Back to Journals » Cancer Management and Research » Volume 13
Cellular Mechanism of Gene Mutations and Potential Therapeutic Targets in Ovarian Cancer
Authors Guo T, Dong X, Xie S, Zhang L, Zeng P, Zhang L
Received 25 November 2020
Accepted for publication 19 March 2021
Published 8 April 2021 Volume 2021:13 Pages 3081—3100
DOI https://doi.org/10.2147/CMAR.S292992
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Tao Guo,1 Xue Dong,2 Shanli Xie,3 Ling Zhang,4 Peibin Zeng,5 Lin Zhang6
1Department of Gynecology and Obstetrics, West China Second Hospital, Sichuan University, Chengdu, Sichuan, 610041, People’s Republic of China; 2Department of Gynecology, Cheng Du Shang Jin Nan Fu Hospital, West China Hospital, Sichuan University, Chengdu, Sichuan, 610041, People’s Republic of China; 3First People’s Hospital of Guangyuan, Guangyuan, Sichuan, 628000, People’s Republic of China; 4Department of Gynecology and Obstetrics, Guangyuan Central Hospital, Guangyuan, Sichuan, 628000, People’s Republic of China; 5West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan, 610041, People’s Republic of China; 6Department of Forensic Biology, West China School of Preclinical and Forensic Medicine, Sichuan University, Chengdu, Sichuan, 610041, People’s Republic of China
Correspondence: Peibin Zeng
West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan, 610041, People’s Republic of China
Email [email protected]
Lin Zhang
Department of Forensic Biology, West China School of Preclinical and Forensic Medicine, Sichuan University, Chengdu, Sichuan, 610041, People’s Republic of China
Email [email protected]
Abstract: Ovarian cancer is a common and complex malignancy with poor prognostic outcome. Most women with ovarian cancer are diagnosed with advanced stage disease due to a lack of effective detection strategies in the early stage. Traditional treatment with cytoreductive surgery and platinum-based combination chemotherapy has not significantly improved prognosis and 5-year survival rates are still extremely poor. Therefore, novel treatment strategies are needed to improve the treatment of ovarian cancer patients. Recent advances of next generation sequencing technologies have both confirmed previous known mutated genes and discovered novel candidate genes in ovarian cancer. In this review, we illustrate recent advances in identifying ovarian cancer gene mutations, including those of TP53, BRCA1/2, PIK3CA, and KRAS genes. In addition, we discuss advances in targeting therapies for ovarian cancer based on these mutated genes in ovarian cancer. Further, we associate between detection of mutation genes by liquid biopsy and the potential early diagnostic value in ovarian cancer.
Keywords: ovarian cancer, gene mutation, TP53, PIK3CA, BRCA1/2, KRAS, targeted therapy, liquids biopsy
Introduction
Ovarian cancer (OC) is well recognized as the most lethal gynecologic malignancy, with an estimated 295,414 newly diagnosed cases, resulting in 184,799 deaths in 2018 worldwide.1 Epithelial ovarian cancer is a heterogeneous disease comprising of five main subtypes including: high-grade serous ovarian carcinoma (HGSOC), low-grade serous ovarian carcinoma (LGSOC), endometrioid ovarian cancer (EnOC), ovarian clear cell carcinoma (OCCC), and mucinous. HGSOC is most commonly observed, accounting for approximately 70% among all cases.2 Sixty percent of patients with OC are diagnosed at an advanced stage because of asymptomatic status and limited screening marker, and the rate of 5-year overall survival is less than 30%.3 The commonly adopted strategy for OC primary treatment is surgical removal of the tumor, followed by consistent chemotherapy. It has been found that 20–30% of patients have no response to initial treatment or progress within 6 months after primary chemotherapy due to being insensitive to the chemotherapeutic drugs.4 Therefore, there is a crucial need to develop newer and more effective therapeutic regimens to overcome chemoresistance in metastatic or recurrent ovarian cancer and to achieve durable clinical prognosis. Research has revealed that most advanced patients expressed different genetic abnormalities. Those gene mutations will guide treatment decisions and novel effective chemotherapeutic agents that target these aberrant genes, to improve the poor prognosis in OC.
Liquid biopsy, based on minimally invasive and serial blood tests, has the advantage of following tumor evolution in real time, offering novel insights on precision medicine. The major components of liquid biopsy analysis involve circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), circulating cell-free microRNAs (miRNAs), and circulating exosomes.5 As a biomarker, it has already been implemented in ovarian cancer diagnosis, prognosis, and response to treatment. The aim of this review is to discuss the recent advances of TP53, BRCA1/2, PIK3CA, and KRAS gene mutations in ovarian cancer. In addition, the potential functional targeted therapy and the biomarker that might eventually be clinically actionable and applied in liquid biopsy are also reviewed.
Ovarian Cancer and Gene Mutation
Multiple studies have reported the significant association between gene mutations and clinical phenotype of cancers, implying the prospect to use the loci of gene mutations as prognosis and therapeutic targets.6–9 Four gene mutations are most commonly reported to be highly associated with epithelial OC, including: TP53, BRCA1/2, PIK3CA, and KRAS. The frequency of these mutations varies among different subtypes of epithelial OC (Table 1). The expression of P53 mutation is the most common mutation in HGSOC. The P53 mutation rate increases to 54.5% in HGSOC. BRCA1/2 genes are responsible for the majority of hereditary OC. The BRCA mutation rate increases to 40% in recurrent HGSOC. PIK3CA mutations have a high frequency in OCCC and the EnOC in relation to endometriosis. The KRAS mutation plays a key role in the LGSOC and mucinous OC. The potential mechanisms between the mutations and OC are described as: loss of function of genes regulating tumor suppression, abnormalities of DNA repair genes, apoptosis, gain in function of oncogenes, and epigenetic inactivation.10
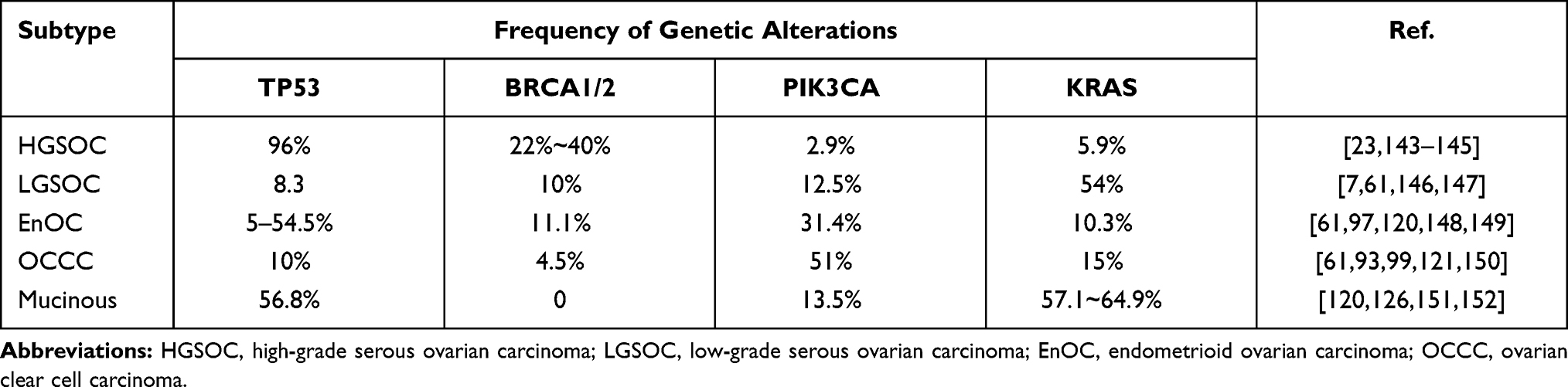 |
Table 1 The Frequency of the Four Gene Mutations Among Epithelial Ovarian Cancer |
Tumor Suppressor Gene TP53 in Ovarian Cancer
The TP53, located chromosome 17P13.1, is composed of 19,198 nucleotides spanning 11 exons.11 TP53, as a “the guardian of the genome” or “cellular gatekeeper”,12,13 plays an important roles in tumor suppression, by regulating the expression of downstream genes to induce a series of cellular responses, such as cell cycle arrest or apoptosis in different types of stress (eg, nutrient deprivation, telomere erosion, hypoxia, DNA damage, ribosomal stress, and oncogene activation)6,14 (Figure 1). In general, protein levels of p53 keep low or undetectable owing to its negative regulator MDM2, which binds to the amino terminus of p53 and targets p53 for proteasome-mediated degradation.6 DNA damage and stresses disrupt P53-MDM2 binding to increase p53 levels.15 Loss of its apoptosis activity can cause tumor development and chemoresistance.16
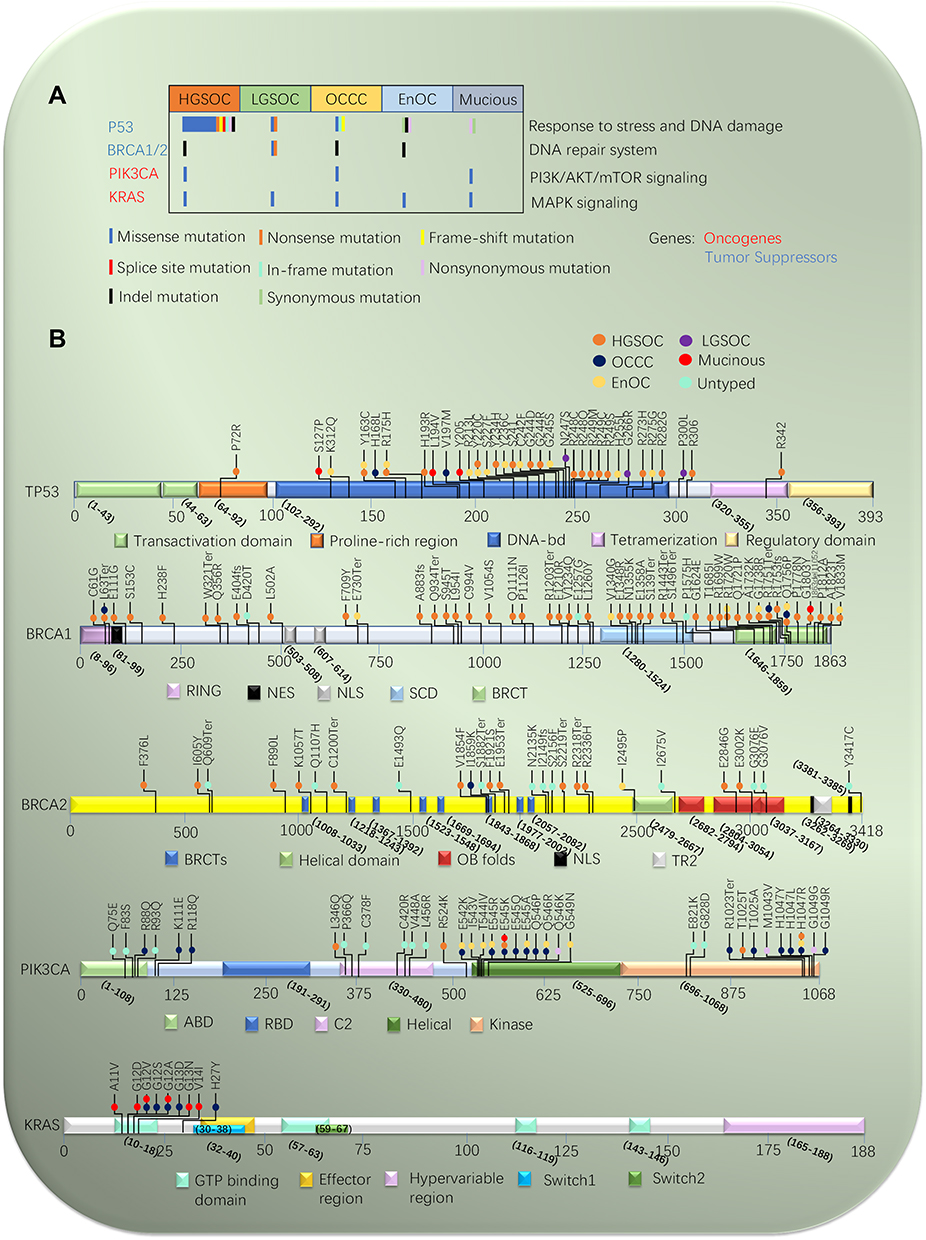 |
Figure 1 Mutational landscape of epithelial ovarian cancer. (A) Mutations in significantly mutated genes in epithelial ovarian cancer and selected known oncogenes and tumor suppressors. Genes mutations are shown in subtype of epithelial ovarian cancer. (B) Variants for P53, BRCA1/2, PIK3CA, and KRAS, color-coded by subtype of epithelial ovarian cancer. Splice site mutations are indicated as involving the acceptor site (exon – nucleotide position of mutation). |
P53 is a tetramer formed by four p53 molecules which self-assemble on two DNA half-sites. It contains three major functional domains, including N terminus contains a transactivation domain, C terminus consists of oligomerization and regulatory domains. The core domain contains a sequence-specific DNA binding domain.17 The next generation sequencing (NGS) has revealed that approximately 95% of the tumor-associated mutation is located in the core DNA-binding domain.18 P53 mutations lead to inactivity of wild-type (WT) P53 function, at the same time it can produce a new protein with new functions, which is called gain-of-function (GOF). These GOF P53 mutations result in mutation P53 accumulating at high levels in cells, contributing to carcinogenesis, multidrug resistance, poor prognosis, and metastasis.19,20 P53 mutated tumor cells are the absence of a functional G1-checkpoint and depend completely on their G2-checkpoint for cell cycle arrest and DNA repair.21
In OC, a recently whole-genome sequencing of DNA found mainly P53 mutation is missense mutation. The missense mutation occurs predominantly in exons 5–10.22 There have been some hotspots (R175, G245, R248, R249, R273, Y220 and R282) identified, and four (R273, R248, R175, and Y220) of those are the most frequent mutations. The most common codons of mutation are R273C, R273H, and R273L mutants.23 The R273 and R248 mutants seem more effective than the other mutants on the migration and invasion of OC14,24 (Figure 1B). In research on 245 primary OC patients, 68 revealed active function of p53 and 177 inactive function of p53, in all 177 patients with P53 mutations, 128 harbored missense, 30 frameshift, 11 nonsense, and only eight splice variants (Figure 1A). One hundred and thirty-four patients had p53 splice variants in 245 ovarian cancers and the mutations expressions were associated with the functional p53 status.25 Using NGS, more than 90% of HGSOC reveal expression of P53 mutation, which is associated with metastatic progression and resistance of chemotherapy.26 More remarkably, there was a high prevalence of the P53 mutations in stage 1 or 2 HGSOC.23 Interestingly, LGSOC, that have poor response to platinum-based chemotherapy, is more typically WT- P53 than HGSOC.27 However, the WT-P53 is found to be dysfunctional by indirectdegradation through several different mechanisms. Restoring the function of WT-P53 can inhibit tumor growth, but the effect of TP53 recovery on tumor growth seems to depend on the stage of cancer progression.18
Advances in New Strategies in Targeting P53 for Ovarian Cancer Treatment
Currently, most reports emphasize that mutations of P53 are associated with progression and prognosis of OC.23,28,29 Mutated p53 protein is an effective target for tumor-specific therapies because of its absence on normal tissue. Consequently, we focus on reviewing the attempts which have been made to exploit P53 as a target to treat ovarian cancer.
Restoring Mutant P53 to WT-P53 Function
Firstly, restoring function of p53 protein could make efficient eradication of tumors in liver carcinomas, sarcomas, and OC.30 Several small molecules have been considered to be able to reverse the oncogenic function of mutant P53. P53 re-activation and induction of massive apoptosis (PRIMA-1, also known as APR-017) and PRIMA-1Met (also known as APR-246) are widely studied, and have been confirmed to induce apoptosis and inhibit tumor growth by refolding and restoring of WT P53 function31,32 (Figure 2). They can rescue P53 function by chemical modifications or ligand binding to stabilizing the active conformation. Meanwhile, in vitro, both APR-017 and APR-246 can inhibit the growth of mutant P53-expressing malignant cells. The WT-P53 downstream target genes could be changed by APR-017 and APR-246, including p21, Noxa, Puma, GAD45, specific caspases (caspase 2, 3 and 9), and MDM2. APR-246 is the only molecule that has been tested in clinical phase. It is also found to be well tolerated with little toxicity; the most commonly adverse effects were dizziness, headache, fatigue, and confusion. Currently, APR-246 is undergoing a Phase I/II clinical trial in recurrent HGSOC patients (Clinical-Trials.gov Identifier: NCT02098343). ReACP53, as a cell-penetrating peptide, has also been designed to inhibit p53 amyloid formation and to rescue TP53 function. ReACP53 can mask the segment 252–258 in mutant P53 aggregates, preventing further aggregation and shifting the equilibrium toward the WT-P53 function. In vitro and vivo, ReACP53 is effective in treating two of the three most common P53 hotspot mutations in HGSOC (R175 and R248) because of the aggregation-prone mutations. Besides, some studies suggested ReACP53 combined with carboplatin therapy may be a valuable therapeutic option for HGSOC.33 CDB3, a small molecules dived from p53-binding protein 2, transactivated P53 target genes MDM2 and P21 through restoring the transcriptional activity of R273H and R175H. There are many small molecules that have been revealed to reactivate and restore the activity of WT-P53, including CP-31398, WR-1065, and P53R3.6 Research has shown that suppression of P13K/mTOR signaling may activate and potentiate P53 function through inducting apoptotic cell death. Therefore, either activation of P53 or inhibition of P13K/mTOR signaling can be recognized as promising molecular-targeted therapies.34 Some other molecules have been indicated to reactivate WT-P53 in other tumor types, such as PK7088, MIRA-3, STIMA-1, CP31398, and SCH529074, which may also have potential applications in ovarian cancer.33
 |
Figure 2 Cellular functions of P53 in ovarian cancer. P53 is activated to regulate the expression downstream genes to induce a series of cellular responses, such as cell cycle arrest, apoptosis, senescence, autophagy, DNA repair, angiogenesis, migration and metabolism in different types of extracellular and intracellular stress (eg, nutrient deprivation, telomere erosion, hypoxia, DNA damage, ribosomal stress, and oncogene activation). The primary treatment strategy for patients with ovarian cancer with P53 mutations is focused on restoring WT-P53 function to mutant P53, Blocking the interaction of WT P53 with MDM2/MDM4, and gene therapy with P53. |
Blocking the Interaction of WT-P53 with MDM2/MDM4
Not all of tumors contain P53 mutations. In some tumors that retain WT-P53 function, the retained protein can lose function by interaction with MDM2/MDM4. So, blocking the interaction of WT-P53 with MDM2/MDM4 is a worthy strategy for targeting P53. MDM2 and MDM4 are major negative regulators of P53 and tightly control P53 activity.13 MDM2/MDM4, which are the downstream target genes, precisely block P53 degradation by preventing the binding of WT-P53. Especially MDM2, which is a transcriptional target and a negative regulator of P53,35 is phosphorylated and activated by AKT. The MDM2 inhibitor can restore P53 function or activate the expression of P53-dependent DNA repair genes to lead to tumor growth inhibition and induct apoptosis.35,36 On the contrary, P53 can stimulate expression of MDM2 and MDM4 mRNA. MDM2 and MDM4 can also inhibit P53 by inhibiting stability and activity and negating P53 transcriptional activity of P53, respectively. Remarkably, cancers often take advantage of this dual action of MDM2 and MDM4 on P53 to stimulate their growth.37 Hence, targeting the P53-MDM2-MDM4 loop is an efficient strategy for ovarian cancer therapy with mutation P53. Here are several small molecule compounds and stapled peptides which can inhibit the interaction between P53 and MDM2 or P53 and MDM4. Currently, the most widely investigated low molecular weight compounds are the nutlins. The nutlins are the cis-imidazoline group of molecules, its function of blocking the interaction between P53 and MDM2 is to mimicking the three critical amino acid residue (Phe19,Trp23, and Leu26) to involve in the connection of P53 to MDM2’s N-terminal end.38 Indeed, P53 can be replaced from MDM2 with nanomolar potencies by specific nutlins, such as nutlin3a.39 Nutlin-3, an analog of the nutlin series, acts as an activator of WT-P53 to induce P53 levels, activate P53 transcription activity, and sensitize WT-P53 OC cell lines to cisplatin. RG7388 is a second-generation MDM2 inhibitor with superior potency to repress the MDM2-P53 interaction and activate the P53 pathway. Nutlin-3/RG7388 synergizes with cisplatin in OC cell lines with WT-P53.6,36 HLI98, MPD, and MEL23/24, small compounds, restrain the E3 ubiquitin ligase to prevent P53 degradation.40,41 JNJ-26854165, TDP521252, TDP665759, PXN727, and PXN822, small molecules, inhibit the interaction of the MDM2-P53 complex to prevent P53 degradation.42,43 Additionally, small stapled peptides can prevent both MDM2 and MDM4 to interact with P53, such as SAH-P53-8, ATSP-704, and PMI. At present, stapled peptides targeting P53 have not been tested in clinical trials. However, MDM2/MDM4 antagonists may have some possible side-effects on inducing inappropriate apoptosis and cell death, because of accumulating WT P53 in normal cells. Another further concern is that if mutant P53 is to appear in any premalignant lesions, stabilizing P53 may increase the possibility of progression to invasive cancer.
Gene Therapy with Wild-Type P53
The retroviral P53 expression vector which is the earliest strategy for P53 targeted therapy is gene therapy. Then, because retroviruses were integrated into the host genome and have been shown to induce malignancy, further trials with retrovirals were abandoned. With the advantage that they do not integrate into the host genome, replication-defective adenoviral vectors are broadly used in gene therapy with P53, especially adenovirus (AD) P53.38,44 Recent studies have suggested that cross-reacting material 197 (CRM197), a mutant form of the diphtheria toxin with weak toxicity, exerted an experimental antitumor effect on OC. In the OC research, a novel gene therapy of AD-mediated CRM197 (ADCRM197) demonstrated that A2780 cells (P53 wild-type cells) were sensitive to ADCRM197 and SKOV3 cells (P53 deletion cells) were resistant to it. But A2780 with knockdown P53 became resistant to ADCRM197 and SKOV3 with restoring P53 became sensitive to ADCRM197. Excitingly, a combination of ADCRM197 and AD-P53 may effectively overcome the resistance of P53-deficient OC.45 In addition, the research found that the expression of P53 upregulated modulator of apoptosis (PUMA), a potent pro-apoptosis protein that was obviously repressed in the TAX(Taxol)-resistant ovarian cancer cell line SKOV3/TAX. Nevertheless, AD-P53 infection upregulated and restored the expression of PUMA and re-sensitized the resistant ovarian cancer cells to TAX.46 The AD type 12 E1B 55-kilodalton oncoprotein tightly associates with P53, and accelerates P53-mediated apoptosis response of OC to cisplatin.47
Recently, a novel gene-editing technology, commonly named CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9, has been used to establish a cancer model.48 CRISPR/Cas9 leads to a rapid expansion of the biomedical field. CRISPR spacers direct the system to the target and Cas9 protein controls spacer acquisition and defense. This system can remove or correct gene mutations that promote the progression of cancer and can be confirmed as of great value in the therapy of genetic disorders through directly editing disease-related mutations.49 Several studies were really successful to use CRISPR/Cas9 to modulate disease-causing alleles in animal models and induced pluripotent stem cell, providing a new strategy for therapeutic genome editing in the clinics.50 A latest research using CRISPR/Cas9 gene editing generated sublines of ID8 (the most widely-used transplantable model of ovarian cancer) bearing loss-of-function of P53, and indicated that these changed tumor growth in the peritoneal cavity.51 Besides, they also suggested that the tumor microenvironment can be changed by single gene mutations. The loss of P53 greatly increases the expression of CCL2 and immunosuppressive myeloid populations within solid tumor and ascites. The expression of CCL2, a critical chemokine for attraction of monocyte populations, can be suppressed by wildtype P53 direct binding to the CCL2 5´UTR. On the whole, using the CRISPR/Csa9 gene editing tool, we can clearly understand the function of a single gene mutation or multiple gene mutation in OC to provide proof for developing an advanced therapeutic strategy.
The clinical trials targeting P53 are still in an early stage (Table 2). Many of the above-mentioned P53 target therapies were conducted in other tumors, and we should pay more attention to P53 of mutation as a top priority target for anticancer therapy in OC. Some of these drugs which are being tested will show significant efficacy.
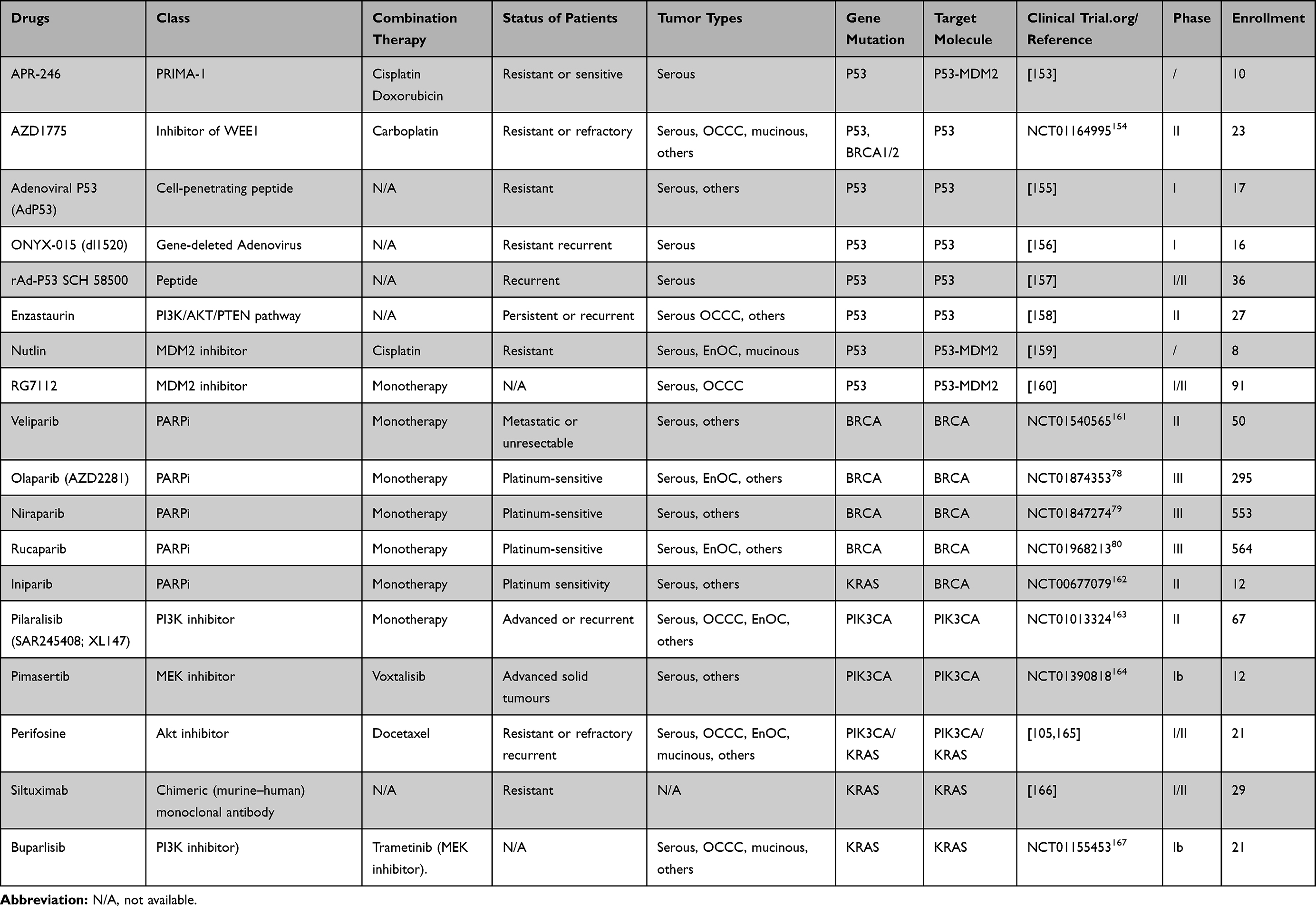 |
Table 2 Trials Using Target Agents on Gene Mutation in Ovarian Cancer |
BRCA1/2 Gene
BRCA1/2 genes locate on chromosomes 17 (17q21) and 13 (13q12.3), as tumor suppressor genes, which play an important role in regulating the cell cycle and DNA repair system.52 BRCA1 is a pleiotropic DNA damage response protein with checkpoint activation, DNA repair, and is involved in pro-survival and apoptotic pathways. BRCA2 is a mediator of the core mechanism of homologous recombination. Both BRCA genes have distinctive primary sequences. BRCA mutation leads to similar pathophysiological effects and cancer spectra and to increased cancer predisposition.53,54 For large regions of the human genome, the mutation in different regions are associated with different types of malignancies. A previous study shows that the 3ʹ region mutation of BRCA1 is related to a lower risk of OC, while mutation in the 3ʹ region downstream is related to a higher risk.4,55
In recent years, studies of BRCA mutation have been carried out all over the world. Germline and somatic BRCA mutations were detected in plasma ctDNA of OC by using NGS technology. The indels mutation is the most common in the BRCA genes in OCs (Figure 1A), pathogenic germline variants of BRCA1/2 in patients of OC are described in Figure 1B. In a study of eastern England, approximately 8% of HGSOC and EnOC were identified with mutation in BRCA1/2, and the prevalence increased to 12% in patients diagnosed under the age of 70 years, but fell to 1% in those aged over 70 years.56 In a Scottish study, the prevalence of pathogenic BRCA1/2 mutation among non-mucinous epithelial OC fell from 13.1% to 8.2% in patients diagnosed over the age of 70 years.57 In a study across the North West of England, the prevalence of BRCA1/2 mutation in epithelial OC by testing germline DNA exceeded 10%, and was consistently over 10% in patients diagnosed under the age of 60 years and over the age of 60 years with either breast and/or OC family history.58 In a Europe series approximately 20% of epithelial OC were shown to a mutation of BRCA1/2, and the prevalence increased to 31.9% in women with a family history of breast or ovarian cancer, but fell to 10.6% in women diagnosed over 60 years old.59 So, the age at diagnosis, family history of breast and/or OC, breast cancer history or a Manchester BRCA Score of ≥15 points are related to a >10% prevalence of BRCA1/2 mutation in epithelial OC.58 Another newstudy assessed the frequency and predictors of BRCA1/2 mutation by using NGS in HGSOC in Serbia.60 Factors that predicted BRCA1/2 mutations included breast and OC in the same patient, age of epithelial OC, menstrual status, and family history of cancer. Family history of breast or OC diagnosed <50 years among first/second-degree relatives was the most significant factor associated with BRCA1/2 in HGSOC patients. Moreover, it indicated a negative family history will not safely exclude all germline BRCA1/2 mutations and that more than 10% of BRCA1/2 mutation carriers would not be identified. Using those to predict the appropriate risk, then BRCA mutation testing is important to assess the strategy of treatment and prognosis for epithelial OC patients.
A great number of studies reported that BRCA1 mutation displayed lower BRCA1 and higher BRCA2 expression. Low BRCA1-expression showed a favorable overall survival in OC. The expression of BRCA2 was associated with poor tumor differentiation as it increases with tumor grade. In contrast to patients with no residual disease, the expression of BRCA2 is higher in patients with any residual disease. Low expression of BRCA1/2 in OC reduces DNA damage repair ability via homologous recombination to result in a better response to platinum-based chemotherapy and Poly (ADP-ribose) polymerase (PARP) inhibition.61 The patients of OC with mutation of BRCA2 are particularly sensitive to platinum. Therefore, the platinum-based chemotherapeutic regimens are a widely recommended treatment in BRCA-related OC. However, other studies revealed that some BRCA-related OC with previous platinum-sensitivity can become platinum resistant, owing to a reversion of the BRCA mutation by secondary intragenic mediating.62,63 Currently, as an encouraging but complex research field in targeted therapy for OC, PARP inhibitors are in a variety of clinical testing as part of Phase I, II, or III study. The present opinions of PARP inhibitors are discussed later.
Finally, the prognosis of BRCA-related OC is optimistic. BRCA mutated OC have a better prognostic outcome and higher chemotherapy sensitivity than those cancers without BRCA dysfunction.64 A retrospective study showed that OC patients with BRCA2 mutation appeared to have higher progression-free survival rates than ovarian cancer patients with a BCRA1 mutation or without BRCA-related dysfunction.65 But the controversial conclusion needs to be further explored.
Advances in BRCA1/2 Gene-Targeted Therapy
The DNA repair system keeps the genetic stability, and its deficiency may lead to diseases generating, including cancer. PARP, a family of nuclear proteins, is a key pathway in DNA repair. In normal circumstances, single-strand breaks (SSBs) occur and repair with subsequent formation of double-strand breaks (DSBs). Then the primary function of PARP is to detect SSBs, recruit DNA repair proteins, and promote the DNA repair system based on ADP-ribosylation, which typically requires the reaction with nicotinamide adenine dinucleotide (NAD+) and then release of nicotinamide.66 Inhibition of the PARP enzyme results in persistence of spontaneously occurring SSBs. The SSBs stall and collapse replication forks, which subsequently causes the formation of DSBs.67 In addition, PARP inhibitors (PARPi) can trap PAPR-1 and PARP-2 enzymes on damaged DNA to form PARP-DNA complexes.68 Then, the DNA duplication may be disturbed by these newly-generated complexes, which could result from the loss of PARP activity and further promote sustained DNA damage.68,69 Preclinical data suggested that the trapped PARP-DNA complexes have stronger cytotoxicity than the unrepaired SSBs. On the other hand, DSBs can be repaired by an homologous recombination (HR) pathway or nonhomologous end joining (NHEJ) pathway.70 HR deficiency could be induced by genetic abnormalities, such as BRCA1/2, PALB2, ATM/ATR, Fanconi anemia gene and so on.71 In the presence of a mutated BRCA gene, DSBs are repaired through a less effective, error-prone pathway such as single-strand annealing and/or non-homologous end joining, which can lead to genomic instability, cell cycle arrest, and subsequent apoptosis. The absence of PARP activity does not completely repair SSBs, leading to an increase in deleterious DSB, thereby preventing BRCA1/2 mutations or HR cells from being effectively repaired. Therefore, loss of function in the repair genes BRCA1/2 and PARP result in cell death, a concept called synthetic lethality72,73 (Figure 3).
 |
Figure 3 The mechanisms of PARP inhibitors in BRCA-related ovarian cancer. PARP-1 mediates the repair of SSBs through the activation and recruitment of repair enzymes. Counterclockwise: activated PARP-1 detects damaged SSB in DNA and binds to adjacent DNA. Once bound, PARP-1 catalyzes the cleavage of the coenzyme nicotinamide adenine dinucleotide (NAD +) to nicotinamide and ADP-ribose to produce a highly charged branch of high poly (ADP-ribose) (PAR). Repair proteins are recruited to the site of injury to repair damaged DNA. After finishing repair, the PAR chain is degraded by PAR glycohydrolase (PARG). BRCA1/2 genes, located on chromosomes 17 (17q21) and 13 (13q12.3), play an important role in regulating cell cycle and the DNA repair system. The mutated BRCA gene loses its function of repairing DNA, and PARP inhibitors also inhibit the repair of DNA by PARP, thereby promoting tumor cell death. |
The defective HR pathway in BRCA-mutated cells is associated with OC tumorigenesis, due to DNA DSBs failure to repair. So, previous study investigated PARPi, such as Olaparib (AZD2281) which induces synthetic lethality in homozygous BRCA-mutated cells, had anti-cancer activity in germline BRCA mutated cancer.74 PARPi also have the same anticancer activity in HGSOC patients without germline BRCA1/2 mutations due to other DNA repair pathways that may be present. The NHEJ pathway, another DNA repair pathway and less accurate, plays a crucial role in DSBs repair when HR defected, which may lead to an increased risk of genetic instability.67,75
As an anticancer agent, there are already five different PARPi tested in OC patients or undergoing clinical testing, such as Olaparib (AZD2281), Veliparib (ABT-888), Niraparib (MK4827), Rucaparib (CO338, AG014699, and PF01367338), and BMN 673.67,76 Recently, the FDA has approved Olaparib monotherapy for the treatment of BRCA-mutated advanced OC patients as maintenance for HGSOC with platinum-sensitivity.77,78 Recently, the pivotal Phase III NOVA trial (NCT 01847274) indicated Niraparib had unprecedented results as maintenance therapy for platinum-sensitive recurrent OC in with BRCA and non-BRCA.79 The Phase III ARIEL2 and ARIEL3 trails suggested that Rucaparib monotherapy can be used for the treatment of BRCA-mutated advanced OC and maintenance treatment for recurrent OC after response to platinum therapy. The study has shown that Rucaparib maintenance treatment significantly improved progression-free survival in those patients.80
A large number of preclinical studies demonstrated the synthetic lethal theory, which is a phenomenon to combine PARPi with other biologic agents, such as anti-angiogenic agents and PI3-kinase (PI3K) inhibitors.81 Antigenic agents can lead to cell hypoxia, which can enhance PARPi sensitivity. The combined effect of anti-angiogenic agents and PARPi can downregulate the homologous recombination repair protein.82 However, PARPi are regarded as a new class of targeted agents in OC treatment. There are still several challenges for the clinical development. PARPi are not only well tolerated, but also have some adverse events and acquired resistance. The adverse events include nausea, fatigue, vomiting, and anemia. More than that, PARPi may lead to myelodysplastic syndrome and acute myeloid leukemia. The newresearch indicated a key resistance mechanism to PARPi in BRCA-mutant OC is the acquisition of BRCA reversion mutation detected by sequencing of cell-free DNA (cfDNA). BRCA reversion mutations are related to poor response to platin-based therapy and PARPi therapy.83,84 The BRCA reversion mutations were also assessed by using targeted massively parallel sequencing in ctDNA samples of PARPi resistant and/or refractory ovarian cancers. Putative BRCA1 or BRCA2 somatic reversion mutations or intragenic deletions were detected in 21% of ctDNA samples from patients, and further confirmed these results using droplet digital PCR (ddPCR).85 Another potential mechanism of PARPi and platinum resistance for BRCA1-mutant is an increased expression of a BRCA1-Δ11q splice variant that lacks the majority of exon 11 but still has residual BRCA1 activity. Inhibition of the spliceosome reduces BRCA1-Δ11q levels and sensitizes exon 11–mutant cell lines to PARPi.86 This is a great challenge for when and how PAPRi agents should be incorporated in the management of OC. Further studies for a better understanding of the potential toxicity and drug interactions are warranted.
A study on BRCA mutated breast cancer of the mouse model showed that the combination of PARPi and BKM120 (an oral PI3K inhibitor) has a synergistic effect, resulting in delayed tumor cell doubling compared with either agent alone.87 As has been mentioned before, PI3/AKT/mTOR pathway inhibitors combined with PARPi also have the synthetic lethal in the treatment of OC. Moreover, FDA trails combined PARPi and PI3K inhibitors in OC patients are ongoing (including [NCT02338622] and [NCT01623349]) (Table 2).
PIK3CA Gene
The PIK3CA gene is located at the chromosomes 3 (3q26.3) and encodes the p110α catalytic subunit of the PI3K. The PI3K pathway is a family of lipid kinases in the early stages of a signaling cascade, which is frequently altered in cancer.88,89 The overexpression of mutation PIK3CA activates its downstream effector AKT that leads to increased activity of mTOR, promoting cell survival, proliferation, oncogenic transformation, and suppressing apoptosis.88,90,91 The PI3K/AKT plays a central role in glucose metabolism, and mTOR is a serine/threonine kinase which acts as an effector in the PI3K/AKT pathway.89,92 The PIK3CA mutation clearly identified as mechanisms of inducing oncogenic PI3K signaling (Figure 4).
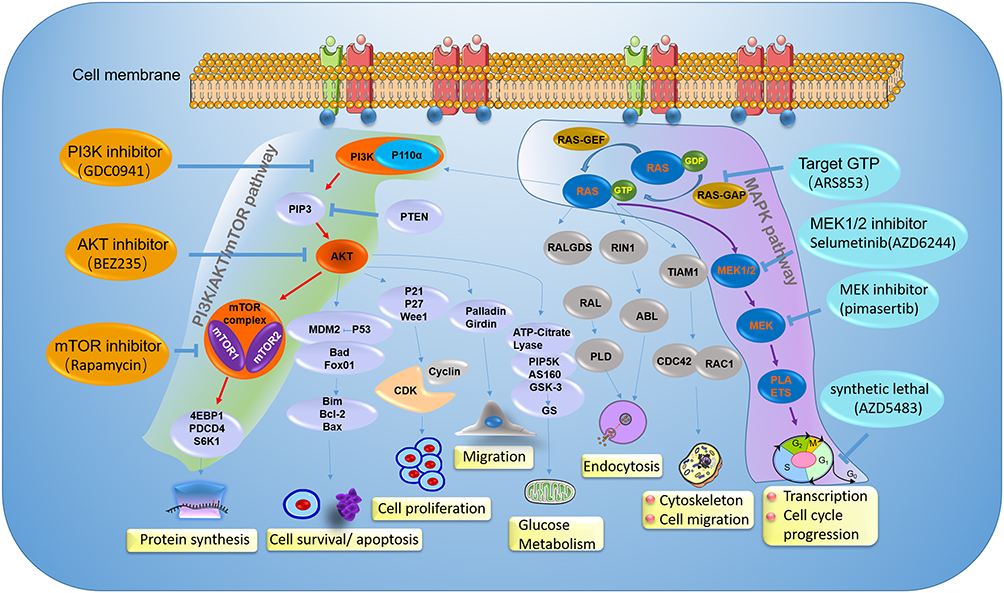 |
Figure 4 The mechanisms of PI3K/Akt/mTOR pathway and MAPK pathway, and inhibitors in ovarian cancer clinical development. Illustration the therapy strategy via inhibiting the PI3K/Akt/mTOR pathway (green) and MAPK pathway (amaranth) in ovarian cancer patients with PIK3CA and KRAS gene mutation. The orange represents a different inhibitory effect of repressing tumor growth by targeting different sites on the PI3K/Akt/mTOR pathway. For patients with PIK3CA gene mutation, clinical treatment drugs are mainly divided into PI3K inhibitor, AKT inhibitor, and mTOR inhibitor. The blue represents a different inhibitor target MAPK pathway in ovarian cancer patients with KRAS gene mutation. The therapy strategy includes restricting KRAS bound to GTP and targeting its downstream signaling pathway. |
The PIK3CA gene results in somatic mutations in a majority of human cancer, including OC. The research evidence revealed that the PI3K/AKT signaling was deregulated in a significant fraction of OC and associated with a poor survival rate. The NGS technology has revealed that most of the PIK3CA mutations were confined to exons 9 and 20.93,94 The most common codons of mutation are H1047R, E545G, E545GK, and E545A mutants (Figure 1B). Interestingly, PIK3CA mutations are much more prevalent in the rare subtypes of OC. The previous reports showed activating PIK3CA mutations was high frequency in OCCC and EnOC in relation to endometriosis95,96 (Table 1). A research suggested 10 of 11 endometriosis-associated EnOC had PIK3CA mutations in exon 9 and eight of 10 OCCC in exon 20.97 Oncogenic mutations are rare, with only 2.9% in HGSOC, while oncogenic amplifications in PIK3CA occur in 25% cases. The researcher examined the results of a multiplatform profiling panel, such as DNA sequencing, immunohistochemistry, fluorescent or chromogenic in situ hybridization, and RNA fragment analysis, confirming that the PIK3CA/Akt/mTOR pathway was altered in 61% OCCC.98 Whole-genome sequencing was performed in 55 Japanese women diagnosed with OCCC. Twenty-three cases had alteration in these genes, including mutations of PIK3CA (35%), PIK3R1 (7%), and PTEN (2%), and amplifications of PIK3R2 (5%), AKT1 (4%), and AKT2 (9%).99 These differences between OC subtypes suggested that subtype-specific treatment strategies might be needed to improve OC outcomes. Some studies suggested the mutation of PIK3CA was considered an early event in the transformation of endometriosis into OCCC.96 The higher frequent expression of PIK3CA mutation in OCCC is regarded as its specific biological behavior with foci of endometriosis.98,100–102
The PI3K/Akt pathway has been reported as a collaboration with other gene expression in tumorigenesis. A study showed that mutations of PIK3CA were detected in 40% (17/42) of OCCC and a majority (71%) of these were found in ARID1A-deficient (which encodes a member of the SWI/SNF family protein BAF250a) carcinomas.103 Remarkable, a study suggested that P53 suppresses PIK3CA transcription through the direct junction with its promoter in ovarian surface epithelial cells. Intriguingly, this study revealed that cisplatin simultaneously attenuated PIK3CA expression and activated P53 expression in sensitive tumors but not in the resistant tumors, which only expressed a low level of P53 activation. But the precise principle of P53-PIK3CA remains elusive at molecular level.90
Advances in PIK3CA Gene-Targeted Therapy
PI3K/AKT/mTOR signal pathway, an important signaling pathway in cell proliferation and survival, is associated with human tumorigenesis. The pathway inhibitors activity is observed in more than 70% of OCs.104 PIK3CA mutations have been shown to initiate in mice of OCs, and inhibition of the PI3K/AKT/mTOR pathway was found to delay tumor growth and prolong survival. Targeting the PI3K/AKT/mTOR signaling pathway has been widely utilized in clinical studies. A series number of clinical trials demonstrated that these pathway inhibitors were a candidate for OC therapeutics (including AKT inhibitor perifosine, mTORC1 inhibitor temsirolimus).105,106 OCCCs and the EnOCs are two of five classified subtypes, and approximately 20% in epithelial OC. The relatively high frequency of PIK3CA mutations in OCCCs and the EnOCs hold promise for novel therapeutic strategies through targeting PI3K or the pathway including PI3K by some inhibitors (Figure 4).
Rapamycin and its analogs (temsirolimus, everolimus, and ridaforolimus) resulted in activation of proliferative and pro-survival effectors such as AKT by stopping a mTORC1-dependent negative feedback loop on PI3K signaling from the p70S6K to the PI3K signaling pathway.89 The mechanism of Rapamycin analogs is bound to the mTORC1 complex but does not affect mTORC2. The mTORC2 is a positive regulator of Akt, which could selectively inhibit mTORC1 results in Akt phosphorylation at serine. Dual mTORC1/mTORC2 inhibitors, such as DS3078a, INK128, AZD8055, OSI027, and AZD2014, have been developed to completely eliminate the entire mTOR complex. This may achieve a powerful antitumor effect.107 Rapamycin, the first inhibitor of the pathway to enter the clinic, acts as a mTOR inhibitor and has been investigated as its activity in a variety of tumor types, including OC. Temsirolimus failed to meet the efficacy endpoint in a Phase II trial of OC treatment. The trial included only a few OCCCs (three of 54) and EnOCs (four of 54). The only one OCCC observed the objective partial response.108 Meanwhile, other encouraging data showed that one of five OCCCs observed objective response for 14 months and one stable disease.109 Although the data could not draw a persuasive conclusion, it can provide a promising strategy for OCCCs targeting therapy.
Pictilisib (GDC0941) is a potent, selective inhibitor of class I PI3K, which has shown clinical activity in a Phase I trial of ovarian cancers. Besides, Pictilisib achieved 80% growth inhibition in PI3K pathway activated OC.110 A dose-escalation phase I trial of BKM120, another oral PI3K inhibitor, has shown promising results with well-tolerated dosing and good tumor control. The BKM120 has been carried forward into phase II trials with endometrial cancer, nonsmall-cell lung cancer (NSCLC), prostate cancer, breast cancer, thyroid cancers, and recurrent glioblastoma (NCT01953445, NCT01790932, NCT01339052).87
Similarly, other PI3K inhibitors, such as PX-866, copanlisib (BAY80-6946), and XL147 (SAR245408), are still ongoing clinical evaluation for different tumors including ovarian cancer. The combination of PI3K inhibitor PX-866 and docetaxel in the treatment of OC was conducted. The combination plan was well tolerated and had no cumulative toxicity.111 The effect of PX-866 combination with carboplatin to reduce proliferation and survival of OC cells was enhanced by Ca2+/calmodulin (CaM)-dependent protein kinase kinase2 (β)(CaMKK2) silencing.112 First-in-human phase I study of copanlisib, a novel, intravenous, potent, highly selective, pan-class I PI3K inhibitor, has demonstrated potent antitumor and pro-apoptotic activity in patients with advance solid tumor, including OC.113 Other PI3K inhibitors have not adopted in clinical trials of OC, but these promising outcomes in observed malignancies may indicate that the PI3K inhibitors have the potential application in the treatment of OC patients with PIK3CA mutation.
BEZ235 is an imidazoquinoline derivative that suppresses expression of AKT and competes at its ATP-binding site to inhibit the four class I PI3K isoforms and the downstream effectors mTORC1/2. In breast cancer cell lines and mouse xenograft models, BEZ235 has shown activity. In addition, some trials indicated BEZ235 reduces tumor growth in OC, but the tumor sensitivity of BEZ235 was independent of the PIK3CA mutation status in the subtype of OC.89 The GSK2141795 is a potent, oral, adenosine triphosphate–competitive pan-kinase inhibitor. The study found GSK2141795 inhibited expression of AKT to cause growth arrest as alone agent, enhanced cisplatin-induced apoptosis, and reduced tumor volume in combination with platinum in platinum-resistant OC with PIK3CA mutation.114 These agents may serve as an effective agent in further clinical trials of OC (Table 2).
KRAS Gene Mutations
KRAS locates on chromosome12p12 and encodes a 21-KD protein (p21RAS) which involves in MAP-kinase signal transduction pathway. As a member of the Ras gene family and an important oncogene, the KRAS gene plays an essential role in cellular proliferation, apoptosis, and carcinogenesis. KRAS mutations promote tumorigenesis and result in uncontrolled proliferation and differentiation of cells by activating the MAPK/ERK pathway which is triggered by MEK, MAPK/ERK-kinase.7,8 The KRAS mutations are the most common RAS isoforms, including KRas4A and KRas4B, which are encoded by alternative fourth exons and the common activating mutations occur in exons 1 or 2.115,116 The inherited variant which is located in the 3ʹUTR of KRAS gene (rs61764370 T > G) is associated with an increased risk of OC, breast cancer, and lung cancer.117
KRAS mutation plays a key role in LGSOC and mucinous OC subtypes. The sequencing analysis showed KRAS gene mutation was the most frequent in borderline serous tumor, LGSOC, and mucinous carcinomas. KRAS mutation rate was reported as 33~41%, 35~54%, and 57.1%, respectively, in contrast with their low or absent expression in HGSOC (Table 1).7,118 The mutations of KRAS were even more than 70% in the recurrent LGSOC. The frequent occurrence of KRAS mutations in this subtype of tumors led to the presumption that the development of LGSOC begins in a stepwise mode from serous cystadenoma or adenofibroma, borderline serous tumor, and serous carcinoma, and its carcinogenic processes are closely related to RAS signaling.7 KRAS mutation in borderline serous OC may be associated with peritoneal implant.119
In research of 142 patients of primary epithelial OC without borderline tumor detected KRAS in exon 2 and 3, 9.9% KRAS mutations were observed, 13 mutations in exon 2 and only one in exon 3. In those mutations, six patients included both P53 and KRAS mutations, and all 14 KRAS mutations were missense mutations to lead to an exchange of the coding amino-acid.120 Another recent study of 15 Korean patients with OCCC detected 20% KRAS mutations.121 In OCs, KRAS mutations occur mostly on codon 12, the most common being the G12V point mutation, followed by G12D and G12S. A study on 63 Rome patients with OCCC also detected 13% KRAS mutations, and the results showed a higher incidence in codon 12 mutations (90%), G12V (43%), G12D (29%), G12S (14%), and G12A (14%), respectively. There is only one mutation at G13D of codon 1393 (Figure 1B).
Mucinous OCs begin in slow stepwise fashion: a mucinous adenoma progresses to mucinous borderline tumor to mucinous carcinoma.122 In mucinous OCs, KRAS mutations occur frequently, which is more frequently than non-mucinous OCs.123 Besides, the KARS mutations, as a tumorigenic, have been detected in adjacent mucinous cystadenoma and mucinous borderline tumor areas of mucinous carcinoma.124 This may lead to the formation of ovarian borderline mucinous cystadenoma, but not advance the result to cystadenocarcinoma.125 The direct sequencing method was used to analyze the mutation of KRAS exon 2, codons 11–14 in ovarian tissues. In normal ovarian tissue, there was no detected KRAS mutation. While, in mucinous neoplastic tissues, codon 12 mutations were detected about 53.45%, codon 13 mutations were detected about 24.14%, and codon 14 mutations were detected about 3.45%. Besides, previous studies have indicated that codon 12 was the most ordinary KRAS mutation in mucinous borderline tumor and mucinous carcinoma, codon 13 was the most ordinary KRAS mutation in mucinous adenoma.123,126 In general, KRAS mutations were associated with better differentiated carcinomas in all types and not related with the patient overall survival.120
Advances in KRAS Gene-Targeted Therapy
Currently, no therapies that directly target KRAS oncoprotein are available in the clinic because of the high affinity for GTP.127 Previously, KRAS has been known as difficult to target for cancer treatment. Recently, the detection of KRAS mutations in numerous tumors has led to the development of new therapeutic agents that aimed to either directly inhibit mutated-KRAS, target its downstream signaling pathway, or exploit synthetic lethality partners of mutant KRAS128,129 (Figure 4).
Targeting KRAS Mutations
In studies on direct inhibition of KRAS activity, strategies of directly restricting and binding KRAS to its functional domains have been proposed. ARS853 could specifically target combination to the G12C mutant of KRAS, significantly restricting the binding of KRAS to GTP, then reducing the phosphorylation level of KRAS and inhibiting the interaction between KRAS and downstream signaling molecules.130 Meanwhile, the exosomes secreted by normal fibroblast-like mesenchymal cells are engineered and encapsulated with siRNA or shRNA for delivery of KRAS G12D mutants. By targeting wild-type KRAS, the growth of lung cancer and colorectal cancer can be significantly inhibited. This provides a reliable method for direct targeting therapy of KRAS mutant tumors.131,132
Targeting Downstream Signaling Pathway of KRAS Gene
The KRAS related downstream signaling contains the RAF-MEK-MEK pathway. Besides, KRAS mutated tumor cells are often accompanied with other signaling molecules mutations, such as PIK3CA, PTEN, P53, which play a key role in tumorigenesis. Selumetinib (AZD6244) is the second generation of MEK1/2 inhibitors, which can specifically inhibit the phosphorylation of ERK1/2, a direct substrate of MEK1/2, thereby inhibiting cell growth.133 A study of 15 patients with epithelial OC genotype-matched in phase I or II trials, including 14 patients with KRAS mutation treatment in combination to selumetinib, suggested seven partial responses, seven with stable disease and one with disease progression.134 Moreover, the higher sensitivity to MEK inhibitors was observed in the OC patients with KRAS mutation. A phase II trial of Selumetinib suggested that the response rate for recurrent low-grade serous cancer was 15%.135
Synthetic Lethal Strategy
Synthetic lethal strategy, which is raised to inhibit both downstream active pathway and feedback regulation pathway of KRAS, so as to achieve the therapeutic effect of inhibiting tumor cell growth. Cyclin dependent kinase 1(CDK1), such as AZD5483, has a synthetic lethal effect on KRAS-mutated tumor by blocking cells at G0/G1 phase. The anticancer effect has been further confirmed in colorectal cancer and pancreatic cancer in vivo.136 Recently research reported that frequent mutation of KRAS have been observed in ovarian mucinous carcinomas. The results suggested that combined MEK inhibitor (pimasertib) and PI3K/mTOR inhibitor (SAR245409, voxtalisib) exhibited synergistic anti-tumor effects in ovarian mucinous carcinomas with KRAS and/or PIK3CA mutation.137 Recently, a great number of synthetic lethal sites have been identified, including serine/threonine kinase 33, PLK1 (polo like kinase 1), Bcl-xL (B-cell lymphoma-extra-large), AK1 (TGF-beta activated kinase 1), and GATA2 (GATA binding protein 2).138 All of these provide new strategies and options for the treatment of KRAS mutant tumors (Table 2).
Liquid Biopsy
A number of studies on ctDNA, representing a small percentage of cfDNA that is shed in circulation by tumor cells and carries tumor specific mutations, attempted to evaluate its clinical value in OC. The research demonstrated 44% of the OCs involved in the study had detected P53 mutations in tumor tissue.
In OCs with advanced disease, the rate of P53 mutation was 28.6%.20 P53 mutants were undetectable in plasma after surgery, but in one patient the P53 mutant again became detectable 16 months after surgery and the patient died 2 months later.5,139 The detection of mutant P53 in cfDNA might be an important strategy for future diagnosis and monitoring of the treatment efficacy. Another recent study attempted to evaluate somatic P53 mutations in patients with serous OC by ddPCR. The research has suggested that P53 mutations were investigated in serial ctDNA samples of HGSOC. Besides, the presence of P53 mutation allele fractions in ctDNA, when compared to serum CA-125 levels, could indicate a much earlier response to chemotherapy.140
Using methylation specific PCR (MSP), the BRCA hypermethylation was detected in cfDNA of early stage (stage I, II) epithelial OC patients. The results showed that the consistency of tumor and plasma/serum DNA methylation pattern in 82% of matched samples was observed. Detecting BRCA1/2 mutation in the patients with OC can provide valuable information in diagnostic, prognostic, and predictive disease progression.5 The reversion of BRCA1/2 mutations in ctDNA was investigated as an indicator of response to platinum-based and PARPi-based chemotherapy or following treatment with PARPi or platinum compounds after disease progression.141,142
PIK3CA mutation is frequent in OCCC, and 16.7% of those had detected in the plasma DNA. PIK3CA-H1047R is a hotspot and can be detected in most of the OCCC patients with PIK3CA mutation. KRAS-G12D can be detected in part of patients with KRAS mutation. Detection of PIK3CA-H1047R and KRAS-G12D in cfDNA by ddPCR would be useful for the early diagnosis of ovarian clear cell carcinoma, to monitor its response to the therapy, and for predicting its recurrence.9 Liquid biopsy, as a promising non-invasive diagnostic, prognostic, and predictive strategy, provides an easily accessible source of DNA derived from the OC.
Conclusion
OC is a complex and polygenic mutation disease. Specific gene mutations have been revealed to drive OC pathogenesis and development. The usability of genome sequencing has provided exact data of gene mutations in ovarian carcinomas to develop some accurate treatment strategies. In a certain extent biologically targeted therapies and some targeted drugs combinations have improved prognosis. Recently, numerous gene mutation studies have offered some potential predictive biomarkers or therapeutics in OC. But few available effective therapies are currently used widely in the clinic. In this review, we highlight the advance in P53, BRAC1/2, PIK3CA, and KRAS gene mutations in OC and summarize the potential targets for novel therapeutic strategies based on the above gene mutant. In the future, to improve OC targeting, we need a deeper delve to identify carcinogenesis and interactions of gene mutations. There is still future research to be done in order to prospect for gene therapy which can solve the treatment of OC. With development of whole gene sequencing technology, liquid biopsy and gene-editing technologies will reveal a more complete genomic landscape which can detect OC in the early stage and establish novel treatments.
Abbreviations
OC, Ovarian cancer; HGSOC, High-grade serous ovarian carcinoma; LGSOC, low-grade serous ovarian carcinoma; EnOC, Endometrioid ovarian cancer; OCCC, Ovarian clear cell carcinoma; CTCs, Circulating tumor cells; ctDNA, Circulating tumor DNA; WT, Wild-type; GOF, Gain-of-function; NGS, Next-generation sequencing; PARP, Poly (ADP-ribose) polymerase; SSBs, Single-strand breaks; DSBs, Double-strand breaks; HR, Homologous recombination; NHEJ, Nonhomologous end joining; NAD+, Nicotinamide adenine dinucleotide; PARG, PAR glycohydrolase; PI3K, Phosphatidylinositol 3-kinase; PARPi, PARP inhibitors; cfDNA, cell-free DNA; CDK1, Cyclin dependent kinase 1; PLK1, Polo like kinase 1; Bcl-xL, B-cell lymphoma-extra-large; AK1, TGF-beta activated kinase 1; GATA2, GATA binding protein 2; ddPCR, Droplet digital PCR; MSP, Methylation specific PCR.
Funding
This research was funded by National Key R&D Program of China, grant number 2017YFC0907305.
Disclosure
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Morgan RD, Clamp AR, Evans DGR, Edmondson RJ, Jayson GC. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemother Pharmacol. 2018;81(4):647–658. doi:10.1007/s00280-018-3532-9
3. Merritt MA, Rice MS, Barnard ME, et al. Pre-diagnosis and post-diagnosis use of common analgesics and ovarian cancer prognosis (NHS/NHSII): a cohort study. Lancet Oncol. 2018;19(8):1107–1116. doi:10.1016/S1470-2045(18)30373-5
4. Quinn JE, Carser JE, James CR, Kennedy RD, Harkin DP. BRCA1 and implications for response to chemotherapy in ovarian cancer. Gynecol Oncol. 2009;113(1):134–142. doi:10.1016/j.ygyno.2008.12.015
5. Giannopoulou L, Kasimir-Bauer S, Lianidou ES. Liquid biopsy in ovarian cancer: recent advances on circulating tumor cells and circulating tumor DNA. Clin Chem Lab Med. 2018;56(2):186–197. doi:10.1515/cclm-2017-0019
6. Wang Z, Sun Y. Targeting p53 for novel anticancer therapy. Transl Oncol. 2010;3(1):1–12. doi:10.1593/tlo.09250
7. Della Pepa C, Tonini G, Santini D, et al. Low grade serous ovarian carcinoma: from the molecular characterization to the best therapeutic strategy. Cancer Treat Rev. 2015;41(2):136–143. doi:10.1016/j.ctrv.2014.12.003
8. Sadlecki P, Antosik P, Grzanka D, Grabiec M, Walentowicz-Sadlecka M. KRAS mutation testing in borderline ovarian tumors and low-grade ovarian carcinomas with a rapid, fully integrated molecular diagnostic system. Tumour Biol. 2017;39(10):1010428317733984. doi:10.1177/1010428317733984
9. Morikawa A, Hayashi T, Shimizu N, et al. PIK3CA and KRAS mutations in cell free circulating DNA are useful markers for monitoring ovarian clear cell carcinoma. Oncotarget. 2018;9(20):15266–15274. doi:10.18632/oncotarget.24555
10. Testa U, Petrucci E, Pasquini L, Castelli G, Pelosi E. Ovarian cancers: genetic abnormalities, tumor heterogeneity and progression, clonal evolution and cancer stem cells. Medicines. 2018;5:1. doi:10.3390/medicines5010016
11. Cole AJ, Dwight T, Gill AJ, et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep. 2016;6:26191. doi:10.1038/srep26191
12. Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000;60:5.
13. Zak K, Pecak A, Rys B, et al. Mdm2 and MdmX inhibitors for the treatment of cancer: a patent review (2011 – present). Expert Opin Ther Pat. 2013;23(4):425–448. doi:10.1517/13543776.2013.765405
14. Lee JG, Ahn JH, Jin Kim T, Ho lee J, Choi JH. Mutant p53 promotes ovarian cancer cell adhesion to mesothelial cells via integrin beta4 and Akt signals. Sci Rep. 2015;5:12642. doi:10.1038/srep12642
15. Davaadelger B, Shen H, Maki CG. Novel roles for p53 in the genesis and targeting of tetraploid cancer cells. PLoS One. 2014;9(11):e110844. doi:10.1371/journal.pone.0110844
16. Wu GS, Ding Z. Caspase 9 is required for p53-dependent apoptosis and chemosensitivity in a human ovarian cancer cell line. Oncogene. 2002;21:1–8. doi:10.1038/sj.onc.1205020
17. Kitayner M, Rozenberg H, Kessler N, et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell. 2006;22(6):741–753. doi:10.1016/j.molcel.2006.05.015
18. Duffy MJ, Synnott NC, Crown J. Mutant p53 as a target for cancer treatment. Eur J Cancer. 2017;83:258–265. doi:10.1016/j.ejca.2017.06.023
19. Padmanabhan A, Candelaria N, Wong KK, et al. USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat Commun. 2018;9(1):1270. doi:10.1038/s41467-018-03599-w
20. Yang-Hartwich Y, Soteras MG, Lin ZP, et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene. 2015;34(27):3605–3616. doi:10.1038/onc.2014.296
21. Alkema NG, Wisman GB, van der Zee AG, van Vugt MA, de Jong S. Studying platinum sensitivity and resistance in high-grade serous ovarian cancer: different models for different questions. Drug Resist Updat. 2016;24:55–69. doi:10.1016/j.drup.2015.11.005
22. Kang HJ, Chun SM, Kim KR, Sohn I, Sung CO. Clinical relevance of gain-of-function mutations of p53 in high-grade serous ovarian carcinoma. PLoS One. 2013;8(8):e72609. doi:10.1371/journal.pone.0072609
23. Ahmed AA, Etemadmoghadam D, Temple J, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221(1):49–56. doi:10.1002/path.2696
24. Ahn JH, Kim TJ, Lee JH, Choi JH. Mutant p53 stimulates cell invasion through an interaction with Rad21 in human ovarian cancer cells. Sci Rep. 2017;7(1):9076. doi:10.1038/s41598-017-08880-4
25. Hofstetter G, Berger A, Fiegl H, et al. Alternative splicing of p53 and p73: the novel p53 splice variant p53delta is an independent prognostic marker in ovarian cancer. Oncogene. 2010;29(13):1997–2004. doi:10.1038/onc.2009.482
26. Alaee M, Danesh G, Pasdar M. Plakoglobin reduces the in vitro growth, migration and invasion of ovarian cancer cells expressing N-cadherin and mutant p53. PLoS One. 2016;11(5):e0154323. doi:10.1371/journal.pone.0154323
27. Emmanuel C, Chiew YE, George J, et al. Genomic classification of serous ovarian cancer with adjacent borderline differentiates RAS pathway and TP53-mutant tumors and identifies NRAS as an oncogenic driver. Clin Cancer Res. 2014;20(24):6618–6630. doi:10.1158/1078-0432.CCR-14-1292
28. Sonego M, Schiappacassi M, Lovisa S, et al. Stathmin regulates mutant p53 stability and transcriptional activity in ovarian cancer. EMBO Mol Med. 2013;5(5):707–722. doi:10.1002/emmm.201201504
29. Brachova P, Thiel KW, Leslie KK. The consequence of oncomorphic TP53 mutations in ovarian cancer. Int J Mol Sci. 2013;14(9):19257–19275. doi:10.3390/ijms140919257
30. Machado-Silva A, Perrier S, Bourdon JC. p53 family members in cancer diagnosis and treatment. Semin Cancer Biol. 2010;20(1):57–62. doi:10.1016/j.semcancer.2010.02.005
31. Pujade-Lauraine E. New treatments in ovarian cancer. Ann Oncol. 2017;28(suppl_8):viii57–viii60. doi:10.1093/annonc/mdx442
32. Bykov VJ, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of mutant p53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front Oncol. 2016;6:21. doi:10.3389/fonc.2016.00021
33. Soragni A, Janzen DM, Johnson LM, et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell. 2016;29(1):90–103. doi:10.1016/j.ccell.2015.12.002
34. Oda K, Ikeda Y, Kashiyama T, et al. Characterization of TP53 and PI3K signaling pathways as molecular targets in gynecologic malignancies. J Obstet Gynaecol Res. 2016;42(7):757–762. doi:10.1111/jog.13018
35. Amin A, Karpowicz PA, Carey TE, et al. Evasion of anti-growth signaling: a key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin Cancer Biol. 2015;35(Suppl):S55–S77.
36. Zanjirband M, Edmondson RJ, Lunec J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget. 2016;7:26. doi:10.18632/oncotarget.9499
37. Zhang Q, Zeng SX, Lu H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell Biochem. 2014;85:281–319.
38. Duffy MJ, Synnott NC, McGowan PM, Crown J, O’Connor D, Gallagher WM. p53 as a target for the treatment of cancer. Cancer Treat Rev. 2014;40(10):1153–1160. doi:10.1016/j.ctrv.2014.10.004
39. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi:10.1126/science.1092472
40. Yang Y, Ludwig RL, Jensen JP, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7(6):547–559. doi:10.1016/j.ccr.2005.04.029
41. Herman AG, Hayano M, Poyurovsky MV, et al. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 2011;1(4):312–325. doi:10.1158/2159-8290.CD-11-0104
42. Kojima K, Burks JK, Arts J, Andreeff M. The novel tryptamine derivative JNJ-26854165 induces wild-type p53- and E2F1-mediated apoptosis in acute myeloid and lymphoid leukemias. Mol Cancer Ther. 2010;9(9):2545–2557. doi:10.1158/1535-7163.MCT-10-0337
43. Schilling D, Düwel M, Molls M, Multhoff G. Radiosensitization of wildtype p53 cancer cells by the MDM2-inhibitor PXN727 is associated with altered heat shock protein 70 (Hsp70) levels. Cell Stress Chaperones. 2012;18(2):183–191. doi:10.1007/s12192-012-0369-8
44. Roth JA. Adenovirus p53 gene therapy. Expert Opin Biol Ther. 2005;6(1):55–61. doi:10.1517/14712598.6.1.55
45. Dai L, Pan Q, Peng Y, et al. p53 plays a key role in the apoptosis of human ovarian cancer cells induced by adenovirus-mediated CRM197. Hum Gene Ther. 2018;29(8):916–926. doi:10.1089/hum.2017.186
46. Liu Q, Sui R, Li R, Miao J, Liu J. Biological characteristics of Taxolresistant ovarian cancer cells and reversal of Taxol resistance by adenovirus expressing p53. Mol Med Rep. 2015;11(2):1292–1297. doi:10.3892/mmr.2014.2784
47. Wang J, Gao Q, Li Q. Adenovirus type 12 E1B 55-kilodalton oncoprotein promotes p53-mediated apoptotic response of ovarian cancer to cisplatin. Tumor Biol. 2015;36(8):6569–6577. doi:10.1007/s13277-015-3351-0
48. Chira S, Gulei D, Hajitou A, Zimta AA, Cordelier P, Berindan-Neagoe I. CRISPR/Cas9: transcending the reality of genome editing. Mol Ther Nucleic Acids. 2017;7:211–222. doi:10.1016/j.omtn.2017.04.001
49. Liu C, Zhang L, Liu H, Cheng K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J Control Release. 2017;266:17–26. doi:10.1016/j.jconrel.2017.09.012
50. Savic N, Schwank G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl Res. 2016;168:15–21. doi:10.1016/j.trsl.2015.09.008
51. Walton J, Blagih J, Ennis D, et al. CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high-grade serous carcinoma. Cancer Res. 2016;76(20):6118–6129. doi:10.1158/0008-5472.CAN-16-1272
52. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12(1):68–78. doi:10.1038/nrc3181
53. Girolimetti G, Perrone AM, Santini D, et al. BRCA-associated ovarian cancer: from molecular genetics to risk management. Biomed Res Int. 2014;2014:787143. doi:10.1155/2014/787143
54. Gadducci A, Guarneri V, Peccatori FA, et al. Current strategies for the targeted treatment of high-grade serous epithelial ovarian cancer and relevance of BRCA mutational status. J Ovarian Res. 2019;12(1):9. doi:10.1186/s13048-019-0484-6
55. Lubinski J, Phelan CM, Ghadirian P, et al. Cancer variation associated with the position of the mutation in the BRCA2 gene. Fam Cancer. 2004;3(1):1–10. doi:10.1023/B:FAME.0000026816.32400.45
56. Plaskocinska I, Shipman H, Drummond J, et al. New paradigms for BRCA1/BRCA2 testing in women with ovarian cancer: results of the genetic testing in epithelial ovarian cancer (GTEOC) study. J Med Genet. 2016;53(10):655–661. doi:10.1136/jmedgenet-2016-103902
57. Rust K, Spiliopoulou P, Tang CY, et al. Routine germline BRCA1 and BRCA2 testing in patients with ovarian carcinoma: analysis of the Scottish real-life experience. BJOG. 2018;125(11):1451–1458. doi:10.1111/1471-0528.15171
58. Morgan RD, Burghel GJ, Flaum N, et al. Prevalence of germline pathogenic BRCA1/2 variants in sequential epithelial ovarian cancer cases. J Med Genet. 2019;56(5):301–307. doi:10.1136/jmedgenet-2018-105792
59. Harter P, Hauke J, Heitz F, et al. Prevalence of deleterious germline variants in risk genes including BRCA1/2 in consecutive ovarian cancer patients (AGO-TR-1). PLoS One. 2017;12(10):e0186043. doi:10.1371/journal.pone.0186043
60. Krivokuca A, Boljevic I, Jovandic S, et al. Germline mutations in cancer susceptibility genes in high grade serous ovarian cancer in Serbia. J Hum Genet. 2019;64(4):281–290. doi:10.1038/s10038-019-0562-z
61. Tsibulak I, Wieser V, Degasper C, et al. BRCA1 and BRCA2 mRNA-expression prove to be of clinical impact in ovarian cancer. Br J Cancer. 2018;119(6):683–692. doi:10.1038/s41416-018-0217-4
62. Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. doi:10.1038/nature06548
63. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi:10.1038/nature06633
64. Biglia N, Sgandurra P, Bounous VE, et al. Ovarian cancer in BRCA1 and BRCA2 gene mutation carriers: analysis of prognostic factors and survival. Ecancermedicalscience. 2016;10:639. doi:10.3332/ecancer.2016.639
65. Hyman DM, Zhou Q, Iasonos A, et al. Improved survival for BRCA2-associated serous ovarian cancer compared with both BRCA-negative and BRCA1-associated serous ovarian cancer. Cancer. 2012;118(15):3703–3709. doi:10.1002/cncr.26655
66. Liu X, Shi Y, Maag DX, et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide PARP inhibitor. Clin Cancer Res. 2012;18(2):510–523. doi:10.1158/1078-0432.CCR-11-1973
67. Liu JF, Konstantinopoulos PA, Matulonis UA. PARP inhibitors in ovarian cancer: current status and future promise. Gynecol Oncol. 2014;133(2):362–369. doi:10.1016/j.ygyno.2014.02.039
68. Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599. doi:10.1158/0008-5472.CAN-12-2753
69. Brown JS, Kaye SB, Yap TA. PARP inhibitors: the race is on. Br J Cancer. 2016;114(7):713–715. doi:10.1038/bjc.2016.67
70. Damia G, Sessa C. Successes and limitations of targeted cancer therapy in ovarian cancer. Prog Tumor Res. 2014;41:89–97.
71. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi:10.1038/nature10166
72. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi:10.1038/nature03445
73. Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–698. doi:10.1038/nrc1691
74. Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. doi:10.1016/S0140-6736(10)60893-8
75. Meehan RS, Chen AP. New treatment option for ovarian cancer: PARP inhibitors. Gynecol Oncol Res Pract. 2016;3:3. doi:10.1186/s40661-016-0024-7
76. Ledermann JA. PARP inhibitors in ovarian cancer. Ann Oncol. 2016;27(Suppl 1):i40–i44. doi:10.1093/annonc/mdw094
77. Ledermann JA, Harter P, Gourley C, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17(11):1579–1589. doi:10.1016/S1470-2045(16)30376-X
78. Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–1284. doi:10.1016/S1470-2045(17)30469-2
79. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
80. Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. doi:10.1016/S0140-6736(17)32440-6
81. Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585(1):1–6. doi:10.1016/j.febslet.2010.11.024
82. Miller RE, Ledermann JA. The status of poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors in ovarian cancer, part 1: olaparib. Clin Adv Hematol Oncol. 2016;14(8):619–627.
83. Christie EL, Fereday S, Doig K, Pattnaik S, Dawson SJ, Bowtell DDL. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high-grade serous ovarian cancer. J Clin Oncol. 2017;35(12):1274–1280. doi:10.1200/JCO.2016.70.4627
84. Lin KK, Harrell MI, Oza AM, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9(2):210–219. doi:10.1158/2159-8290.CD-18-0715
85. Weigelt B, Comino-Mendez I, de Bruijn I, et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin Cancer Res. 2017;23(21):6708–6720. doi:10.1158/1078-0432.CCR-17-0544
86. Wang Y, Bernhardy AJ, Cruz C, et al. The BRCA1-Delta11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76(9):2778–2790. doi:10.1158/0008-5472.CAN-16-0186
87. Juvekar A, Burga LN, Hu H, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048–1063. doi:10.1158/2159-8290.CD-11-0336
88. Wang D, Wang M, Jiang N, Zhang Y, Bian X. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget. 2016;7:14.
89. Oishi T, Itamochi H, Kudoh A, et al. The PI3K/mTOR dual inhibitor NVP-BEZ235 reduces the growth of ovarian clear cell carcinoma. Oncol Rep. 2014;32(2):553–558. doi:10.3892/or.2014.3268
90. Thakur B, Ray P. p53 loses grip on PIK3CA expression leading to enhanced cell survival during platinum resistance. Mol Oncol. 2016;10(8):1283–1295. doi:10.1016/j.molonc.2016.06.006
91. Heudel PE, Fabbro M, Roemer-Becuwe C, et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: a stratified type I-type II study from the GINECO group. Br J Cancer. 2017;116(3):303–309. doi:10.1038/bjc.2016.430
92. Gungor H, Saleem A, Babar S, et al. Dose-finding quantitative 18F-FDG PET imaging study with the oral pan-AKT inhibitor GSK2141795 in patients with gynecologic malignancies. J Nucl Med. 2015;56(12):1828–1835. doi:10.2967/jnumed.115.156505
93. Zannoni GF, Improta G, Pettinato A, et al. Molecular status of PI3KCA, KRAS and BRAF in ovarian clear cell carcinoma: an analysis of 63 patients. J Clin Pathol. 2016;69(12):1088–1092. doi:10.1136/jclinpath-2016-203776
94. Wang D, Li C, Zhang Y, et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol Oncol. 2016;142(3):548–556. doi:10.1016/j.ygyno.2016.07.092
95. Despierre E, Yesilyurt BT, Lambrechts S, et al. Epithelial ovarian cancer rationale for changing the one-fits-all standard treatment regimen to subtype-specific treatment. Int J Gynecol Cancer. 2014;24:10. doi:10.1097/IGC.0000000000000089
96. Samartzis EP, Noske A, Dedes KJ, et al. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int J Mol Sci. 2013;14:18824–18849.
97. Matsumoto T, Yamazaki M, Takahashi H, et al. Distinct β-Catenin and PIK3CA mutation profiles in endometriosis-associated ovarian endometrioid and clear cell carcinomas. Am J Clin Pathol. 2015;144(3):452–463. doi:10.1309/AJCPZ5T2POOFMQVN
98. Friedlander ML, Russell K, Millis S, Gatalica Z, Bender R, Voss A. Molecular profiling of clear cell ovarian cancers: identifying potential treatment targets for clinical trials. Int J Gynecol Cancer. 2016;26(4):648–654. doi:10.1097/IGC.0000000000000677
99. Murakami R, Matsumura N, Brown JB, et al. Exome sequencing landscape analysis in ovarian clear cell carcinoma shed light on key chromosomal regions and mutation gene networks. Am J Pathol. 2017;187(10):2246–2258. doi:10.1016/j.ajpath.2017.06.012
100. Zannoni GF, Improta G, Chiarello G, et al. Mutational status of KRAS, NRAS, and BRAF in primary clear cell ovarian carcinoma. Virchows Arch. 2014;465(2):193–198. doi:10.1007/s00428-014-1599-1
101. Despierre E, Yesilyurt BT, Lambrechts S, et al. Epithelial ovarian cancer: rationale for changing the one-fits-all standard treatment regimen to subtype-specific treatment. Int J Gynecol Cancer. 2014;24(3):468–477.
102. Samartzis EP, Noske A, Dedes KJ, Fink D, Imesch P. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int J Mol Sci. 2013;14(9):18824–18849. doi:10.3390/ijms140918824
103. Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol. 2012;25(4):615–624. doi:10.1038/modpathol.2011.189
104. Kolasa IK, Rembiszewska A, Felisiak A, et al. PIK3CA amplification associates with resistance to chemotherapy in ovarian cancer patients. Cancer Biol Ther. 2009;8(1):21–26. doi:10.4161/cbt.8.1.7209
105. Fu S, Hennessy BT, Ng CS, et al. Perifosine plus docetaxel in patients with platinum and taxane resistant or refractory high-grade epithelial ovarian cancer. Gynecol Oncol. 2012;126(1):47–53. doi:10.1016/j.ygyno.2012.04.006
106. Behbakht K, Sill MW, Darcy KM, et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a gynecologic oncology group study. Gynecol Oncol. 2011;123(1):19–26. doi:10.1016/j.ygyno.2011.06.022
107. Zhang Y-J, Duan Y, Zheng XFS. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today. 2011;16(7–8):325–331. doi:10.1016/j.drudis.2011.02.008
108. Cheaib B, Auguste A, Leary A. The PI3K/Akt/mTOR pathway in ovarian cancer: therapeutic opportunities and challenges. Chin J Cancer. 2015;34(1):4–16. doi:10.5732/cjc.014.10289
109. Takano M, Kikuchi Y, Kudoh K, et al. Weekly administration of temsirolimus for heavily pretreated patients with clear cell carcinoma of the ovary: a report of six cases. Int J Clin Oncol. 2011;16(5):605–609. doi:10.1007/s10147-010-0177-z
110. Sarker D, Ang JE, Baird R, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(1):77–86. doi:10.1158/1078-0432.CCR-14-0947
111. Bowles DW, Ma WW, Senzer N, et al. A multicenter phase 1 study of PX-866 in combination with docetaxel in patients with advanced solid tumours. Br J Cancer. 2013;109(5):1085–1092. doi:10.1038/bjc.2013.474
112. Gocher AM, Azabdaftari G, Euscher LM, et al. Akt activation by Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J Biol Chem. 2017;292(34):14188–14204. doi:10.1074/jbc.M117.778464
113. Patnaik A, Appleman LJ, Tolcher AW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol. 2016;27(10):1928–1940. doi:10.1093/annonc/mdw282
114. Cheraghchi-Bashi A, Parker CA, Curry E, et al. A putative biomarker signature for clinically effective AKT inhibition: correlation of in vitro, in vivo and clinical data identifies the importance of modulation of the mTORC1 pathway. Oncotarget. 2015;6:14. doi:10.18632/oncotarget.6153
115. Tsuchida N, Murugan AK, Grieco M. Kirsten Ras oncogene: significance of its discovery in human cancer research. Oncotarget. 2016;7(29):46717–46733. doi:10.18632/oncotarget.8773
116. Tsai FD, Lopes MS, Zhou M, et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112(3):779–784. doi:10.1073/pnas.1412811112
117. Hollestelle A, Pelletier C, Hooning M, et al. Prevalence of the variant allele rs61764370 T>G in the 3ʹUTR of KRAS among Dutch BRCA1, BRCA2 and non-BRCA1/BRCA2 breast cancer families. Breast Cancer Res Treat. 2011;128(1):79–84. doi:10.1007/s10549-010-1080-z
118. Sakai K, Ukita M, Schmidt J, et al. Clonal composition of human ovarian cancer based on copy number analysis reveals a reciprocal relation with oncogenic mutation status. Cancer Lett. 2017;405:22–28. doi:10.1016/j.canlet.2017.07.013
119. Boyd J, Luo B, Peri S, et al. Whole exome sequence analysis of serous borderline tumors of the ovary. Gynecol Oncol. 2013;130(3):560–564. doi:10.1016/j.ygyno.2013.06.007
120. Rechsteiner M, Zimmermann AK, Wild PJ, et al. TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Exp Mol Pathol. 2013;95(2):235–241. doi:10.1016/j.yexmp.2013.08.004
121. Kim SI, Lee JW, Lee M, et al. Genomic landscape of ovarian clear cell carcinoma via whole exome sequencing. Gynecol Oncol. 2018;148(2):375–382. doi:10.1016/j.ygyno.2017.12.005
122. Koshiyama M, Matsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. Biomed Res Int. 2014;2014:934261. doi:10.1155/2014/934261
123. Lee YJ, Lee MY, Ruan A, et al. Multipoint Kras oncogene mutations potentially indicate mucinous carcinoma on the entire spectrum of mucinous ovarian neoplasms. Oncotarget. 2016;7(50):82097–82103. doi:10.18632/oncotarget.13449
124. Garrett AP, Lee KR, Colitti CR, Muto MG, Berkowitz RS, Mok SC. k-ras mutation may be an early event in mucinous ovarian tumorigenesis. Int J Gynecol Pathol. 2001;20(3):244–251. doi:10.1097/00004347-200107000-00007
125. Li XS, Sun J, He XL. Expression of c-myc and mutation of the KRAS gene in patients with ovarian mucinous tumors. Genet Mol Res. 2015;14(3):10752–10759. doi:10.4238/2015.September.9.14
126. Mackenzie R, Kommoss S, Winterhoff BJ, et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer. 2015;15:415. doi:10.1186/s12885-015-1421-8
127. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25(3):272–281. doi:10.1016/j.ccr.2014.02.017
128. Pang X, Liu M. Defeat mutant KRAS with synthetic lethality. Small GTPases. 2016;8(4):212–219. doi:10.1080/21541248.2016.1213783
129. Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers. 2016;8(4):45. doi:10.3390/cancers8040045
130. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–608. doi:10.1126/science.aad6204
131. Pecot CV, Wu SY, Bellister S, et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther. 2014;13(12):2876–2885. doi:10.1158/1535-7163.MCT-14-0074
132. Kamerkar S, LeBleu VS, Sugimoto H, et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546(7659):498–503. doi:10.1038/nature22341
133. Yeh TC, Marsh V, Bernat BA, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13(5):1576–1583. doi:10.1158/1078-0432.CCR-06-1150
134. Spreafico A, Oza AM, Clarke BA, et al. Genotype-matched treatment for patients with advanced type I epithelial ovarian cancer (EOC). Gynecol Oncol. 2017;144(2):250–255. doi:10.1016/j.ygyno.2016.12.002
135. Kaldawy A, Segev Y, Lavie O, Auslender R, Sopik V, Narod SA. Low-grade serous ovarian cancer: a review. Gynecol Oncol. 2016;143(2):433–438. doi:10.1016/j.ygyno.2016.08.320
136. Costa-Cabral S, Brough R, Konde A, et al. CDK1 is a synthetic lethal target for KRAS mutant tumours. PLoS One. 2016;11(2):e0149099. doi:10.1371/journal.pone.0149099
137. Inaba K, Oda K, Aoki K, et al. Synergistic antitumor effects of combination PI3K/mTOR and MEK inhibition (SAR245409 and pimasertib) in mucinous ovarian carcinoma cells by fluorescence resonance energy transfer imaging. Oncotarget. 2016;7(20):29577–29591. doi:10.18632/oncotarget.8807
138. de Castro Carpeno J, Belda-Iniesta C. KRAS mutant NSCLC, a new opportunity for the synthetic lethality therapeutic approach. Transl Lung Cancer Res. 2013;2(2):142–151. doi:10.3978/j.issn.2218-6751.2013.02.07
139. Otsuka J, Okuda T, Sekizawa A, et al. Detection of p53 mutations in the plasma DNA of patients with ovarian cancer. Int J Gynecol Cancer. 2004;14(3):459–464. doi:10.1111/j.1048-891x.2004.014305.x
140. Parkinson CA, Gale D, Piskorz AM, et al. Exploratory analysis of TP53 mutations in circulating tumour DNA as biomarkers of treatment response for patients with relapsed high-grade serous ovarian carcinoma: a retrospective study. PLoS Med. 2016;13(12):e1002198. doi:10.1371/journal.pmed.1002198
141. Mayor P, Gay LM, Lele S, Elvin JA. BRCA1 reversion mutation acquired after treatment identified by liquid biopsy. Gynecol Oncol Rep. 2017;21:57–60. doi:10.1016/j.gore.2017.06.010
142. Giannopoulou L, Zavridou M, Kasimir-Bauer S, Lianidou ES. Liquid biopsy in ovarian cancer: the potential of circulating miRNAs and exosomes. Transl Res. 2019;205:77–91. doi:10.1016/j.trsl.2018.10.003
143. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. doi:10.1158/1078-0432.CCR-13-2287
144. Nakamura K, Nakayama K, Ishikawa N, et al. Reconstitution of high-grade serous ovarian carcinoma from primary fallopian tube secretory epithelial cells. Oncotarget. 2018;9(16):12609–12619. doi:10.18632/oncotarget.23035
145. Marchetti C, De Leo R, Musella A, et al. BRCA mutation status to personalize management of recurrent ovarian cancer: a multicenter study. Ann Surg Oncol. 2018;25(12):3701–3708. doi:10.1245/s10434-018-6700-6
146. Singer G, Stohr R, Cope L, et al. Patterns of p53 mutations separate ovarian serous borderline tumors and low- and high-grade carcinomas and provide support for a new model of ovarian carcinogenesis: a mutational analysis with immunohistochemical correlation. Am J Surg Pathol. 2005;29(2):218–224. doi:10.1097/01.pas.0000146025.91953.8d
147. Jones S, Wang TL, Kurman RJ, et al. Low-grade serous carcinomas of the ovary contain very few point mutations. J Pathol. 2012;226(3):413–420. doi:10.1002/path.3967
148. Liu J, Westin SN. Rational selection of biomarker driven therapies for gynecologic cancers: the more we know, the more we know we don’t know. Gynecol Oncol. 2016;141(1):65–71. doi:10.1016/j.ygyno.2016.01.003
149. Jean S, Li J, Katsaros D, et al. Paclitaxel is necessary for improved survival in epithelial ovarian cancers with homologous recombination gene mutations. Oncotarget. 2016;7(30):48577–48585. doi:10.18632/oncotarget.9373
150. Ho ES, Lai CR, Hsieh YT, et al. p53 mutation is infrequent in clear cell carcinoma of the ovary. Gynecol Oncol. 2001;80(2):189–193. doi:10.1006/gyno.2000.6025
151. Shi T, Wang P, Tang W, et al. Survival benefit of germline BRCA mutation is associated with residual disease in ovarian cancer. Cell Physiol Biochem. 2018;47(5):2088–2096. doi:10.1159/000491477
152. Zhang S, Royer R, Li S, et al. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol Oncol. 2011;121(2):353–357. doi:10.1016/j.ygyno.2011.01.020
153. Fransson A, Glaessgen D, Alfredsson J, Wiman KG, Bajalica-Lagercrantz S, Mohell N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant high-grade serous ovarian cancer. J Ovarian Res. 2016;9(1):27. doi:10.1186/s13048-016-0239-6
154. Leijen S, van Geel RM, Sonke GS, et al. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J Clin Oncol. 2016;34(36):4354–4361. doi:10.1200/JCO.2016.67.5942
155. Wolf JK, Bodurka DC, Gano JB, et al. A phase I study of Adp53 (INGN 201; ADVEXIN) for patients with platinum- and paclitaxel-resistant epithelial ovarian cancer. Gynecol Oncol. 2004;94(2):442–448. doi:10.1016/j.ygyno.2004.05.041
156. Vasey PA, Shulman LN, Campos S, et al. Phase I trial of intraperitoneal injection of the E1B-55-kd-gene-deleted adenovirus ONYX-015 (dl1520) given on days 1 through 5 every 3 weeks in patients with recurrent/refractory epithelial ovarian cancer. J Clin Oncol. 2002;20(6):1562–1569. doi:10.1200/JCO.2002.20.6.1562
157. Buller RE, Runnebaum IB, Karlan BY, et al. A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Ther. 2002;9(7):553–566. doi:10.1038/sj.cgt.7700472
158. Usha L, Sill MW, Darcy KM, et al. A gynecologic oncology group phase II trial of the protein kinase C-beta inhibitor, enzastaurin and evaluation of markers with potential predictive and prognostic value in persistent or recurrent epithelial ovarian and primary peritoneal malignancies. Gynecol Oncol. 2011;121(3):455–461. doi:10.1016/j.ygyno.2011.02.013
159. Mir R, Tortosa A, Martinez-Soler F, et al. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance in TP53 wild-type ovarian cancer cells. Int J Cancer. 2013;132(7):1525–1536. doi:10.1002/ijc.27832
160. Makii C, Oda K, Ikeda Y, et al. MDM2 is a potential therapeutic target and prognostic factor for ovarian clear cell carcinomas with wild type TP53. Oncotarget. 2016;7(46):75328–75338. doi:10.18632/oncotarget.12175
161. Coleman RL, Sill MW, Bell-McGuinn K, et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation - an NRG oncology/gynecologic oncology group study. Gynecol Oncol. 2015;137(3):386–391. doi:10.1016/j.ygyno.2015.03.042
162. Bell-McGuinn KM, Konner JA, Tew WP, et al. A phase 2, single arm study of iniparib in patients with BRCA1 or BRCA2 associated advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer. Int J Gynecol Cancer. 2016;26(2):255–260. doi:10.1097/IGC.0000000000000591
163. Matulonis U, Vergote I, Backes F, et al. Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol Oncol. 2015;136(2):246–253. doi:10.1016/j.ygyno.2014.12.019
164. Schram AM, Gandhi L, Mita MM, et al. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer. 2018;119(12):1471–1476. doi:10.1038/s41416-018-0322-4
165. Hasegawa K, Kagabu M, Mizuno M, et al. Phase II basket trial of perifosine monotherapy for recurrent gynecologic cancer with or without PIK3CA mutations. Invest New Drugs. 2017;35(6):800–812. doi:10.1007/s10637-017-0504-6
166. Angevin E, Tabernero J, Elez E, et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2014;20(8):2192–2204. doi:10.1158/1078-0432.CCR-13-2200
167. Bedard PL, Tabernero J, Janku F, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21(4):730–738. doi:10.1158/1078-0432.CCR-14-1814
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.