer xenograft experiments, 0.1 mL of PBS containing a total number of 1×106 MKN28 (BGC823) cells that had been cocultured with different concentrations (0.01 and 10 μg/mL) of CNPs was injected subcutaneously into the right axillae of 4-week-old male nude mice. The mice were divided into two groups (a 0.01 μg/mL group and a 10 μg/mL group). In the control group, 1×106 of MKN28 (BGC823) cells that were not cocultured with CNPs were injected into the left axillae. A volume of 50 μL of PBS that contained CNPs (0.01 or 10 μg/mL) was injected into the tumor site every 3 days. A volume of 50 μL of PBS was injected at the same time into the left axillae. After 6 weeks, the mice were euthanized, and the tumors were harvested, weighed, and measured.
Gastric cancer metastasis assays
For the peritoneal metastasis assays, 4-week-old male nude mice were peritoneally injected with 0.1 mL of a single cell suspension of 1×106 MKN28 (BGC823) cells that had been cocultured with different concentrations of CNPs (0, 0.01, and 10 μg/mL). A total of 30 mice were divided into two groups, each containing three subgroups (0, 0.01, and 10 μg/mL groups). A volume of 50 μL of PBS that contained CNPs (0, 0.01, or 10 μg/mL) was injected into the peritoneal cavity of each group every 3 days. After 6 weeks, the mice were euthanized, and tumors were counted.
Statistical analysis
All data are presented as the mean ± standard error (SE), and analyses were performed using Prism 6.0 software (GraphPad). Independent-samples t-tests were used to compare two groups, and one-way analysis of variance (ANOVA) was used to compare three or more groups. P<0.05 was considered statistically significant.
Results
CNPs reduced migration and proliferation in gastric cancer cells in vitro
In our study, we first explored whether CNPs would inhibit the migration ability of gastric cancer cells. We chose 0.01, 0.1, 0.5, 1, and 10 μg/mL CNP concentrations for these experiments. Both MKN28 and BGC823 cells were cocultured with different concentrations of CNPs for 3 days. The CNPs inhibited the migratory ability of MKN28 and BGC823 cells compared to the results in the control group (Figure 1A). Moreover, we observed that this inhibitory effect was dose-independent (Figure 1B and C).
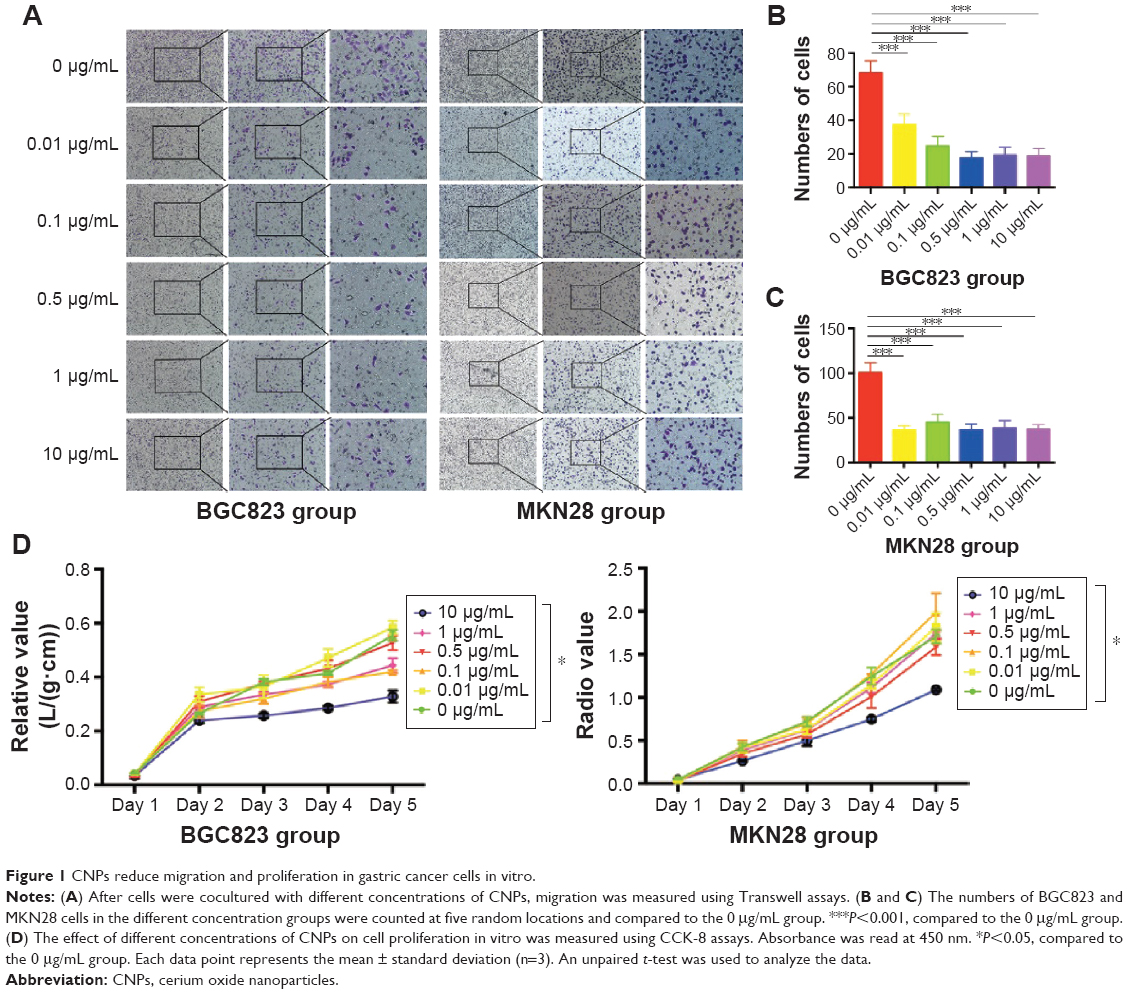 | Figure 1 CNPs reduce migration and proliferation in gastric cancer cells in vitro. |
We next measured proliferation in gastric cancer cells that were cocultured with different concentrations of CNPs. Interestingly, the results of these assays were different from those in the migration assays. We found that cell proliferation was inhibited only in the group treated with a high concentration of CNPs (10 μg/mL) (Figure 1D). The other treatment groups (0.01, 0.1, 0.5, and 1 μg/mL) showed no significant changes toward either promotion or inhibition compared to the control group (0 μg/mL). Recently, Hussain et al18 reported that CNPs induced apoptosis in human peripheral blood monocytes. We therefore sought to determine whether CNPs would influence apoptosis in gastric cancer cells. We analyzed the cells using flow cytometry and found that CNPs had no effect on apoptosis in either the MKN28 or the BGC823 gastric cancer cells (Supplementary figures; Figure S2).
These data suggest that CNPs inhibit the migratory ability of gastric cancer cells at a broad range of concentrations, whereas only a high concentration (10 μg/mL) of CNPs inhibits proliferation in these cells.
The location of CNPs in gastric cancer cells
We have found that CNPs inhibited migration and proliferation in gastric cancer cells, but the mechanism underlying this effect remained unclear. We therefore sought to determine whether CNPs can enter gastric cancer cells. As shown in Figure 1, we focused on two concentrations (0.01 and 10 μg/mL) of CNPs in the experiments aimed at exploring the localization of CNPs in cocultured gastric cancer cells. Using TEM, we found that the CNPs were localized in the lysosomes of the cells (Figure 2). We did not observe CNPS that were localized in other parts of the cells in our samples. This result indicates that CNPs localize in the lysosomes of gastric cancer cells in cocultures.
 | Figure 2 The localization of CNPs in gastric cancer cells. |
CNPs increase the expression of DHX15
To further explore the mechanism by which CNPs inhibit proliferation and migration in gastric cancer cells, we used an mRNA expression microarray (HTA 2.0) to determine which genes showed change in their levels. According to the microarray results (Gene Ontology [GO] analysis and Pathway analysis) (Figure 3A and B), the genes associated with CNPs mainly related to the Spliceosome and MAPK pathways. We then selected the genes that were upregulated or downregulated in both groups (0.01 μg/mL group and 10 μg/mL group) compared to the control group (Figure 3C). According to the observed fold changes and significance (P<0.05), we chose the first three genes (DHX15, TRIP12, and EIF4G2) as the candidate genes. In addition, qRT-PCR showed that only DHX15 was upregulated in gastric cancer cells that were cocultured with CNPs (0.01 and 10 μg/mL) (Figure 3D). Western blot analysis also showed that the expression of DHX15 was upregulated (Figure 3E and F). These results indicate that CNPs activate the expression of DHX15 while simultaneously inhibiting proliferation and migration in gastric cancer cells.
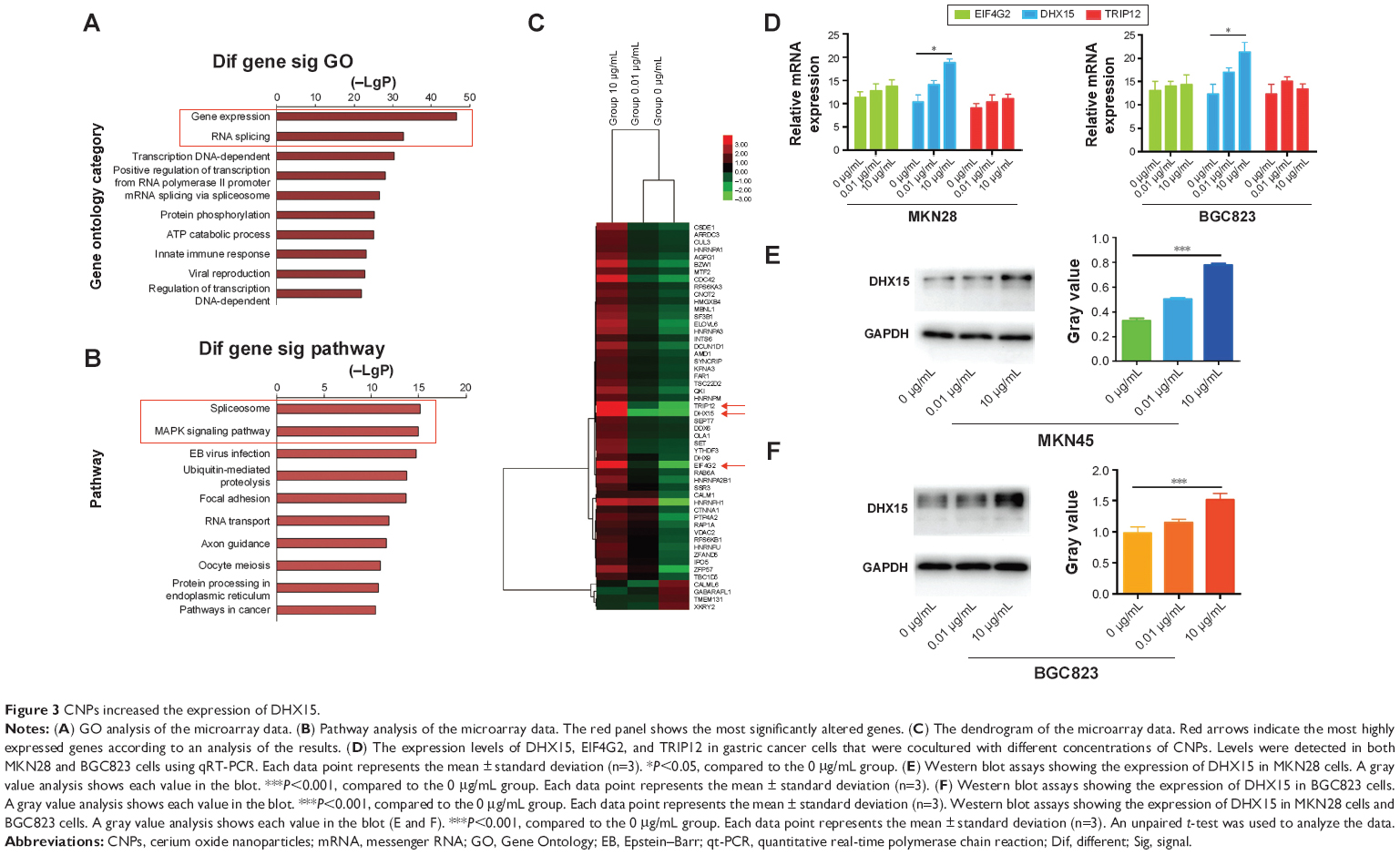 | Figure 3 CNPs increased the expression of DHX15. |
Genes downstream of DHX15 were activated by CNPs
We found that CNPs upregulated the expression of DHX15, which is a type of putative ATP-dependent RNA helicase that has been implicated in pre-mRNA splicing. Currently, no other studies have described a nanomaterial that influences the expression of DHX15.
Studies have shown that DHX15 activates the MAPK signaling pathway during antiviral responses.19 We therefore sought to determine whether the MAPK signaling pathway was also activated according to the results of our microarray analysis (Figure 3B). As expected, we found that the expression of p38 was increased after cells were cocultured with CNPs (Figure 4A and B). In addition, we also found that CNPs increased the expression of DHX15 and p38 in immunofluorescence experiments (Figure 4E and F). These results indicated that CNPs may increase the expression of DHX15 and its downstream signal pathways, including the MAPK signal pathway. Interestingly, despite the increase in the total expression of p38, we also observed that the phosphorylation status of p38 was decreased (Figure 4C and D), and p38 was more localized in the cytosol, after being cocultured with the CNPs (Supplementary figures; Figure S3).
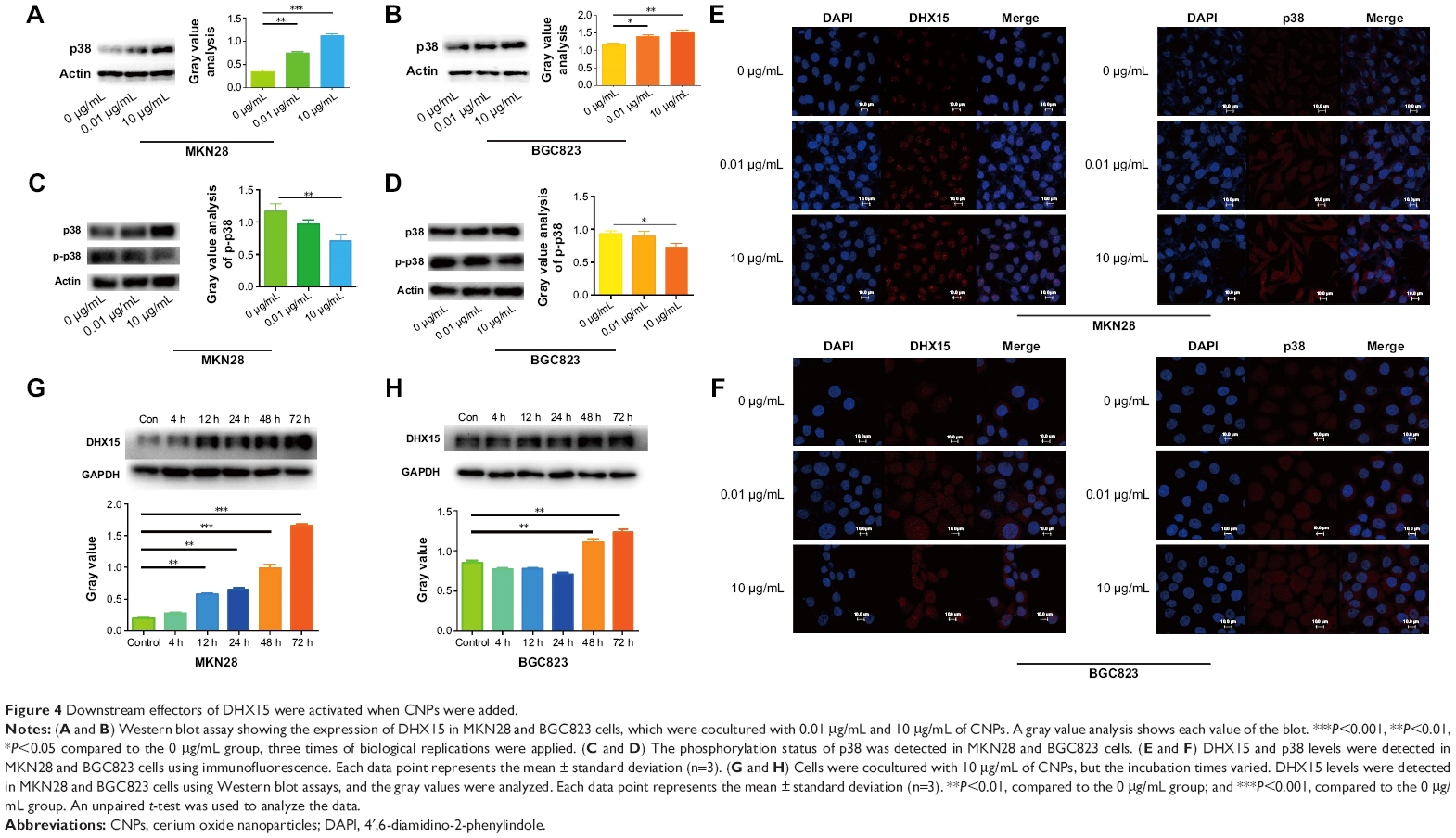 | Figure 4 Downstream effectors of DHX15 were activated when CNPs were added. |
As many reports have shown, CNPs influence ROS. We therefore explored whether the increase of DHX15 induced by CNPs was caused by an increase of ROS. However, using immunofluorescence and flow cytometry, we found that ROS levels in gastric cancer cells were not changed after the cells were cocultured with different concentrations of CNPs (Supplementary figures; Figures S4 and S5). These data suggested that the increase in DHX15 in gastric cancer cells was not associated with ROS levels.
We then explored the time point at which DHX15 was activated after CNPs were added to the cells. We found that the DHX15 activation time points were different in MKN28 and BGC823 cells (Figure 4G and H). In MKN28 cells, the expression of DHX15 was activated at 12 hours after CNPs were added, whereas in BGC823 cells, DHX15 was activated at 48 hours after CNPs were added. This difference may be due to differences in the uptake of CNPs in different gastric cancer cell lines.
A high concentration of CNPs reduced tumor growth in vivo, while low concentration of CNPs had no effect
Our results showed that a high concentration of CNPs (10 μg/mL) modulated gastric cancer cell growth in vitro. We therefore investigated whether CNPs could inhibit gastric cancer cells in vivo. MKN28 (BGC823) cell suspensions (1×106) that were cocultured with different concentrations of CNPs (0.01 and 10 μg/mL) were injected into the right axillae of nude mice (Figure 5A, B, D, and E). The same number of untreated (not incubated with CNPs) cells were injected into the corresponding left axillae of nude mice in the control group. As the results show, a high concentration of CNPs inhibited gastric cancer growth and reduced tumor volumes (Figure 5B and E), while a low concentration of CNPs had no effect (Figure 5A and D). Tumor weights were also measured, and we found that only a high concentration of CNPs reduced tumor weights (Figure 5C and F). We also found that the expression of DHX15 was higher in the tumors, which were treated with different concentrations of CNPs (Figure 5G). The data were in agreement with the results of in vitro proliferation assays, suggesting that a high concentration of CNPs inhibits gastric cancer cell growth, while a low concentration of CNPs has no effect.
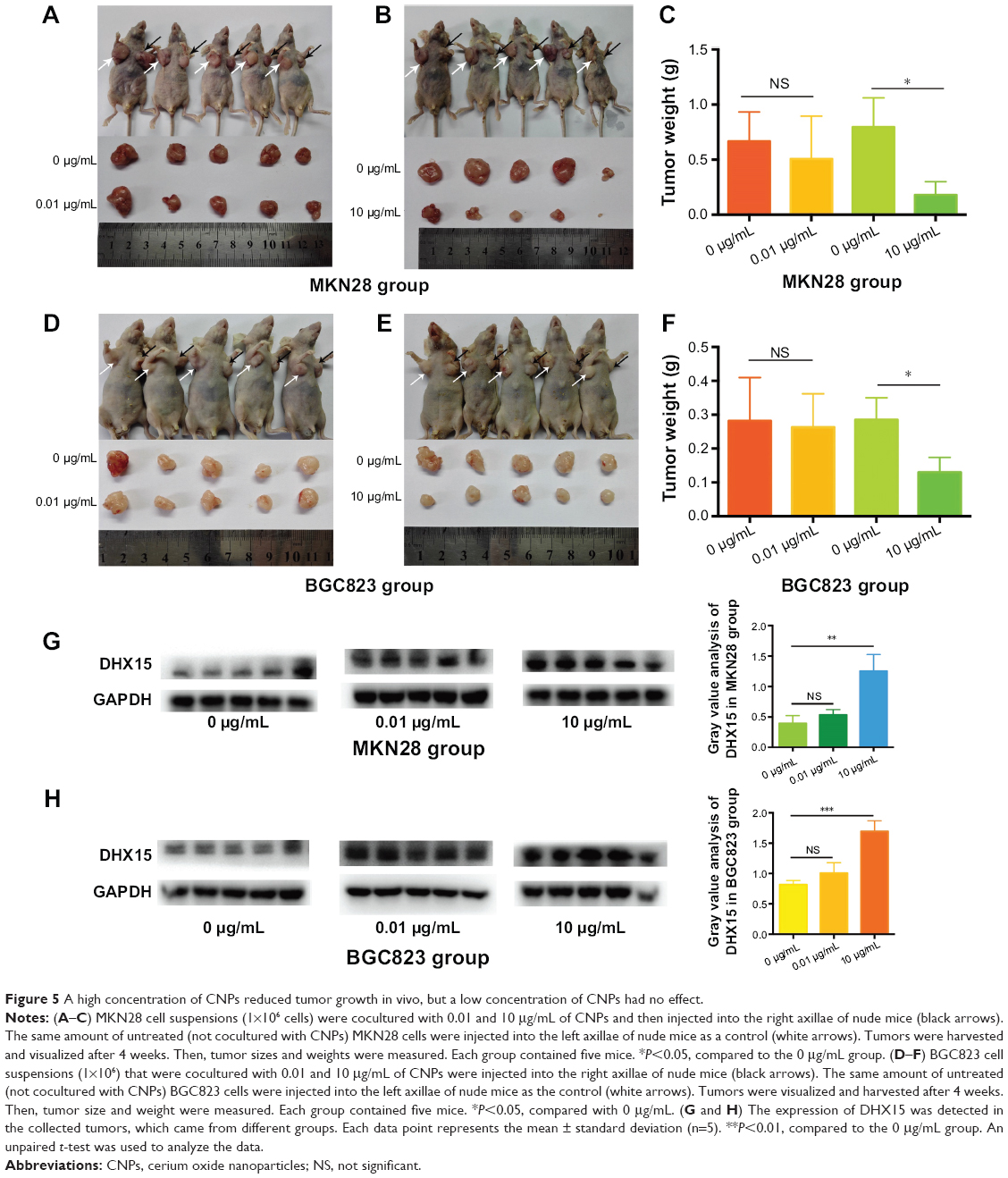 | Figure 5 A high concentration of CNPs reduced tumor growth in vivo, but a low concentration of CNPs had no effect. |
CNPs inhibit GC cell metastasis in vivo
We further explored whether CNPs inhibit migration in gastric cancer cells in vivo. MKN28 (or BGC823) cells that were cocultured with CNPs (0.01 and 10 μg/mL) were injected into nude mice via peritoneal injection. The same amount of untreated gastric cancer cells (not cocultured with CNPs) were injected into the other five nude mice in the control group (Figure 6A). The nude mice were euthanized 6 weeks later, and the number of metastatic tumors was counted (Figure 6B and C). We found that there were fewer metastatic tumors in the CNP-treated groups (both the high-concentration group and the low-concentration group) than there were in the control group. This assay suggested that the peritoneal metastatic ability of MKN28 and BGC823 cells was inhibited after the cells were cocultured with CNPs. We conclude that CNPs suppress the metastatic potential of gastric cancer cells.
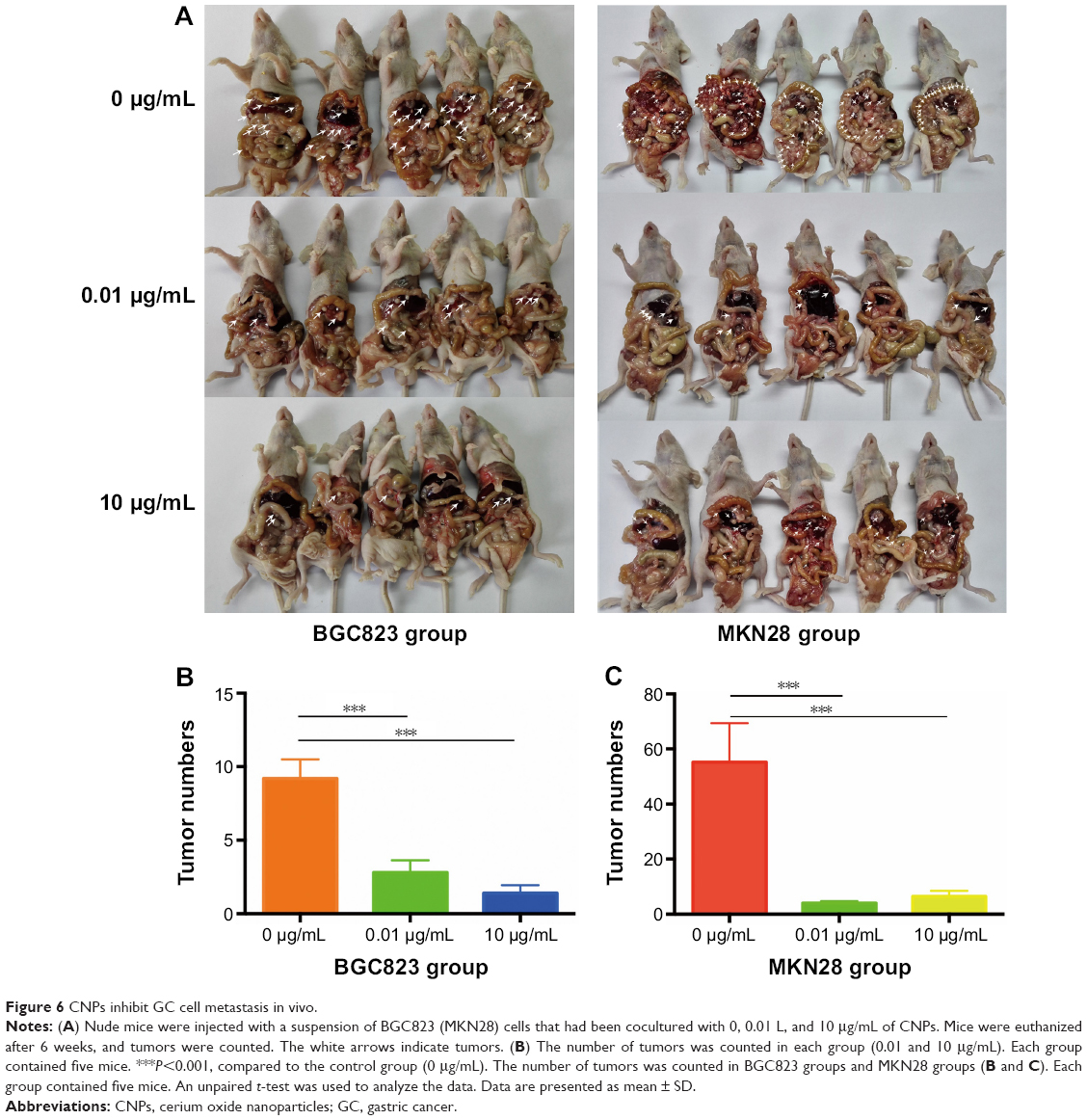 | Figure 6 CNPs inhibit GC cell metastasis in vivo. |
Discussion
Because the effects and mechanism of CNPs in organic systems are not fully understood, the enthusiasm of researchers remains high. Asati et al20 reported that the surface charge of a CNP could determine its localization and function in a cellular environment. Babu et al14 reported that CNPs could be used as a potentially antibacterial nanomaterial in vivo. Moreover, CNPs have been shown to be potentially effective drugs for treating different types of cancer, such as melanoma,21 pancreatic cancer,22 and colorectal cancer.16 It has been reported that CNPs may protect gastrointestinal epithelial cell lines (normal human colon cells, CRL 1541) from radiation-induced damage by reducing the levels of ROS.23 However, few reports have explored the effects of CNPs on gastric cancer cells.
In the present study, we have shown that CNPs inhibit human gastric cancer cell migration in vitro and in vivo, which is not a dose-dependent effect because no significant difference exists between the low-concentration group (0.01 μg/mL) and the high-concentration group (10 μg/mL) in the results of Transwell assays and the peritoneal metastasis assays. Interestingly, we also found that only the high concentration (10 μg/mL) of CNPs suppressed proliferation of gastric cancer cells according to the result of CCK8 assays and xenograft assays. A low concentration (0.01 μg/mL) of CNPs had no effect on the proliferative ability of the cells. These data suggested that CNPs could be potentially effective agents to treat gastric cancer.
We also found that CNPs were localized in the lysosomes of gastric cancer cells (Figure 2). According to Asati et al,20 positively charged and negatively charged CNPs are more likely to be taken up into lysosomes, whereas CNPs that are neutral in charge were localized in the cytoplasm when the CNPs were cocultured with cells. In this study, we focused on the CNPs that carry a negative charge, and our results are therefore in agreement with those of Asati et al.20
Most reports have shown that CNPs influence biological systems through redox reactions. Pagliari et al24 reported that CNPs protect cardiac progenitor cells from oxidative stress without affecting cell growth and functions. Rubio et al25 also found that CNPs could be used as a pharmacological agent to combat oxidative stress, because it has regenerative free radical–scavenging properties. However, our study shows that the ROS levels in gastric cancer cells did not change after the cells were cocultured with CNPs (Figures S4 and S5), indicating that a ROS-independent mechanism may be involved in the effects that were induced when gastric cancer cells were cocultured with CNPs. This result is similar to the results in Hussain et al.18 Hussain et al18 found that CNPs increased apoptosis-inducing factor (AIF) but did not increase the production of ROS.18 We used a microarray to explore the details of the implicated mechanism. We found, for the first time, that CNPs increase the expression of DHX15 based on the results of our microarray and Western blot analyses. DHX15 is an RNA helicase that has been implicated in pre-mRNA splicing. Recently, Wang et al26 reported that NIrp6-bound viral RNA via DHX15 during intestinal antiviral innate immune responses. Their study also showed that RBM5, a tumor suppressor, directly interacted with DHX15 and stimulated its activity.27 These results indicate that DHX15 is very important to biological systems, because it is a component of spliceosomes. In our study, we observed that the expression of DHX15 was increased after cells were cocultured with different concentrations of CNPs (Figure 3E and F). In addition, we found that the activation of DHX15 occurred earlier in MKN28 cells than in BGC823 cells (Figure 4G and H). We also observed that the downstream effectors of DHX15 were activated, including an increase in the expression of p38 MAPK (Figure 4A and B). A study showed that DHX15 activates the p38 MAPK and NF-κB signaling pathways during the RNA virus response.19 We also found that the level of p65 was increased after cells were cocultured with CNPs, but the phosphorylation status of p65 was low, and p65 was more localized in the cytosol (Supplementary figures; Figures S3 and S6). Furthermore, AKT-mTOR-S6RP and Jnk were also involved in regulating cell proliferation, and we found that there was no change in AKT and c-jun expression after coculturing with CNPs (Supplementary figures; Figure S7).
The role of p38 MAPK signaling pathway in cancer is complicated. It can act as either a tumor suppressor or a tumor activator in different types of cancer.28,29 Recently, Zhong et al30 reported that the activation of the p38 MAPK pathway induced autophagy in HeLa cells. The activation of the p38 MAPK signaling pathway was also shown to inhibit the proliferation and migration of gastric cancer cells, as reported by Hao et al.31 Interestingly, Wang et al32 also reported that carbon nanotubes that carried a potent anticancer ruthenium polypridyl complex antagonize multidrug resistance in cancer cells by activating the downstream p38 MAPK signaling pathway. Moreover, recent studies have also shown that neuropilin-1-targeted gold nanoparticles enhanced the effect of platinum(IV) in prostate cancer by upregulating the protein level of NF-KB (p65) and its DNA binding ability.33 According to the results of this and other recent studies, we believe that CNPs inhibit proliferation and migration of cancer cells by activating the expression of DHX15 and its downstream signal pathway.
However, there are still some limitations in our study. We did not determine the mechanisms by which the CNPs increase the expression of DHX15 in gastric cancer cells. Further studies are needed to explore the underlying mechanisms.
Conclusion
In summary, we found that CNPs inhibited migration of gastric cancer cells at both low and high concentrations, but suppressed cell proliferation at a high concentration only. We also found that CNPs performed this function by increasing the expression of DHX15. Our study suggests that CNPs have a potential to be used for gastric cancer therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | ||
Allemani C, Weir HK, Carreira H, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015;385(9972):977–1010. | ||
Bleeker EA, de Jong WH, Geertsma RE, et al. Considerations on the EU definition of a nanomaterial: science to support policy making. Regul Toxicol Pharmacol. 2013;65(1):119–125. | ||
Wang J, Gu H. Novel metal nanomaterials and their catalytic applications. Molecules. 2015;20(9):17070–17092. | ||
Goris B, Bals S, Van den Broek W, et al. Atomic-scale determination of surface facets in gold nanorods. Nat Mater. 2012;11(11):930–935. | ||
Baumann J, Sakka Y, Bertrand C, Koser J, Filser J. Adaptation of the Daphnia sp. acute toxicity test: miniaturization and prolongation for the testing of nanomaterials. Environ Sci Pollut Res Int. 2014;21(3):2201–2213. | ||
Veiseh O, Tang BC, Whitehead KA, Anderson DG, Langer R. Managing diabetes with nanomedicine: challenges and opportunities. Nat Rev Drug Discov. 2015;14(1):45–57. | ||
Mao HY, Laurent S, Chen W, et al. Graphene: promises, facts, opportunities, and challenges in nanomedicine. Chem Rev. 2013;113(5):3407–3424. | ||
Thakor AS, Gambhir SS. Nanooncology: the future of cancer diagnosis and therapy. CA Cancer J Clin. 2013;63(6):395–418. | ||
Li C, Chen T, Ocsoy I, et al. Gold-coated Fe3O4 nanoroses with five unique functions for cancer cell targeting, imaging and therapy. Adv Funct Mater. 2014;24(12):1772–1780. | ||
Liu Y, Bai J, Jia X, Jiang X, Guo Z. Fabrication of multifunctional SiO2@GN-serum composites for chemo-photothermal synergistic therapy. ACS Appl Mater Interfaces 2015;7(1):112–121. | ||
Wang Y, Chen J, Irudayaraj J. Nuclear targeting dynamics of gold nanoclusters for enhanced therapy of HER2+ breast cancer. ACS Nano. 2011;5(12):9718–9725. | ||
Babu KS, Anandkumar M, Tsai TY, Kao TH, Stephen Inbaraj B, Chen BH. Cytotoxicity and antibacterial activity of gold-supported cerium oxide nanoparticles. Int J Nanomedicine. 2014;9:5515–5531. | ||
Lin W, Huang YW, Zhou XD, Ma Y. Toxicity of cerium oxide nanoparticles in human lung cancer cells. Int J Toxicol. 2006;25(6):451–457. | ||
Pesic M, Podolski-Renic A, Stojkovic S, et al. Anti-cancer effects of cerium oxide nanoparticles and its intracellular redox activity. Chem Biol Interact. 2015;232:85–93. | ||
Yang ZY, Luo SL, Li H, et al. Alendronate as a robust anchor for ceria nanoparticle surface coating: facile binding and improved biological properties. RSC Adv. 2014;4(104):59965–59969. | ||
Hussain S, Al-Nsour F, Rice AB, et al. Cerium dioxide nanoparticles induce apoptosis and autophagy in human peripheral blood monocytes. ACS Nano. 2012;6(7):5820–5829. | ||
Mosallanejad K, Sekine Y, Ishikura-Kinoshita S, et al. The DEAH-box RNA helicase DHX15 activates NF-kappaB and MAPK signaling downstream of MAVS during antiviral responses. Sci Signal. 2014;7(323):ra40. | ||
Asati A, Santra S, Kaittanis C, Perez JM. Surface-charge-dependent cell localization and cytotoxicity of cerium oxide nanoparticles. ACS Nano. 2010;4(9):5321–5331. | ||
Alili L, Sack M, von Montfort C, et al. Downregulation of tumor growth and invasion by redox-active nanoparticles. Antioxid Redox Signal. 2013;19(8):765–778. | ||
Wason MS, Colon J, Das S, et al. Sensitization of pancreatic cancer cells to radiation by cerium oxide nanoparticle-induced ROS production. Nanomedicine. 2013;9(4):558–569. | ||
Colon J, Hsieh N, Ferguson A, et al. Cerium oxide nanoparticles protect gastrointestinal epithelium from radiation-induced damage by reduction of reactive oxygen species and upregulation of superoxide dismutase 2. Nanomedicine. 2010;6(5):698–705. | ||
Pagliari F, Mandoli C, Forte G, et al. Cerium oxide nanoparticles protect cardiac progenitor cells from oxidative stress. ACS Nano. 2012;6(5):3767–3775. | ||
Rubio L, Annangi B, Vila L, Hernandez A, Marcos R. Antioxidant and anti-genotoxic properties of cerium oxide nanoparticles in a pulmonary-like cell system. Arch Toxicol. 2016;90(2):269–278. | ||
Wang P, Zhu S, Yang L, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science. 2015;350(6262):826–830. | ||
Niu Z, Jin W, Zhang L, Li X. Tumor suppressor RBM5 directly interacts with the DExD/H-box protein DHX15 and stimulates its helicase activity. FEBS Lett. 2012;586(7):977–983. | ||
Grossi V, Peserico A, Tezil T, Simone C. p38alpha MAPK pathway: a key factor in colorectal cancer therapy and chemoresistance. World J Gastroenterol. 2014;20(29):9744–9758. | ||
Gupta J, del Barco Barrantes I, Igea A, et al. Dual function of p38alpha MAPK in colon cancer: suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell. 2014;25(4):484–500. | ||
Zhong W, Zhu H, Sheng F, et al. Activation of the MAPK11/12/13/14 (p38 MAPK) pathway regulates the transcription of autophagy genes in response to oxidative stress induced by a novel copper complex in HeLa cells. Autophagy. 2014;10(7):1285–1300. | ||
Hao W, Yuan X, Yu L, et al. Licochalcone A-induced human gastric cancer BGC-823 cells apoptosis by regulating ROS-mediated MAPKs and PI3K/AKT signaling pathways. Sci Rep. 2015;5:10336. | ||
Wang N, Feng YX, Zeng LL, Zhao ZN, Chen TF. Functionalized multiwalled carbon nanotubes as carriers of ruthenium complexes to antagonize cancer multidrug resistance and radioresistance. ACS Appl Mater Interfaces. 2015;7(27):14933–14945. | ||
Kumar A, Huo S, Zhang X, et al. Neuropilin-1-targeted gold nanoparticles enhance therapeutic efficacy of platinum(IV) drug for prostate cancer treatment. ACS Nano. 2014;8(5):4205–4220. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.