Back to Journals » OncoTargets and Therapy » Volume 9
Cell-free DNA as a diagnostic marker for cancer: current insights
Authors Salvi S, Gurioli G, De Giorgi U, Conteduca V, Tedaldi G ![]() , Calistri D, Casadio V
, Calistri D, Casadio V
Received 12 July 2016
Accepted for publication 3 September 2016
Published 25 October 2016 Volume 2016:9 Pages 6549—6559
DOI https://doi.org/10.2147/OTT.S100901
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Samanta Salvi,1 Giorgia Gurioli,1 Ugo De Giorgi,2 Vincenza Conteduca,2 Gianluca Tedaldi,1 Daniele Calistri,1 Valentina Casadio1
1Biosciences Laboratory, 2Department of Medical Oncology, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, Meldola, Italy
Abstract: The increasing knowledge of the molecular pathogenesis of cancer and the rapid development of new molecular techniques are promoting the study of early molecular alterations involved in cancer development in body fluids. Specific genetic and epigenetic alterations could be found in plasma, serum, and urine cell-free DNA (cfDNA) and could potentially be used as diagnostic biomarkers for several types of cancers. This review focuses on the role of cfDNA in diagnosis: a PubMed search was performed by selecting papers according to journal impact factor and robustness of statistical analysis. A comprehensive evaluation of “liquid biopsy”, including cfDNA analysis, will be one of the critical challenges to better understand the early mechanisms of cancer development.
Keywords: cell-free DNA, diagnosis, cancer, liquid biopsy
Introduction
With a total of 1,658,370 cases each year in the US, cancer represents a prominent worldwide public health problem.1 Screening programs and early diagnosis have an important impact in improving disease-free survival and reducing mortality in cancer patients. As noninvasive approaches for early diagnosis foster patient compliance, they can be included in screening programs.
Currently, noninvasive serum-based biomarkers widely used in clinical practice include carcinoma antigen 125 (CA 125), carcinoembryonic antigen, carbohydrate antigen 19-9 (CA19-9), and prostate-specific antigen (PSA) for the detection of ovarian, colon, and prostate2,3 cancers, respectively.
These biomarkers generally have low specificity (high number of false-positive results): new noninvasive biomarkers have been relentlessly developed by researchers in the last years.
The increasing knowledge of the molecular pathogenesis of cancer and the rapid development of new molecular techniques are promoting the study of early molecular alterations in body fluids. Cell-free DNA (cfDNA) can be found in serum, plasma, urine, and other body fluids,4 representing a “liquid biopsy”, which is a circulating picture of a specific disease.5
The existence of cfDNA was firstly demonstrated about 70 years ago by Mandel and Metais;6 cfDNA originates from necrotic or apoptotic cells, and it is generally released by all types of cells. About 40 years after the discovery of cfDNA, Stroun et al showed that specific cancer alterations could be found in the cfDNA of patients.7 A number of following papers confirmed that cfDNA contains specific tumor-related alterations, such as mutations, methylation, and copy number variations (CNVs), thus confirming the existence of circulating tumor DNA (ctDNA).8,9
cfDNA in plasma or serum is the best characterized, while urine cfDNA (ucfDNA) is less known. However, some recent studies demonstrated that ucfDNA could also be a promising source of biomarkers.10
In blood, apoptosis seems to be the most frequent event that determines the amount of cfDNA. In cancer patients, however, the amount of cfDNA seems to be also influenced by necrosis.11,12 Since apoptosis seems to be the main release mechanism, circulating cfDNA has a size distribution which reveals an enrichment in short fragments of about 167 bp,13,14 corresponding to nucleosomes generated by apoptotic cells.
The present review focuses on the role of cfDNA in the diagnosis of tumors; a PubMed search was performed using the following phrases: cell-free DNA, liquid biopsy, diagnosis, early detection, ucfDNA, and circulating cfDNA. We selected the most recent and important studies on this topic according to journal impact factor and accuracy of statistical analysis.
Plasma/serum cfDNA
The evaluation of circulating cfDNA provides information regarding intratumor heterogeneity, reasons for primary resistance, detection of minimal residual disease, and disease evolution. In particular, the evaluation of ctDNA could allow for the reconstruction of the rearrangements and the epigenetic status of the tumor genome, and the identification of potentially metastatic clonal and subclonal cells.15 The noninvasive sample collection and the easy-to-perform circulating cfDNA analysis could make a good diagnostic tool. ctDNA analysis could replace tissue biopsies as it can be easily repeated over time, allowing tumor burden and treatment response monitoring, and early identification of relapse.
Noteworthy, the circulating cfDNA has a specific profile. Indeed, a study on prenatal cfDNA showed a specific length of about 167 bp due to nuclease-cleaved nucleosomes,14 suggesting that the major fragments were caused by apoptotic events both in healthy individuals and cancer patients. Interestingly, a recent study analyzing plasma DNA from 32 patients with stage IV colorectal cancer (CRC) showed a subset of patients (34.4%) with a biphasic size distribution (166 bp and 332 bp) of plasma DNA fragments associated with increased circulating tumor cell (CTC) numbers and elevated concentration of mutated plasma DNA component.16 Distribution of different fragments could associate with tumor content in plasma samples, with a potential diagnostic significance. However, a more sensitive test is needed for early tumor stage detection.
We reviewed studies aiming to characterize cfDNA for concentration, cancer-related genetic alterations (such as mutations, CNV, and microsatellite instability [MSI]), and epigenetic alterations. Table 1 summarizes the sensitivity and specificity of the markers reported.
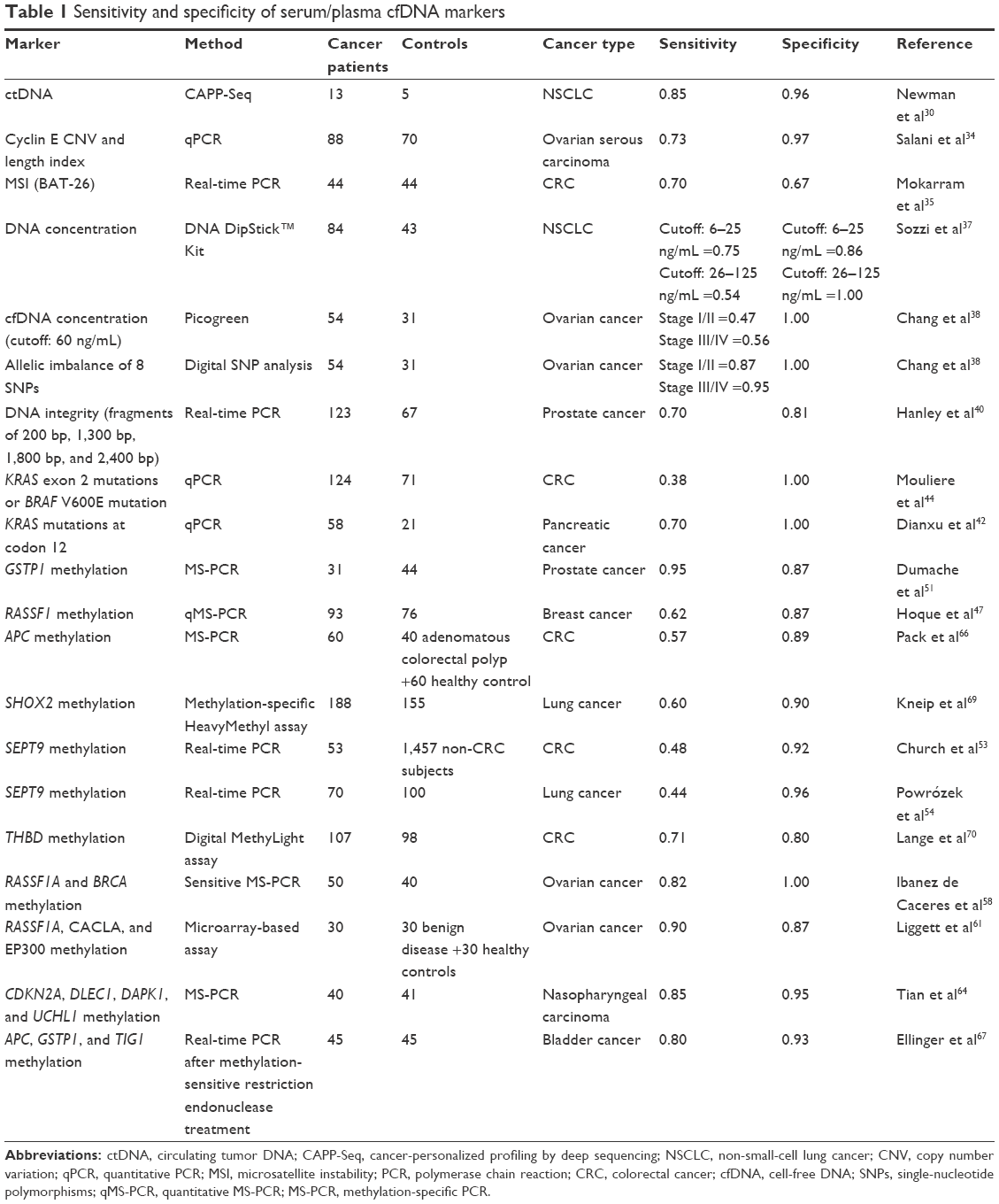 | Table 1 Sensitivity and specificity of serum/plasma cfDNA markers |
Circulating cfDNA concentration as a diagnostic marker
The amount of circulating cfDNA in serum and plasma seems to be significantly higher in patients with tumors than in healthy controls, especially in those with advanced-stage tumors than in early-stage tumors.17–19 The variability of the amount of circulating cfDNA is higher in cancer patients than in healthy individuals,16 and the amount of circulating cfDNA is influenced by several physiological and pathological conditions, including proinflammatory diseases.20,21 However, in a study conducted on 50 patients with resectable non-small-cell lung cancer (NSCLC), 101 patients with chronic respiratory inflammation, and 40 healthy volunteers, the authors found a significantly higher amount of circulating cfDNA in plasma of NSCLC patients than in subjects with chronic respiratory inflammation and healthy individuals, with 90% sensitivity and 80.5% specificity in discriminating NSCLC patients from healthy individuals (area under the curve [AUC] =0.90).22
Noteworthy, circulating cfDNA amount was influenced by various issues. Firstly, cfDNA amount was significantly higher in serum than in plasma,23,24 due to clotting of white blood cells in serum,23 suggesting that serum is a worse source for tumor-specific DNA analysis because of the possible presence of wild-type DNA.
Besides, circulating cfDNA is less stable, with a variable half-life in the circulation ranging from 15 minutes to several hours.25 For these reasons, diagnostic studies based on the amount of circulating cfDNA provide insufficiently robust and consistent results.
Cancer-related genetic alterations
Deep-sequencing data
Several studies aimed at correlating rearrangements in matched tissue and plasma samples were conducted to confirm that circulating cfDNA analysis can be used as a diagnostic tool.
A next-generation sequencing (NGS) evaluation on 50 cancer genes covering 2,800 COSMIC mutations in 60 tumor tissues and 31 plasma samples from 17 metastatic breast cancer patients showed a 76% concordance between tissue and plasma. The authors concluded that plasma can be prospectively tested as an alternative to metastatic biopsies.26 These results were confirmed in an independent cohort of 34 patients with 18 different tumor types: 46 genes covering more than 6,800 COSMIC mutations in tissue and plasma samples were analyzed. Twenty-seven out of 34 patients showed a 97% concordance between mutations found in tissue and in ctDNA.27 ctDNA-based NGS analyses could revolutionize the management of patients with potentially curable or metastatic disease.28
In a recent extensive study, Bettegowda et al used a digital polymerase chain reaction (PCR)-based method to identify ctDNA for early detection. They evaluated 640 plasma samples from patients with various cancer types, showing differences in the ctDNA samples: more than 75% of patients with advanced pancreatic, ovarian, colorectal, bladder, gastroesophageal, breast, melanoma, hepatocellular, and head-and-neck cancers presented evaluable ctDNA. However, ctDNA was found only in 48%–73% of localized tumors, such as colorectal, gastroesophageal, pancreatic, and breast adenocarcinoma.29 Newman et al performed an ultrasensitive and economical method called cancer-personalized profiling by deep sequencing (CAPP-Seq) for quantifying ctDNA.30 CAPP-Seq was implemented by the capture of recurrent mutations or gene fusions in NSCLC by complementary hybridization in solution. After evaluating 1 plasma sample from 5 controls, and 35 plasma samples from 13 patients with NSCLC, the authors identified ctDNA in 85% of patients with stage II–IV disease (sensitivity), while 96% of controls had no ctDNA (specificity). Stage I patients had a ctDNA/cfDNA ratio tenfold lower than stage II–IV patients, suggesting that DNA is less released into circulation at an early stage of disease, due to less apoptotic events or less vascularization. In conclusion, they found a significant correlation between ctDNA levels and the tumor volume.
Another study on EGFR deep sequencing in 288 plasma and matched biopsies from NSCLC patients showed a good concordance between the 2 sample types. However, in about 50% of patients, the alterations were found in tissues but were not confirmed in plasma samples.31 Sensitivity in finding plasma alterations was even lower in early stages.
Copy number variation
A study conducted on 90 primary breast cancer patients, 30 metastatic patients, and 98 female controls compared CNV of HER2 in tissue and plasma samples. HER2 copy number was found in 95% of plasma-matched tissue. No amplification was found in circulating cfDNA of the controls, but only 8 out of 68 patients with primary tumor and 5 out of 30 patients with metastatic tumor showed HER2 amplification in circulating cfDNA, suggesting a poor role in diagnosis for low-positive cases.32
Clonal and subclonal CNV was recently evaluated in ctDNA using massively multiplexed PCR and NGS. The authors matched tissue and plasma samples from 11 patients with stage II breast cancer for CNV detection. They showed that this technique was able to detect subclonal mutations in plasma, which were otherwise missed in tumor tissue biopsies.33 The authors found CNV with very low allelic values in ctDNA in 8 out of 11 (72.7%) patients with stage II breast cancer, concluding that this technique is suitable for early diagnosis.
Salani et al evaluated cyclin E CNV in the plasma of 88 ovarian serous carcinoma patients and 70 controls. The authors also studied the integrity index of cyclin E. Combination of cyclin E copy number (400 bp) and DNA length index had an AUC value of 0.936.34 Since the analysis of cyclin E had a higher sensitivity (95.6%) than cytology (74%), it may be suitable for false-negative cytology cases.
Microsatellite instability
BAT-25 and BAT-26 mononucleotide microsatellites were tested in tissue and serum from 44 healthy individuals and 44 CRC patients by real-time PCR and high-performance liquid chromatography (HPLC). BAT-26 resulted more sensitive than BAT-25 in identifying MSI tumors, and real-time PCR methods showed higher specificity than HPLC.35 Sensitivity and specificity values are reported in Table 1.
A study on serum from 34 patients with primary (n=8) and metastatic (n=24) breast cancers evaluated 4 microsatellite loci of chromosomes 10q22-23, 16q22-23, 17q11-12, and 17q21. The authors showed loss of heterozygosity at different loci in 16 patients and MSI in only 1 patient suggesting that genomic aberrations on chromosomes 10, 16, and 17 are detectable in circulating cfDNA and could represent diagnostic information.36 However, further evaluations on healthy controls are needed to confirm the diagnostic role of genetic aberrations.
A study on 84 patients with NSCLC and 43 healthy controls evaluated DNA concentration and MSI at loci located at 3p14.2 (D3S1300, FHIT locus), 3p21 (D3S1289), 3p23 (D3S1266), 3p24.2 (D3S2338), and 3p25–26 (D3S1304), which are hotspots of deletions in lung cancer. DNA concentration was higher even in stage I patients than in controls with an AUC of 0.844. Moreover, all patients except 3 with MSI in the plasma had a stage I tumor, suggesting a potential diagnostic use in lung cancer screening.37 None of these alterations were found in healthy controls.
Circulating cfDNA concentration and allelic status were evaluated as diagnostic markers by 8 single-nucleotide polymorphisms in combination with CA 125 levels in plasma DNA from 54 ovarian cancer patients and 31 controls. For circulating cfDNA concentration and allelic imbalances, the authors showed an area under the receiver operating characteristic (ROC) curve of 0.90 and 0.95 for patients and controls, respectively. Moreover, combination of the serum CA 125 level and the circulating cfDNA concentration increased the area under the ROC curve from 0.78 (CA 125 alone) to 0.84.38
Circulating cfDNA integrity
Circulating cfDNA was also evaluated in terms of integrity. Madhavan et al evaluated circulating cfDNA from breast cancer patients (n=383) and from healthy controls (n=100) by measuring ALU and LINE1 repetitive DNA elements using quantitative PCR. They observed a hierarchical decrease in circulating cfDNA integrity and an increase in circulating cfDNA concentration from healthy controls to primary and to metastatic breast cancer patients.39 They reported an AUC of 0.75.
Hanley et al evaluated circulating cfDNA integrity in plasma from 123 prostate cancer (PCa) patients and 67 controls. The authors analyzed 4 different fragment sizes (200 bp, 1,300 bp, 1,800 bp, and 2,300 bp), providing a score of integrity status. Circulating cfDNA integrity was positive in 86 out of 123 patients and in 13 out of 67 controls. Moreover, circulating cfDNA integrity identified 63% of the PCa patients negative for PSA levels.40
Mutations
KRAS mutations are the most studied alterations in different types of cancers, in tissue and body fluids.
Analysis of KRAS mutations in plasma of 44 pancreatic patients vs 37 controls showed a 27% sensitivity and a 100% specificity. Moreover, patients with mutation showed a significantly shorter survival than patients with wild-type KRAS.41
A following study on KRAS mutations at codon 12 in plasma samples from 58 pancreatic cancer patients and 21 healthy controls showed good test accuracy. In particular, the authors found KRAS mutations in 71% of the patients and none in healthy controls. Moreover, when the analysis was combined with CA19-9 evaluation, sensitivity rose from 73% (CA19-9 alone) to 90% (both tests), suggesting a better early detection tool.42
Spindler et al evaluated plasma cfDNA levels and tumor-specific KRAS mutations in a cohort of 229 metastatic CRC patients and 100 healthy individuals. They found higher levels of cfDNA in CRC patients than healthy individuals with an AUC of 0.9486 and a 85% overall concordance of KRAS mutations in plasma and tissue.43
An extensive study was conducted by Mouliere et al on the plasma of 124 CRC patients and 71 healthy individuals aiming at evaluating multi-markers, such as the total circulating cfDNA concentration, the presence of point mutations, the proportion of mutated allele, and the circulating cfDNA integrity index. They showed that the point mutations of KRAS (exon 2 mutations: G12V, G12A, G12D, G12S, G12C, G12R, and G13D) and BRAF (V600E) were found in 42 out of the 124 plasma samples analyzed. Moreover, they found that more than 80% of circulating cfDNA was shorter than 145 bp, with higher – but more fragmented – DNA concentration in plasma from patients than controls.44 BRAF mutation (V600E) was also informative for monitoring melanoma in serum of patients.45
Though the KRAS mutations analysis reported low sensitivity in circulating cfDNA, the analysis comparing tissue and circulating cfDNA plasma samples revealed higher sensitivity. Bettegowda et al analyzed matched tissue and plasma samples from 206 patients with metastatic CRCs, showing a sensitivity of 87.2% and a specificity of 99.2% of ctDNA detection.29
Epigenetics
Epigenetic events, such as DNA methylation in CpG islands, occur early in cancer development suggesting a potential role of DNA methylation as a biomarker for early diagnosis.46 Blood-based test could improve sensitivity and specificity of current screening; to this aim, a number of potential methylation biomarkers from plasma or serum were tested.47,48 DNA methylation is the most investigated event in cfDNA, and several markers have been proposed.
Global genomic hypomethylation is a hallmark of cancer in humans. Chen et al investigated the role of hypomethylation of Alu elements in tumor tissue and matched serum of glioma patients and healthy controls by bisulfite sequencing. They showed a correlation of Alu hypomethylation between tumor and serum samples; ROC curve analysis showed an AUC for diagnosis of 0.861, suggesting that the detection of Alu hypomethylation in serum may be used in clinical practice for the diagnosis of glioma.49
GSTP1 methylation in PCa is an early event in carcinogenesis, representing one of the most studied epigenetic markers with a diagnostic role. The meta-analysis conducted by Wu et al on GSTP1 methylation in urine, plasma, and serum showed 0.50%–0.75% sensitivity and 0.80%–0.95% specificity.50 Another important recent study showed 95% sensitivity and 87% specificity.51 Although it is not approved by the US Food and Drug Administration (FDA), it could nevertheless be associated with PSA in serum.52
Methylation of SEPT9 promoter region in circulating cfDNA of CRC gave promising results at first assessment (72%–90% sensitivity and 88%–90% specificity), but it presented low sensitivity in the subsequent PRESEPT study.53 Even though the test still requires some improvements, it is under review for FDA approval for clinical use. SEPT9 test for CRC detection has been used for lung cancer with 44% sensitivity and 96% specificity.54
Hypermethylation of the promoter regions of RASSF1A has a role in different cancer types. Hoque et al showed that RASSF1A methylation in plasma of breast cancer patients reached 62% sensitivity and 87% specificity.47 Analysis of RASSF1A and RARbeta2 methylation provided 95% of diagnostic coverage in breast cancer patients and 60% in patients with benign lesions and did not present false-positive results in healthy women.55
Cassinotti et al highlighted that methylation of RASSF1A combined with CYCD2, HIC1, PAX5, RB1, and SRBC distinguished CRC patients and controls with 84% sensitivity and 68% specificity, whereas RASSF1A combined with HIC1 and MDG1 differentiated patients with adenomatous polyps and controls with 55% sensitivity and 65% specificity.56 RASSF1 combined with VHL methylation analyzed in cell-free serum DNA in patients with renal cell carcinoma reached high specificity but low sensitivity for renal cell carcinoma diagnosis.57
Ibanez de Caceres et al identified RASSF1A and BRCA hypermethylation in serum DNA of ovarian cancer patients with 82% sensitivity and 100% specificity with sensitive methylation-specific PCR (MS-PCR).58
MethDet56 is a novel method for identifying new biomarkers, consisting in a microarray panel of frequently methylated genes for measuring the quantity of methylated target sequence following digestion with endonuclease Hin6I and PCR amplification of undigested fragments.59 At first, it was applied to pancreatic cancer, and then to ovarian cancer, selecting several genes able to identify ovarian cancer.60,61 The methylation panel of RASSF1A, CALCA, and EP300 distinguished between patients with ovarian cancer and healthy controls with 90% sensitivity and 87% specificity, whereas methylation of RASSF1A and PGR differentiated between ovarian cancer samples and benign ovarian disease with 80% sensitivity and 73.3% specificity.61 The methylation panel of RASSF1A, UCHL1, NPTX2, SARP2, ppENK, and p16 (CDKN2A) showed that they are able to distinguish between plasma DNA derived from pancreatic cancer samples and control samples by MS-PCR and confirm by direct sequencing after bisulfite treatment. Moreover, CDKN2A was differentially methylated between pancreatic cancer and chronic pancreatitis, which is a recognized risk factor for pancreatic cancer.62 Methylated CDKN2A in plasma is useful for detecting lung cancer, but it seems to work better when it is a part of a biomarker panel rather than as a single gene.63
Promoter hypermethylation of CDKN2A combined with DLEC1, DAPK1, and UCHL1 analyzed with MS-PCR in serum DNA of patients with nasopharyngeal carcinoma achieved 85% sensitivity and 95.1% specificity, which is higher than that achieved by the individual gene.64
Cell-free plasma DNA methylation levels of GSTP1, p16, and APC genes were found significantly higher in PCa patients than in cancer-free controls with pyrosequencing approach by Delgado-Cruzata et al.65
APC is a biomarker investigated in different diseases. Pack et al showed that APC methylation in plasma has 57% sensitivity and 84% specificity for CRC detection. It is also significantly increased in stage I CRC, and the most sensitive (57%) and specific (89%) marker when compared to the promoter methylation of other 4 genes.66 Hypermethylation of APC, GSTP1, and TIG1 in serum cfDNA is able to distinguish bladder cancer and controls with 80% sensitivity and 93% specificity.67 Radpour et al identified a methylation panel of 8 tumor suppressor genes including APC in circulating cfDNA which is higher in patients with breast cancer than in controls. This panel could achieve >90% sensitivity and specificity, for the development of a blood-based test for breast cancer diagnosis.68
APC, RARb, and CDH13 were found to be differentially methylated in cfDNA between patients with lung cancer and healthy controls,63 although with low sensitivity. Also, SHOX2 gene promoter methylation was found to be a potential biomarker for lung cancer detection reaching 60% sensitivity and 90% specificity.69
Other methylated genes could have a role of cancer type-specific biomarkers. CST6 shows a differentially methylated pattern between breast cancer and control plasma samples using bisulfite conversion and MS-PCR in circulating cfDNA. CST6 is also included in an 8-gene biomarker panel which reaches 90% sensitivity and specificity in patients with early-stage breast cancer vs 30 healthy controls as shown using PCR after bisulfite treatment.68
The promoter region methylation of THBD differentiated CRC and control plasma samples with 71% sensitivity and 80% specificity.70
Urine cfDNA
Urine sample is very advantageous for noninvasive detection of cancer. Given its complexity, it could be a source for a variety of biomarkers, from proteins to nucleic acids.71 Some urinary biomarkers for early diagnosis of prostate and bladder cancers are already FDA approved (eg, NMP22, FISH Urovysion, PCA3).72,73 Most of the published data regarding urine focus on biomarkers from exfoliated cells, and very little is known about the role of ucfDNA.
ucfDNA originates from cells coming in direct contact with urine (necrotic or apoptotic cells) or from cfDNA in blood. Glomerular filtration acts as a “dimensional selection”: only small DNA fragments from circulation (about 100 bp) can penetrate through the pores of the glomerular barrier, appearing in urine.74,75 As a consequence, ucfDNA could provide important information on specific alterations of circulating cfDNA and genomic DNA coming from cells shedding into urine, thus being useful for identifying both cancers of the urological tract and other solid tumors.76
All papers published on ucfDNA for diagnostic purposes are preliminary studies conducted on small series of patients and still far from any clinical application. The development of new molecular technologies (eg, NGS or digital PCR), alongside a broader case series analysis, will offer a deeper insight into the practical clinical translation of these promising findings.
Table 2 summarizes the sensitivity and specificity of the markers analyzed in ucfDNA.
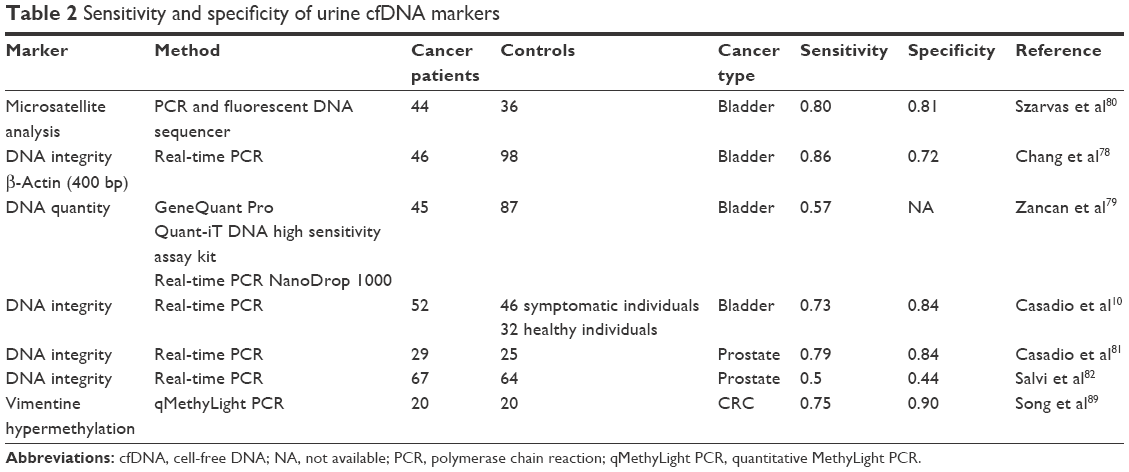 | Table 2 Sensitivity and specificity of urine cfDNA markers |
Cancer-related genetic alterations
ucfDNA can be used for studying genetic alterations from tumors distal to the urological tract. Fifteen years ago, 2 studies demonstrated that specific cancer-associated mutations detectable in plasma and serum were also present in urine,73,75 suggesting that ucfDNA may be studied for detecting specific alterations.
ucfDNA genetic alterations are mostly evaluated for urological cancers, even though some studies demonstrated that urine could effectively produce a picture of DNA alterations coming from circulation.76
Su et al demonstrated that k-RAS gene mutations were detectable in urine of patients with adenomatous polyps or CRC and that mutations in urine better correlate with tissue than plasma samples. In a following study on 20 patients, they further confirmed their previous hypothesis77 that urine could be even more representative of k-RAS-mutated DNA than serum or plasma. They used a restriction-enriched PCR to analyze k-RAS mutations and found 95%, 35%, and 40% mutation incidence in urine, serum, and plasma samples, respectively. Although remarkable, their findings are unusable for diagnostic purposes (low number of analyzed cases), even though 95% mutation incidence in urine samples suggests a good test sensitivity.
A number of studies have been published on ucfDNA characteristics in urological cancers, especially bladder.
Firstly, ucfDNA quantity was evaluated by Chang et al using picogreen,78 though it resulted inaccurate for proper distinction between bladder cancer patients and healthy individuals, as confirmed by Zancan et al.79 On the other hand, Chang et al demonstrated that the detection of a long (400 bp) DNA fragment could be a potential diagnostic marker, with 86% sensitivity and 72% specificity.78 Casadio et al confirmed the ucfDNA integrity, with a real-time PCR approach, detecting 3 long amplicons belonging to 3 oncogenes frequently amplified in bladder cancer (c-MYC, BCAS1, HER2).10 They obtained 73% sensitivity, with 84% and 83% specificity in healthy individuals and in patients with urological symptoms, respectively. Interestingly, Szarvas et al80 analyzed 12 microsatellite markers mapped on 6 different chromosomes. They obtained 80% sensitivity and 81% specificity. After comparing the results obtained in urine supernatant vs urine sediments, they reported higher sensitivity in the cell-free fraction.
ucfDNA integrity was also evaluated for early diagnosis of PCa: despite an initial study with promising results,81 this marker failed to have a good early diagnostic role in the confirmatory study.82
Following the technological advancements, some recent papers83–85 with no diagnostic purposes used NGS approaches, microarray, or digital PCR on ucfDNA. These studies are important because they demonstrated the feasibility of NGS approaches on cfDNA in urine,85 even presenting a higher tumor genome burden in the cell-free fraction than in the cell sediment.84 All these results indicate further applications of ucfDNA in tumors.
Epigenetics
Besides serum and plasma circulating cfDNA, GSTP1 methylation also appears to be a promising diagnostic marker in ucfDNA as shown in a study,86 although it was conducted on a small case series with no robust statistical results.
LINE-1 hypomethylation in bladder cancer is a promising DNA methylation biomarker for diagnostic purposes.87 Ghanjati et al found that DNA methylation profiles of LINE-1 promoter regions in ucfDNA of urothelial carcinoma patients can be detected by bisulfite genomic sequencing. LINE-1 hypomethylation may be used for diagnostic purposes as unmethylated full-length LINE-1 sequences prevail in urine of cancer patients.88
Bisulfite conversion is used for urological tumor detection as DNA fragments are longer than 300 bp. Bisulfite conversion, however, is unsuitable for other types of tumor detection due to further DNA fragmentation. For this reason, Song et al developed a quantitative MethyLight PCR-based assay to detect hypermethylated vimentin in the low-molecular weight (LMW) voided urine of CRC patients. The assay targeted a 39-nucleotide segment of the hypermethylated region of vimentin gene, detecting hypermethylated vimentin in 75% of LMW urine DNA from CRC patients and in 10% of urine samples of healthy controls. As a consequence, a urine test using epigenetic markers may be evaluated for CRC screening.89
Feng et al demonstrated the feasibility of urine hypermethylation of DAPK1, RARB, TWIST1, and CDH13 genes for cervical cancer screening with sensitivity similar to that of an exfoliated cervical cytology.90
Discussion
cfDNA is an undeniable source of biomarkers for assisting clinicians in early cancer detection, monitoring patients under treatment, and predicting drug response or disease progression.91,92 It is noteworthy that the studies on the role of circulating cfDNA in earlier stages of cancer are not as many as those on advanced cancer. This is probably due to the fact that ctDNA in circulation is more representative in patients with advanced and metastatic than local diseases, and that the likelihood of finding alterations increases alongside the aggressiveness of the disease.29 However, we strongly believe that the technological advancements (eg, NGS approaches and digital PCR-based methods) will improve sensitivity in early detection and provide more robust data.
The ideal early diagnostic marker should be noninvasive and highly accurate, with a good cost/benefit ratio and allow simple interpretation of the results. Despite that cfDNA seems to be a good source of markers with such characteristics, and the high number of publications, none of the cfDNA-related markers have yet entered the clinical practice. Marker performance varies widely depending on the population, the sample storage, the test performance, and the result analysis. For these reasons, comparison between cfDNA-based biomarkers is inappropriate, unless within a single study, and translation to clinical practice is still difficult. In addition, pilot studies on the same marker are mostly retrospective, and the sample size and the statistical power are often inadequate for proving the robustness of a cfDNA biomarker. Lastly, studies reporting diagnostic accuracy of early diagnostic markers, including cfDNA, should follow the Standards for Reporting of Diagnostic Accuracy guidelines for publication.93
Epigenetic alterations seem to be the most promising biomarkers based on circulating cfDNA studies. Epigenetic events, especially methylation of specific tumor suppressor genes promoter, are often early phenomena in the process of carcinogenesis. For this reason, they are more suitable for early detection than mutations or other genetic alterations. One example of a highly promising approach is the analysis of GSTP1 in PCa,50 which is detectable in circulation and urine in a high number of cancer patients, without being present in healthy individuals. However, this promising marker also did not enter the clinical practice due to the lack of inter-studies reproducibility; several methylation studies used different approaches such as bisulfite conversion and immunoprecipitation enzymatic digestion, leading to noncomparable results.
ucfDNA has been mostly evaluated for urological cancers. Despite the limited number of studies and that the restricted sample size does not allow for valid conclusions, ucfDNA appears to be a promising source of early diagnostic markers. NGS approach seems to be feasible in urine samples, showing that many efforts have been recently made to improve the technology for the study of ucfDNA. This will lead to further advancements in the employment of ucfDNA.
Clinical applications and future perspectives
Using ctDNA characterization for the early diagnosis of tumors has a great potential for clinical application; however, some limitations have to be considered. First of all, even if ctDNA could be distinguished from total cfDNA using somatic mutations analysis, the very low presence of ctDNA (often only <0.1%) needs more sensitive and reproducible methods. Secondly, cfDNA characteristics could be different among patients, forcing a qualitative analysis and specific optimization procedure for each patient. Despite these limitations and the low number of large studies on diagnostics, there are a number of potential clinical applications encouraging the search for new, sensitive, and robust methods. A very promising application in early diagnosis is adding ctDNA detection to conventional markers used for screening programs; in this context, the detection of somatic mutations might suggest an early development of disease. To reach these clinical applications, several sensitive methods have been already proposed: first of all, massively parallel sequencing or NGS,94,95 and then digital PCR. Nowadays, ctDNA could be analyzed for gene-specific panel or whole exome/genome using NGS with the advantages of multiple mutations data from only 1 analysis. In parallel, digital PCR analysis offers a high level of sensitivity (up to about 0.01%) and specificity for only few molecular targets94 with real quantification of mutated sample percentage compared to total samples. Improvement methods will combine the multiplicity data from NGS results and the more sensitive and precise digital PCR, for moving to real clinical and routine applications.
Moreover, a more detailed picture of disease status is also given by the knowledge about all liquid biopsy aspects such as CTCs, small extracellular vesicles, and cell-free RNA. However, for an early diagnosis, ctDNA seems to be the most promising marker for the real clinical application, thanks to the most easy detection and stability compared to CTCs and cell-free RNA.
The next achievement in cfDNA evaluation will be robustness: larger prospective studies, with more sensitive and reproducible methods, are needed. Also, other laboratories must confirm cfDNA alterations as early diagnostic markers before being translated into clinical practice.
A deeper knowledge of cancer development and evolution will lead to more sensitive and robust analysis methods useful for the characterization of all aspects of liquid biopsy, thus providing answers for each clinical query.
Acknowledgment
The authors wish to thank Veronica Zanoni for editing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Terry KL, Schock H, Fortner RT, et al. A prospective evaluation of early detection biomarkers for ovarian cancer in the European EPIC cohort. Clin Cancer Res. Epub 2016 Apr 8. | ||
Zhang Y, Yang J, Li H, Wu Y, Zhang H, Chen W. Tumor markers CA19-9, CA242 and CEA in the diagnosis of pancreatic cancer: a meta-analysis. Int J Clin Exp Med. 2015;8(7):11683–11691. | ||
Chan AK, Chiu RW, Lo YM; Clinical Sciences Reviews Committee of the Association of Clinical Biochemists. Cell-free nucleic acids in plasma, serum and urine: a new tool in molecular diagnosis. Ann Clin Biochem. 2003;40(Pt 2):122–130. | ||
De Mattos-Arruda L, Caldas C. Cell-free circulating tumour DNA as a liquid biopsy in breast cancer. Mol Oncol. 2016;10(3):464–474. | ||
Mandel P, Metais P. Les acides nucleiques du plasma sanguin chez l’ homme [The nucleic acids in blood plasma in humans]. C R Seances Soc Biol Fil. 1948;142(3–4):241–243. | ||
Stroun M, Anker P, Maurice P, Lyautey J, Lederrey C, Beljanski M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology. 1989;46(5):318–322. | ||
Goessl C, Krause H, Müller M, et al. Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res. 2000;60(21):5941–5945. | ||
Frenel JS, Carreira S, Goodall J, et al. Serial next-generation sequencing of circulating cell-free DNA evaluating tumor clone response to molecularly targeted drug administration. Clin Cancer Res. 2015;21(20):4586–4596. | ||
Casadio V, Calistri D, Tebaldi M, et al. Urine cell-free DNA integrity as a marker for early bladder cancer diagnosis: preliminary data. Urol Oncol. 2013;31(8):1744–1750. | ||
Hao TB, Shi W, Shen XJ, et al. Circulating cell-free DNA in serum as a biomarker for diagnosis and prognostic prediction of colorectal cancer. Br J Cancer. 2014;111(8):1482–1489. | ||
Zonta E, Nizard P, Taly V. Assessment of DNA integrity, applications for cancer research. Adv Clin Chem. 2015;70:197–246. | ||
Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61(1):112–123. | ||
Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2(61):61ra91. | ||
Kaiser J. Medicine. Keeping tabs on tumor DNA. Science. 2010;327(5969):1074. | ||
Heitzer E, Auer M, Hoffmann EM, et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int J Cancer. 2013;133(2):346–356. | ||
Sozzi G, Conte D, Leon M, et al. Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol. 2003;21(21):3902–3908. | ||
Kim K, Shin DG, Park MK, et al. Circulating cell-free DNA as a promising biomarker in patients with gastric cancer: diagnostic validity and significant reduction of cfDNA after surgical resection. Ann Surg Treat Res. 2014;86(3):136–142. | ||
Shao X, He Y, Ji M, et al. Quantitative analysis of cell-free DNA in ovarian cancer. Oncol Lett. 2015;10(6):3478–3482. | ||
Raptis L, Menard HA. Quantitation and characterization of plasma DNA in normals and patients with systemic lupus erythematosus. J Clin Invest. 1980;66(6):1391–1399. | ||
Shapiro B, Chakrabarty M, Cohn EM, Leon SA. Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer. 1983;51(11):2116–2120. | ||
Szpechcinski A, Chorostowska-Wynimko J, Struniawski R, et al. Cell-free DNA levels in plasma of patients with non-small-cell lung cancer and inflammatory lung disease. Br J Cancer. 2015;113(3):476–483. | ||
Lee TH, Montalvo L, Chrebtow V, Busch MP. Quantitation of genomic DNA in plasma and serum samples: higher concentrations of genomic DNA found in serum than in plasma. Transfusion. 2001;41(2):276–282. | ||
Chan KCA, Yeung S-W, Lui W-B, Rainer TH, Lo YMD. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem. 2005;51(4):781–784. | ||
Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer – a survey. Biochim Biophys Acta. 2007;1775(1):181–232. | ||
Rothé F, Laes J-F, Lambrechts D, et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014;25(10):1959–1965. | ||
Lebofsky R, Decraene C, Bernard V, et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol Oncol. 2015;9(4):783–790. | ||
Lianos GD, Mangano A, Cho WC, Dionigi G, Roukos DH. Circulating tumor DNA: new horizons for improving cancer treatment. Future Oncol. 2015;11(4):545–548. | ||
Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24. | ||
Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–554. | ||
Uchida J, Kato K, Kukita Y, et al. Diagnostic accuracy of noninvasive genotyping of EGFR in lung cancer patients by deep sequencing of plasma cell-free DNA. Clin Chem. 2015;61(9):1191–1196. | ||
Page K, Hava N, Ward B, et al. Detection of HER2 amplification in circulating free DNA in patients with breast cancer. Br J Cancer. 2011;104(8):1342–1348. | ||
Kirkizlar E, Zimmermann B, Constantin T, et al. Detection of clonal and subclonal copy-number variants in cell-free DNA from patients with breast cancer using a massively multiplexed PCR methodology. Transl Oncol. 2015;8(5):407–416. | ||
Salani R, Davidson B, Fiegl M, et al. Measurement of cyclin E genomic copy number and strand length in cell-free DNA distinguish malignant versus benign effusions. Clin Cancer Res. 2007;13(19):5805–5809. | ||
Mokarram P, Rismanchi M, Alizadeh Naeeni M, et al. Microsatellite instability typing in serum and tissue of patients with colorectal cancer: comparing real time PCR with hybridization probe and high-performance liquid chromatography. Mol Biol Rep. 2014;41(5):2835–2844. | ||
Schwarzenbach H, Müller V, Stahmann N, Pantel K. Detection and characterization of circulating microsatellite-DNA in blood of patients with breast cancer. Ann N Y Acad Sci. 2004;1022:25–32. | ||
Sozzi G, Conte D, Mariani L, et al. Analysis of circulating tumor DNA in plasma at diagnosis and during follow-up of lung cancer patients. Cancer Res. 2001;61(12):4675–4678. | ||
Chang HW, Lee SM, Goodman SN, et al. Assessment of plasma DNA levels, allelic imbalance, and CA 125 as diagnostic tests for cancer. J Natl Cancer Inst. 2002;94(22):1697–1703. | ||
Madhavan D, Wallwiener M, Bents K, et al. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res Treat. 2014;146(1):163–174. | ||
Hanley R, Rieger-Christ KM, Canes D, et al. DNA integrity assay: a plasma-based screening tool for the detection of prostate cancer. Clin Cancer Res. 2006;12(15):4569–4574. | ||
Castells A, Puig P, Móra J, et al. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J Clin Oncol. 1999;17(2):578–584. | ||
Dianxu F, Shengdao Z, Tianquan H, et al. A prospective study of detection of pancreatic carcinoma by combined plasma K-ras mutations and serum CA19-9 analysis. Pancreas. 2002;25(4):336–341. | ||
Spindler KLG, Pallisgaard N, Andersen RF, Brandslund I, Jakobsen A. Circulating free DNA as biomarker and source for mutation detection in metastatic colorectal cancer. PLoS One. 2015;10(4):1–14. | ||
Mouliere F, El Messaoudi S, Pang D, Dritschilo A, Thierry AR. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol Oncol. 2014;8(5):927–941. | ||
Shinozaki M, O’Day SJ, Kitago M, et al. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin Cancer Res. 2007;13(7):2068–2074. | ||
Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. | ||
Hoque MO, Feng Q, Toure P, et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J Clin Oncol. 2006;24(26):4262–4269. | ||
Guerrero-Preston R, Hadar T, Ostrow KL, et al. Differential promoter methylation of kinesin family member 1a in plasma is associated with breast cancer and DNA repair capacity. Oncol Rep. 2014;32(2):505–512. | ||
Chen J, Gong M, Lu S, et al. Detection of serum Alu element hypomethylation for the diagnosis and prognosis of glioma. J Mol Neurosci. 2013;50(2):368–375. | ||
Wu T, Giovannucci E, Welge J, Mallick P, Tang WY, Ho SM. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: a meta-analysis. Br J Cancer. 2011;105(1):65–73. | ||
Dumache R, Puiu M, Motoc M, Vernic C, Dumitrascu V. Prostate cancer molecular detection in plasma samples by glutathione S-transferase P1 (GSTP1) methylation analysis. Clin Lab. 2014;60(5):847–852. | ||
Van Neste L, Herman JG, Otto G, Bigley JW, Epstein JI, Van Criekinge W. The epigenetic promise for prostate cancer diagnosis. Prostate. 2012;72(11):1248–1261. | ||
Church TR, Wandell M, Lofton-Day C, et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2014;63(2):317–325. | ||
Powrózek T, Krawczyk P, Kucharczyk T, Milanowski J. Septin 9 promoter region methylation in free circulating DNA-potential role in noninvasive diagnosis of lung cancer: preliminary report. Med Oncol. 2014;31(4):917. | ||
Skvortsova TE, Rykova EY, Tamkovich SN, et al. Cell-free and cell-bound circulating DNA in breast tumours: DNA quantification and analysis of tumour-related gene methylation. Br J Cancer. 2006; 94(10):1492–1495. | ||
Cassinotti E, Melson J, Liggett T, et al. DNA methylation patterns in blood of patients with colorectal cancer and adenomatous colorectal polyps. Int J Cancer. 2012;131(5):1153–1157. | ||
de Martino M, Klatte T, Haitel A, Marberger M. Serum cell-free DNA in renal cell carcinoma: a diagnostic and prognostic marker. Cancer. 2012;118(1):82–90. | ||
Ibanez de Caceres I, Battagli C, Esteller M, et al. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 2004;64(18):6476–6481. | ||
Melnikov AA, Scholtens D, Talamonti MS, Bentrem DJ, Levenson VV. Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J Surg Oncol. 2009;99(2):119–122. | ||
Melnikov A, Scholtens D, Godwin A, Levenson V. Differential methylation profile of ovarian cancer in tissues and plasma. J Mol Diagn. 2009;11(1):60–65. | ||
Liggett TE, Melnikov A, Yi Q, et al. Distinctive DNA methylation patterns of cell-free plasma DNA in women with malignant ovarian tumors. Gynecol Oncol. 2011;120(1):113–120. | ||
Park JW, Baek IH, Kim YT. Preliminary study analyzing the methylated genes in the plasma of patients with pancreatic cancer. Scand J Surg. 2012;101(1):38–44. | ||
Warton K, Samimi G. Methylation of cell-free circulating DNA in the diagnosis of cancer. Front Mol Biosci. 2015;2:13. | ||
Tian F, Yip SP, Kwong DL, Lin Z, Yang Z, Wu V. Promoter hypermethylation of tumor suppressor genes in serum as potential biomarker for the diagnosis of nasopharyngeal carcinoma. Cancer Epidemiol. 2013;37(5):708–713. | ||
Delgado-Cruzata L, Hruby GW, Gonzalez K, et al. DNA methylation changes correlate with Gleason score and tumor stage in prostate cancer. DNA Cell Biol. 2012;31(2):187–192. | ||
Pack SC, Kim HR, Lim SW, et al. Usefulness of plasma epigenetic changes of five major genes involved in the pathogenesis of colorectal cancer. Int J Colorectal Dis. 2013;28(1):139–147. | ||
Ellinger J, El Kassem N, Heukamp LC, et al. Hypermethylation of cell-free serum DNA indicates worse outcome in patients with bladder cancer. J Urol. 2008;179(1):346–352. | ||
Radpour R, Barekati Z, Kohler C, et al. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011;6(1):e16080. | ||
Kneip C, Schmidt B, Seegebarth A, et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J Thorac Oncol. 2011;6(10):1632–1638. | ||
Lange CP, Campan M, Hinoue T, et al. Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PLoS One. 2012;7(11):e50266. | ||
Calistri D, Casadio V, Bravaccini S, Zoli W, Amadori D. Urinary biomarkers of non-muscle-invasive bladder cancer: current status and future potential. Expert Rev Anticancer Ther. 2012;12(6):743–752. | ||
Grossman HB, Messing E, Soloway M, et al. Detection of bladder cancer using a point-of-care proteomic assay. JAMA. 2005;293(7):810–816. | ||
Wei JT, Feng Z, Partin AW, et al. Can urinary PCA3 supplement PSA in the early detection of prostate cancer? J Clin Oncol. 2014;32(36):4066–4072. | ||
Botezatu I, Serdyuk O, Potapova G, et al. Genetic analysis of DNA excreted in urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin Chem. 2000;46(8 Pt 1):1078–1084. | ||
Su YH, Wang M, Brenner DE, et al. Human urine contains small, 150 to 250 nucleotide-sized, soluble DNA derived from the circulation and may be useful in the detection of colorectal cancer. J Mol Diagn. 2004;6(2):101–107. | ||
Su YH, Wang M, Aiamkitsumrit B, Brenner DE, Block TM. Detection of a K-ras mutation in urine of patients with colorectal cancer. Cancer Biomark. 2005;1(2–3):177–182. | ||
Su YH, Wang M, Brenner DE, Norton PA, Block TM. Detection of mutated K-ras DNA in urine, plasma, and serum of patients with colorectal carcinoma or adenomatous polyps. Ann N Y Acad Sci. 2008;1137:197–206. | ||
Chang HW, Tsui KH, Shen LC, Huang HW, Wang SN, Chang PL. Urinary cell-free DNA as a potential tumor marker for bladder cancer. Int J Biol Markers. 2007;22(4):287–294. | ||
Zancan M, Galdi F, Di Tonno F, et al. Evaluation of cell-free DNA in urine as a marker for bladder cancer diagnosis. Int J Biol Markers. 2009;24(3):147–155. | ||
Szarvas T, Kovalszky I, Bedi K, et al. Deletion analysis of tumor and urinary DNA to detect bladder cancer: urine supernatant versus urine sediment. Oncol Rep. 2007;18(2):405–409. | ||
Casadio V, Calistri D, Salvi S, et al. Urine cell-free DNA integrity as a marker for early prostate cancer diagnosis: a pilot study. Biomed Res Int. 2013;2013:270457. | ||
Salvi S, Gurioli G, Martignano F, et al. Urine cell-free DNA integrity analysis for early detection of prostate cancer patients. Dis Markers. 2015;2015:574120. | ||
Birkenkamp-Demtröder K, Nordentoft I, Christensen E, et al. Genomic alterations in liquid biopsies from patients with bladder cancer. Eur Urol. 2016;70(1):75–82. | ||
Togneri FS, Ward DG, Foster JM, et al. Genomic complexity of urothelial bladder cancer revealed in urinary cfDNA. Eur J Hum Genet. 2016;24(8):1167–1174. | ||
Xia Y, Huang CC, Dittmar R, et al. Copy number variations in urine cell free DNA as biomarkers in advanced prostate cancer. Oncotarget. Epub 2016 Apr 26. | ||
Bryzgunova OE, Morozkin ES, Yarmoschuk SV, Vlassov VV, Laktionov PP. Methylation-specific sequencing of GSTP1 gene promoter in circulating/extracellular DNA from blood and urine of healthy donors and prostate cancer patients. Ann N Y Acad Sci. 2008;1137:222–225. | ||
Schulz WA, Steinhoff C, Florl AR. Methylation of endogenous human retroelements in health and disease. Curr Top Microbiol Immunol. 2006;310:211–250. | ||
Ghanjati F, Beermann A, Hermanns T, et al. Unreserved application of epigenetic methods to define differences of DNA methylation between urinary cellular and cell-free DNA. Cancer Biomark. 2014;14(5):295–302. | ||
Song BP, Jain S, Lin SY, et al. Detection of hypermethylated vimentin in urine of patients with colorectal cancer. J Mol Diagn. 2012;14(2):112–119. | ||
Feng Q, Hawes SE, Stern JE, et al. Promoter hypermethylation of tumor suppressor genes in urine from patients with cervical neoplasia. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1178–1184. | ||
Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–437. | ||
Schwarzenbach H, Alix-Panabières C, Müller I, et al. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin Cancer Res. 2009;15(3):1032–1038. | ||
Bossuyt PM, Reitsma JB, Bruns DE, et al. STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies. Clin Chem. 2015;61(12):1446–1452. | ||
Chaudhuri AA, Binkley MS, Osmundson EC, Alizadeh AA, Diehn M. Predicting radiotherapy responses and treatment outcomes through analysis of circulating tumor DNA. Semin Radiat Oncol. 2015;25(4):305–312. | ||
Ignatiadis M, Lee M, Jeffrey SS. Circulating tumor cells and circulating tumor DNA: challenges and opportunities on the path to clinical utility. Clin Cancer Res. 2015;21(21):4786–4800. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.