Back to Journals » Cancer Management and Research » Volume 10
Celecoxib in breast cancer prevention and therapy
Authors Li J ![]() , Hao Q, Cao W
, Hao Q, Cao W ![]() , Vadgama JV
, Vadgama JV ![]() , Wu Y
, Wu Y ![]()
Received 28 June 2018
Accepted for publication 23 July 2018
Published 26 October 2018 Volume 2018:10 Pages 4653—4667
DOI https://doi.org/10.2147/CMAR.S178567
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Raphael Catane
Jieqing Li,1,2 Qiongyu Hao,2 Wei Cao,2,3 Jaydutt V Vadgama,2,4,* Yong Wu2,4,*
1Department of Breast Surgery, Tianjin Central Hospital of Gynecology and Obstetrics, Tianjin, China; 2Division of Cancer Research and Training, Department of Internal Medicine, Charles R. Drew University of Medicine and Science, Los Angeles,CA, USA; 3Department of Nuclear Medicine, Union Hospital of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China; 4David Geffen UCLA School of Medicine and UCLA Jonsson Comprehensive Cancer Center, University of California, Los Angeles, CA, USA
*These authors contributed equally to this work
Abstract: Breast cancer has a high incidence worldwide. The results of substantial studis reveal that inflammation plays an important role in the initiation, development, and aggressiveness of many malignancies. The use of celecoxib, a novel NSAID, is repetitively associated with the reduced risk of the occurrence and progression of a number of types of cancer, particularly breast cancer. This observation is also substantiated by various meta-analyses. Clinical trials have been implemented on integration treatment of celecoxib and shown encouraging results. Celecoxib could be treated as a potential candidate for antitumor agent. There are, nonetheless, some unaddressed questions concerning the precise mechanism underlying the anticancer effect of celecoxib as well as its activity against different types of cancer. In this review, we discuss different mechanisms of anticancer effect of celecoxib as well as preclinical/clinical results signifying this beneficial effect.
Keywords: celecoxib, breast cancer, COX-2, inflammation
Introduction
Breast cancer (BC) is the most frequent cancer in women, the second most common cancer worldwide, and the second primary cause of cancer-related deaths.1 One in eight women who live to age 85 years will develop BC over the course of their lifetime.2 Previous studies suggest that inflammation is associated with cancer, and a robust correlation exists between the manifestation of inflammation and the progress of precancerous lesions at a number of anatomic sites.3 On the other hand, cancer cells might exploit components of the inflammatory process to induce angiogenesis, inhibit apoptosis, and enhance proliferation, migration, and metastasis,4 such as NF-κB, cytokines or cytokine receptors, chemokines or chemokine receptors, fibroblast growth factor or receptor (FGF or FGFR), and vascular endothelial growth factor (VEGF). Increasing evidence demonstrates the key role of chronic inflammation markers in increased BC risk5; for example, a meta-analysis suggested a significant dose–response correlation for C-reactive protein (CRP) with BC risk.6 The pro-inflammatory cytokines, such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α, induce BC cells to penetrate the blood vessels, contributing to metastasis.7
Owing to its significant pro-tumor effects, inflammation has become a promising target for cancer prevention and treatment. Among various inflammatory factors, cyclooxygenase 2 (COX-2) is the most commonly studied anti-inflammatory/anticancer target.8,9 Unlike COX-1, COX-2 is undetectable in normal breast tissue, but in tumor tissue, it overexpresses by 40%,10 and in ductal carcinoma in situ (DCIS) it overexpresses by approximately 80%.11 The overexpression of COX-2 has been reported in different tumor cells and neovascular endothelial cells.12 The overexpressed COX-2 converts arachidonic acid (AA) into prostaglandin E2 (PGE2),13 which promotes BC progression through different mechanisms, for instance, suppression of antitumor immunity,14 promotion of invasiveness,15 migration,15 stem-like cell (SLC) formation,16 angiogenesis,17 and lymphangiogenesis.18
Over 20 years ago, NSAIDs were reported to have anti-colon cancer effects.19 Abundant epidemiological and preclinical/clinical studies demonstrated that celecoxib, a specific COX-2 inhibitor, was related to suppression of cancer cell proliferation and decrease in cancer incidents. In this article, different mechanisms underlying anticancer effect of celecoxib as well as preclinical/clinical results signifying this beneficial effect are discussed.
Celecoxib and BC
Celecoxib is the international nonproprietary name of 4-[5-(4-methylphenyl)–3-(trifluoromethyl)-1H-pyrazol-1-yl] benzenesulfonamide, a COX-2-selective NSAID. Its oral capsule form was initially approved by US Food and Drug Administration (FDA) and marketed by Pfizer, Inc. (New York, NY, USA) in 1999. As a selective COX-2 inhibitor, celecoxib is used as an analgesic, anti-inflammatory, and antipyretic drug. Numerous preclinical evidence suggests that celecoxib may provide a strong chemopreventive activity against BC. Celecoxib treatment (500–1,500 mg/kg diet) can significantly decrease incidence, multiplicity, and tumor volume in several animal models of BC.20,21 In addition, metastasis to the lung and brain could also be prevented.22,23 Besides preclinical studies, clinical trials also showed positive results. Two case–control studies, including 32324 and 18,36825 BC cases, respectively, illustrated that a standard dose intake of celecoxib (200 mg/day) for more than 12 months was associated with significant reduced risk of BC. However, in these studies, OR was different,25 and this might be caused by a longer duration of drug intake in the former study (more than 2 years vs more than 1 year).
Mechanisms of celecoxib’s antitumor action
In spite of the encouraging efficacy mentioned earlier, the mechanisms of celecoxib’s antitumor action still need to be explored. COX-2 plays an important role in tumorigenesis and development. Thus, celecoxib, as a selective COX-2 inhibitor, is believed to have several potential antitumor mechanisms, including inhibition of proliferation, induction of apoptosis, immunoregulation, regulation of tumor microenvironment, antiangiogenic effect, and resensitization of other antitumor drugs. Meanwhile, COX-2-independent pathways also contribute to the antitumor effect of celecoxib.
Inhibition of proliferation
Celecoxib represses the proliferation of BC cells in vitro and also prevents the incidence of BC chemically induced by 7,12-dimethylben anthracene (DMBA) in rats. Thus, celecoxib shows an anticancer activity and seems to be effective in anticancer treatment.26 Bocca et al27 assessed the antiproliferative activity of celecoxib on human BC cells with different COX-2 expression levels. Celecoxib treatment induces a robust inhibition of cell growth in estrogen receptor (ER)α (+) MCF-7 cells, which is accompanied by a decrease in expression and activation of aromatase and ERα. The related mechanism may involve ERK and Akt inhibition as well as induction of PP2A and PTEN. In this cell line, celecoxib shows only weak effect on COX-2 level. In contrast, in ERα (–) MDA-MB-231 cells, celecoxib induces a striking suppression of COX-2, which is associated with a decrease in aromatase expression and cell proliferation. These results imply that celecoxib may exert antiproliferative activity in BC cells through COX-2-dependent or COX-2-independent pathways. A study identifying transcriptional changes in BC tissues of patients treated with celecoxib suggested that short-term COX-2 inhibition by the drug stimulates transcriptional programs that facilitate antitumor activity in primary BC tissue. The influence on proliferation-related genes is reflected by a decrease in Ki-67 (+) cells.28 Basu et al29 explored the mechanisms by which celecoxib affects tumor growth of two human BC cell lines such as MDA-MB-231 (highly invasive) and MDA-MB-468 (moderately invasive). They demonstrated that the distinct molecular mechanisms of celecoxib-induced growth suppression depend on the expression level of COX-2 and invasiveness in different human BC cell lines. The studies suggest that COX-2 plays an important role not only in the cancer cell growth but also in activating the angiogenic pathway via modulating levels of VEGF. Taken together, these results provide a theoretical and experimental basis for the clinical anticancer effect of celecoxib.
Induction of apoptosis
Apoptosis is an evolutionary conserved programmed cellular suicide mechanism that is vital for tissue homeostasis in multicellular organisms. It also causes the cytotoxic effects in response to standard genotoxic chemotherapy/radiotherapy. Intriguingly, some researchers found that the tumor growth inhibition effect of celecoxib was mainly caused by inducing apoptosis30 rather than disturbing cell proliferation.31 Celecoxib-resistant cell lines with COX-2 overexpression32 exhibit a reduced level of Bax, a pro-apoptosis protein, and increased levels of anti-apoptosis proteins such as Bcl-2 or Bcl-xL. COX-2 knockdown by its specific siRNA significantly decreases clonogenicity and levels of Bcl-xL and Bcl-2 in these cells. In MDA-MB-23129,30 and MCF-7 cell lines,33 celecoxib induces apoptosis by decreasing phosphorylation of Akt, then increasing the expression of Bax and activation of caspase 3 and caspase 7. Wang et al34 showed that celecoxib induces apoptosis of the BC cell line, MDA-MB-231, by inhibiting the NF-κB pathway. Another mechanism of celecoxib-induced apoptosis in most cellular systems involves p53-independent mitochondrial apoptosis pathway, which is COX-2 independent and could not be inhibited by the overexpression of Bcl-2.35,36
Some studies suggest that the antineoplastic effects of celecoxib are attributed to its unspecific inhibitory actions on β-catenin signaling,37,38 which is a fundamental component of the canonical Wnt pathway. Overactivation of Wnt/β-catenin signaling is associated with the progression of different types of cancer and promotion of cancer cell growth, survival, and malignant phenotype. Furthermore, recent studies suggest that Wnt/β-catenin signaling plays an important role in the modulation of cancer stem cells.39,40 GSK-3β phosphorylates and marks β-catenin for ubiquitination and succeeding proteasomal degradation. Binding of Wnt ligands to their receptors initiates a signaling cascade that averts GSK-3β from tagging β-catenin for degradation, leading to its accumulation, translocation to the nucleus, binding to the T cell factor (TCF) family of transcription factors, and activation of Wnt target gene expression. Celecoxib treatment induces GSK-3β dephosphorylation, contributing to β-catenin phosphorylation induced by GSK-3β and suppression of the Wnt/β-catenin-dependent gene transcription, for example, c-Myc or cyclin D1, COX-2, and VEGF.38,41 It is noteworthy that GSK-3β is a direct downstream target of Akt/PKB and activated Akt/PKB phosphorylates and consequently inactivates GSK-3β. Hence, celecoxib inhibition of Akt/PKB might at least partly be responsible for the reduction in the β-catenin levels.
Survivin is an “inhibitor of apoptosis” (IAP) protein that also functions as a mitotic regulator essential for cell division.42 It represses apoptosis through hindering the activation of caspases. Intriguingly, celecoxib treatment downregulates survivin levels of cancer cells in vitro and in vivo.43–47 The degree of survivin inhibition of celecoxib correlates with its efficacy to impede cancer cell growth and to promote apoptosis among different types of cancer cells. Since prostaglandins elevate survivin expression, the downregulation of survivin may be partly attributable to celecoxib-induced suppression of COX-2.48 In view of the role played by survivin in apoptosis resistance of cancer cells, the inhibitory effects of celecoxib on this protein might be particularly relevant to its use in anticancer treatment.49,50 Whether the induction of apoptosis leads to clinical benefits is still debatable. A study showed that celecoxib increased apoptosis and reduced the levels of PG and VEGF expression.51 However, this effect could not postpone tumor appearance and reduce tumor progression and development.
Immunoregulation
A thorny problem in the process of treating tumors is a repressed cell-mediated immunity, characterized by the failure of immune effector cells to induce effective antitumor responses. CD4+ or CD8+ T cells were mostly involved in local tumor suppression, while natural killer (NK) cells were involved in tumor metastasis.52 Immunosuppressive factors, produced by the tumor, cause this problem according to tolerance. COX-2 plays an important role in BC immune escape. PGE2 has a series of adverse effects on the immune response to tumors in the body, for example, negatively influencing the activity of T/B lymphocytes, NK cells, and dendritic cells, reducing TNF-α synthesis and increasing the activity of immunosuppressive IL-10.53 These unfavorable effects ablate the effectiveness of host defenses in monitoring and eliminating malignant cells, thus resulting in their unrestrained proliferation.54 Previous studies demonstrate that the reduced expression of COX-2 in BC cells promotes tissue infiltration of cytotoxic T lymphocytes (CD8+), implying a role of COX-2 in immunosuppression. COX-2 inhibitors such as celecoxib regulate antitumor immunity in local and metastasis BC individually.55 In addition, the application of celecoxib in human BCs prompts an increased number of immune cells in the tumor microenvironment.54 A study by Gallouet et al56 demonstrated that follicular lymphoma stromal cells release great amounts of PGE2. This production can be abolished by celecoxib treatment that targets the COX-2 isoenzyme associated with PGE2 synthesis. Interestingly, they also found that celecoxib promotes apoptosis in primary follicular lymphoma B cells cocultured with stromal cells, nonetheless, independently of the PGE2/COX-2 axis.
Substantial studies suggested that celecoxib might switch the function of immune cells to a more tumor-killing phenotype via impeding tumors from releasing prostaglandins and via hindering COX activity in immune effector cells. Lang et al57 suggested that cancer cells suppress the physiological function of immune cells and that celecoxib, to a certain extent, recovers this function. These results deliver an in-depth understanding of the anticancer effect of celecoxib and support its prophylactic use in high-risk patients. In a study evaluating the influence of celecoxib administration on tumor-infiltrating lymphocyte (TIL) subsets (CD3[+]CD4[+]CD8[+]CD25[+] and T cell receptor [TCR]-zeta-expressing cells) and tryptase(+) mast cells in human cervical cancers, Ferrandina et al58 provided the first evidence that this drug can restore zeta expression by TIL in primary cervical cancers. Overall, these results suggest that a positive regulation of immune function might serve as a crucial mechanism underlying the antitumor effect of celecoxib.
Regulation of tumor microenvironment
During chronic inflammation, pro-inflammatory molecules including cytokines, ROS, NF-κB, and inducible nitric oxide synthase (iNOS) are increased and provide an advantageous microenvironment for cancer cell growth. Therefore, inflammation might result in the initiation of cancer and provide the suitable environment to support tumor growth.59 The tumor microenvironment consists of a variety of cell and molecular components, such as matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinase (TIMP)-1. Tumor microenvironment has profound impacts on tumor cell proliferation, migration, and apoptosis.60,61 Celecoxib inhibits 12-O-tetradecanoyl phorbol-13-acetate (TPA)-induced MMP-9 expression in a dose-dependent manner by increasing the activity of TIMP-1.62,63
The stimulation of constitutive expression of COX-2 is a crucial factor in the tumorigenic process. Various key risk factors associated with cancer causativeness are capable of stimulating COX-2. These factors comprise certain essential dietary fatty acids, nicotine and its metabolites, growth factors, infectious agents, hypoxia, hormones, ultraviolet B, and free radicals; oncogenic proteins; and endotoxins; etc.64–67 Some microenvironmental stimuli, for example, bacterial lipopolysaccharides, TNF, and by-products of protein synthesis and degradation, also induce constitutive COX-2 expression. In addition, because the COX-2 gene contains various promoter binding sites, nuclear transcription factors including NF-κβ or NF-IL6 might also mediate its upregulation.68 Genetic induction of COX-2 in BC cells triggers local constitutive estrogen synthesis through activating the promoter II region of the aromatase gene (CYP-19) in adjacent fat and muscle cells.69 Terry et al70 revealed a vital relationship between COX-2 overexpression and mammary tumorigenesis induced by estrogen. Hence, COX-2 tumorigenesis seems to involve synergistic interactions between many microenvironmental and genetic cofactors. Accordingly, recent studies suggested that regular use of aspirin and other coxibs has noteworthy therapeutic impact in cancer patients.71,72 Expression of COX-2 can also be increased by large amount of collagen, which contributes to high breast density73 and growing incidence of BC.74,75 This effect can be inhibited by celecoxib through reducing overall collagen deposition and the levels of COX-2, PGE2, and Ki-67 expression.76
Tumor-associated macrophages are associated with cancer cell survival. In a microenvironment study of BC cells, Li et al77 demonstrated that COX-2 is plentifully expressed in breast tumor-associated macrophages, which is associated with poor prognosis in BC patients. Their studies suggested that COX-2 serves as an important cancer-promoting factor through prompting a positive feedback loop between macrophages and BC cells. Apparently, COX-2 inhibitor, celecoxib, is favorable in disturbing this feedback loop in the cancer microenvironment. Accordingly, COX-2 can be exploited as a target for BC prevention and therapy. These findings provide solid molecular evidence to support the anti-BC effect of celecoxib, which has potential to raise positive expectations for clinical use.
Antiangiogenic effect
Angiogenesis refers to the generation of new blood vessels through the extension of preexisting vasculature. It is modulated by an equilibrium between the pro- and antiangiogenic factors. During tumorigenesis, the role of pro-angiogenic factors exceeds that of their counterpart and triggers the growth of new capillaries to supply more blood flow and overcome hypoxia inside the cancer microenvironment, leading to tumor growth and metastasis. At the molecular level, one of the mechanisms underlying COX-2-dependent neoplastic initiation and development in BC involves its proangiogenic activity.78,79 Actually, COX-2 activates MMPs in an intricate mechanism involving NF-κB. This protein also promotes endothelial migration by thromboxane A2 (TXA2).78,80 Moreover, the augmented activity of COX-2 contributes to the release of proangiogenic factors by epithelial and endothelial neoplastic cells, fibroblasts, and macrophages.80,81 In detail, COX-2-dependent angiogenesis begins with the formation of proangiogenic prostaglandins (primarily PGE2) by tumor cells, which enhances the levels of VEGF and bFGF. VEGF directly induces COX-2 in ECs, while bFGF induces COX-2 in fibroblasts to synthesize PGs, which can stimulate the PKA pathway via the EP2 receptor. In addition to their direct pro-angiogenic action, PGs may also induce angiogenesis indirectly, via activating monocytes that infiltrate tumor tissues. Subsequently, the activated vascular COX-2 leads to the elevated permeability, proliferation, and morphogenesis of vasculatures.81 The high microvessel density results in the greater metastatic potential of tumor cells and poor patient prognosis.78,81 Furthermore, PGE2 can also promote angiogenesis through activating EP4 and its second messenger PKA in ECs.82
Celecoxib could inhibit PGE2-induced angiogenesis and lymphangiogenesis and sequentially inhibit tumor growth and metastasis, especially in COX-2-overexpressed cell lines,17,83 together with the reduction in microvessel density, microtubule formation, and serum VEGF level.84,85 This inhibition effect was associated with PGE2 receptor 4 (EP4) and could be reversed by exogenous PGE2.86 Microvascular permeability could also be reduced by celecoxib.87,88 Tamoxifen (TAM) is an ER modulator and widely used in the treatment of BC as an adjuvant therapy against recurrence after surgery. Nevertheless, prolonged TAM administration increases VEGF levels in BC patients, stimulating new blood vessel formation and thus limiting its effectiveness. Kumar et al89 demonstrated that celecoxib can relieve TAM-induced angiogenesis via ROS-dependent VEGF/VEGFR2 autocrine signaling. In addition, Vaish and Sanyal90 reported that celecoxib can inhibit angiogenesis during the early neoplasm of colon through regulating PI3-K/PTEN/Akt and the canonical Wnt/β-catenin signaling pathway. In short, these findings shed light on molecular mechanisms underlying celecoxib’s anticancer effect from another perspective, which enhances the positive anticipation of its clinical application.
Integrating celecoxib into BC treatment
Celecoxib has been examined for improvement in chemotherapy effectiveness in cancer clinical trials. In fact, it has been reported that celecoxib could stimulate sensitivity to chemotherapy of BC cells91–93 via affecting the activation of multidrug resistance protein 1 (MDR1) which induces drug resistance and could be upregulated by COX-2.94,95 Instead of affecting the pump function of MDR1, celecoxib downregulates its expression by inducing hypermethylation of MDR1 gene promoter96 and inhibiting the DNA-binding activity and expression of nuclear transcription factors such as AP-1 and NF-κB, which can combine with putative binding sites of human MDR1 gene promoter.97 In addition to the activation of MDR1, of pertinence to this review, celecoxib was recently demonstrated to significantly sensitize other antitumor drugs with multiple mechanisms.
Combination with chemotherapy
Considering its own antitumor competence and resensitization of other antitumor drugs, celecoxib could be a potential candidate for combination therapy. Preclinical research suggested that the antitumor effect of several agents can be enhanced by combining with celecoxib,98 including doxorubicin99 and 5-fluorouracil (5-FU).100 In several Phase II studies, the combination of celecoxib and capecitabine, an orally administered pro-drug of 5-FU, could provide a clinical benefit rate at 42.1–47.5% and an unexpected lower toxicity than capecitabine alone in metastatic BC (MBC) patients.101,102 In a single-arm, mono-institutional, nonrandomized, Phase II, two-step clinical trial, celecoxib was combined with cyclophosphamide, and the clinical benefit of this combination came to 55% in 20 advanced BC (ABC) patients.103
Celecoxib can also be integrated into multidrug chemotherapy regimens, in which FEC (5-FU, epirubicin, and cyclophosphamide) is the most common one. A study containing 50 patients showed that preoperative FEC with celecoxib (FECC) could provide lower intensity staining for COX-2, Ki-67, and p53 in 90% patients, while no difference was observed on tumor size, grade, or axillary lymph node status.104 In a Phase II, multicenter, open-label, single-arm study (N001),105 64 invasive BC patients received four cycles of FEC (500, 100, 500 mg/m2) followed by four cycles of docetaxel (100 mg/m2) with celecoxib (200 mg twice daily) as neoadjuvant therapy (NAT). After NAT, 43 patients achieved clinical complete response (cCR) and 13 achieved clinical partial response (cPR). In addition, despite potential side effects on cardiac system, the cardiac safety of celecoxib has been declared to be acceptable.106,107
It has also been reported that celecoxib increases the sensitivity of drug-resistant KBV20C cancer cells to antimitotic drugs.108 This sensitization mechanism is independent of the suppression of p-glycoprotein, indicating that the KBV20C cells are sensitized via targeting of signaling pathways by celecoxib. Moreover, it has also been observed that celecoxib intensely sensitizes KBV20C cells to vinblastine and paclitaxel, as indicated by microscopic observation, determination of Annexin V staining, and cleaved poly(ADP-ribose) polymerase (cleaved PARP). These results suggest that COX-2 inhibitors such as celecoxib can be used for cancer patients with potential resistance, without the toxic effects of p-glycoprotein suppression. Of interest, as detailed previously,109 celecoxib promotes (sorafenib + sildenafil) lethality in multiple ovarian cancer cell lines, concomitant with a decrease in the expression of several chaperone proteins in parallel with decreased levels of the drug efflux pumps such as ABCB1/ABCG2. The cytotoxicity by the triple combination was induced by caspase 9-dependent apoptotic pathway and RIP-1/caspases 2, 4/AIF-dependent necroptotic pathway. In addition, the triple combination significantly reverted platinum chemotherapy resistance. Combined with the previous studies substantiating in vivo the combinations of “celecoxib + sildenafil” and “sorafenib + sildenafil” as cytotoxic to various cancer cell types, it has been suggested that the celecoxib/sorafenib/sildenafil combination ought to be investigated in a Phase I trial in ovarian cancer.109 Table 1 summarizes the drugs suggested for use in combination with celecoxib for BC treatment based on preclinical data.
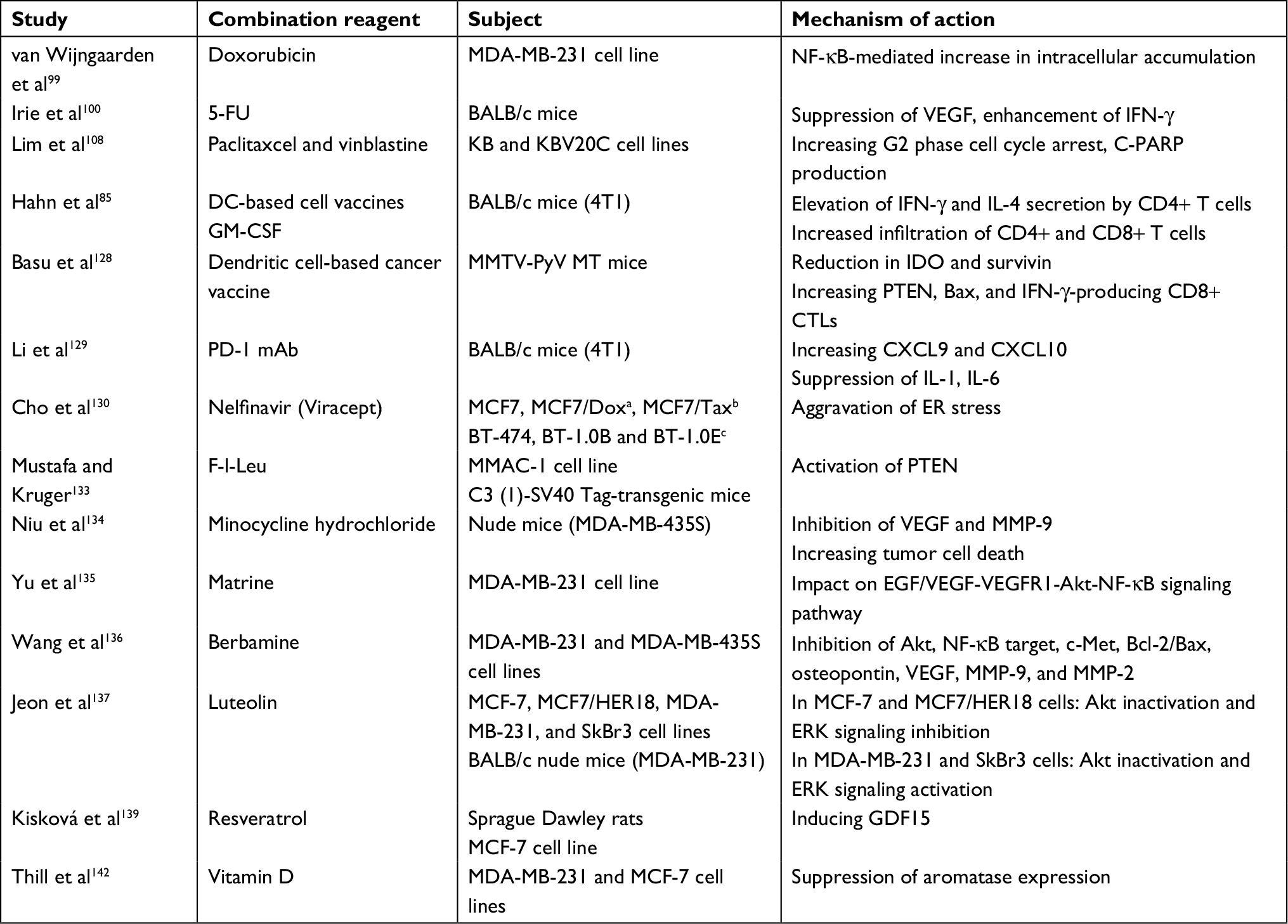 | Table 1 Preclinical studies on combination of celecoxib and other therapeutic drugs in BC Notes: aMCF7/Dox, a doxorubicin/multidrug-resistant variant of MCF7. bMCF7/Tax, a taxol-resistant variant of MCF7. cBT-1.0B and BT-1.0E, two trastuzumab-resistant variants of BT474. Abbreviations: BC, breast cancer; CTLs, cytotoxic T cells; CXCL, C–X–C motif ligand; DC, dendritic cell; ER, estrogen receptor; F-l-Leu, N-(9-fluorenyl-methyloxycarbonyl)-l-leucine; 5-FU, fluorouracil; GDF, growth differentiation factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; IL, interleukin; MMAC, mammary adenocarcinoma cell; MMP, matrix metalloproteinase; PARP, poly(ADP-ribose) polymerase; PD-1 mAb, programmed death 1 monoclonal antibody; VEGF, vascular endothelial growth factor. |
Not all the studies showed positive results, especially in patients with HER2-negative tumor. A multicenter randomized controlled Phase II clinical trial showed that celecoxib did not improve pathological complete response (pCR) rates in addition to epirubicin–cyclophosphamide–docetaxel (EC-D) regimen.110 Moreover, the REMAGUS-02 multicenter randomized Phase II trial111 demonstrated that the addition of celecoxib could not provide an increased pCR rate in HER2-negative patients. The long-term follow-up indicated that, in the HER2-negative subgroup, the addition of celecoxib led to smaller tumor size and lower expression of progesterone receptor (PgR) status, but no association of disease-free survival (DFS) benefit. In BC patients, COX-2 overexpression can be induced by HER2 oncogene activation112 and provide a positive feedback through its product PGE2 which induces HER2 expression.113 However, the combination of celecoxib (400 mg twice daily) and trastuzumab (2 mg/kg intravenous injection weekly or 6 mg/kg intravenous injection every 3 weeks) provided no significant enhancement in a Phase II study.114 Of note, El-Awady et al115 explored the ability of celecoxib to sensitize different types of cancer cells (HeLa, HCT116, HepG2, MCF-7, and U251) to a number of anticancer drugs (5-FU, cisplatin, doxorubicin, and etoposide). Interaction of celecoxib with these chemotherapeutic drugs is antagonistic in the BC cells, MCF-7, but not in other cells, suggesting that celecoxib exerts distinct molecular actions in different cancer cells. Mechanistic investigations demonstrated that celecoxib increases drug-triggered G2/M arrest in MCF-7 cells allowing more time to repair drug-elicited DNA damage before access into mitosis, leading to decrease in cell death and thus contributing to antagonism. These findings, if substantiated in vivo, suggest that celecoxib is not an appropriate chemo-sensitizer for BC. Therefore, the combination of celecoxib with other chemotherapeutic drugs must be customized to the cancer type. To obtain a more accurate conclusion, more in-depth and extensive clinical trials are needed. Table 2 summarizes the clinical trials of celecoxib or celecoxib combined with chemotherapy on BC patients.
 | Table 2 Clinical trials of celecoxib on BC (ClinicalTrial.gov) Abbreviations: BC, breast cancer; ER, estrogen receptor; MBC, metastatic BC; PgR, progesterone receptor; BID, twice a day; QD, once a day. |
Combination with endocrinal therapy
It was demonstrated that the expression of aromatase CYP19 might be potentially regulated by PGE2 through cAMP-mediated pathways, and it makes further influence on aromatase activity116,117 and estrogen biosynthesis.69 Linear positive correlation was shown between CYP19 and COX-2 by semiquantitative reverse transcriptase PCR (RT-PCR), suggesting that the combination of COX-2 and aromatase inhibitors (AIs) could have synergistic effect on hormone-dependent BC.69 The inhibition of aromatase by celecoxib was observed at transcriptional level by real-time PCR and appeared to be dose dependent.118 Anastrozole, an AI, was combined with celecoxib to treat BC in rats.119 The results showed that this combination might be workable for clinical therapy.
Besides laboratory investigations, clinical trials also conducted to combine celecoxib with selective ER modulator (TAM) and AIs (exemestane). In the Celecoxib Anti-Aromatase Neoadjuvant (CAAN) trial,120 a combination of exemestane (25 mg daily) and celecoxib (400 mg twice daily) gained significantly lowered cholesterol and low-density lipoprotein (LDL) levels and higher bone mineral density (BMD) and BC subscale scores compared with single-agent groups of exemestane (25 mg daily) and letrozole (2.5 mg daily), in postmenopausal women with histologically proven local ABC (LABC). So, although the final outcomes showed no statistical difference on clinical response and tumor volume,121 which meant that different neoadjuvant anti-aromatase therapies have similar efficacy, the combination with celecoxib may provide some additional benefits. Some other studies, including a Phase II study122 and a Phase III study,123 positively supported the combination of celecoxib and exemestane in postmenopausal MBC patients. A Phase II trial of neoadjuvant exemestane (25 mg daily) plus celecoxib (400 mg twice daily) demonstrated that the combination was tolerated and anticancer response was observed in the majority of postmenopausal women with BC. Statistically, noteworthy reduction could also be found in the expression of ER, PgR, Ki-67, and COX-2.124 Nevertheless, some other research provided a different opinion. A study established by Dirix et al125 showed that the demographic characteristics, prognostic factors, and time to progression (TTP) were all similar no matter whether celecoxib was added to endocrine therapy or not, and the lack of COX-2 expression may attribute to this result.126 Moreover, it was suggested that the anticancer effect of combination therapy might have mainly resulted from exemestane instead of celecoxib.127
TAM is extensively used in BC therapy as a preventive drug against recurrence after surgical treatment, but the long-term TAM treatment enhances patients’ VEGF levels, stimulates neovascularization, and thus restrains its effectiveness. Kumar et al89 demonstrated that the combination of TAM and celecoxib at nontoxic concentrations exerted antiangiogenic effects via explicitly targeting VEGF–VEGFR2 pathway through ROS formation. In addition, their preclinical studies suggested that the TAM/celecoxib combination is a feasible strategy for the treatment of BCs with VEGF/VEGFR2 overexpression. This inventive combination exhibits encouraging effect in anti-metastasis and stimulation of apoptosis and may be a superior personalized clinical regimen vs TAM alone for BC treatment. The clinical trials on the combination of endocrine therapy and celecoxib are summarized in Table 3.
 | Table 3 Summary of clinical trials on the combination of endocrine therapy and celecoxib Note: aMeasured by European Organization for Research and Treatment of Cancer Quality of Life Questionnaire core - 30 version 3.0 (EORTC QLQ C-30 v3.0) and European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Breast-Cancer-Specific 23(EORTC QLQ-BR23). Abbreviations: ABC, advanced BC; BC, breast cancer; CEL, celecoxib; CR, complete response; CTC, common toxicity criteria; ER, estrogen receptor; EXE, exemestane; LET, letrozole; MBC, metastatic BC; n, number of patients; NR, not reassessed; PD, progressive disease; PgR, progesterone receptor; PR, partial response; QOL, quality of life; R, randomized study; SD, stable disease; TAM, tamoxifen; TTP, time to progression. |
Combination with other antitumor treatments
Dendritic cell-based cancer vaccine, from tumor lysate-pulsed dendritic cell, is a popular candidate for cancer immunotherapy. Hahn et al85 tested the antitumor immune response using the combination of celecoxib, vaccine, and GM-CSF in 4T1 cells, a cell line with COX-2 expression, poorly immunogenic, and highly metastatic ability. The triple combination therapy successfully suppressed primary tumor growth and significantly reduced the incidence of lung metastases. This effect was achieved by a tumor-specific immune response which could be observed as increased interferon (IFN)-γ and IL-4 secretion by CD4+ T cells and infiltration of CD4+ and CD8+ T cells to the tumor site. Basu et al128 also combined celecoxib with dendritic cell-based cancer vaccine and reconfirmed that the combination gained its antitumor effect by downregulating the expression of indoleamine 2,3-dioxygenase (IDO), a negative regulator of T cell activity. A recent study by Li et al129 using alginate hydrogel system to locally deliver celecoxib and programmed death 1 (PD-1) monoclonal antibody (mAb) to treat 4T1 MBC mouse model demonstrated a significant improvement in the anticancer activities of celecoxib, PD-1 mAb, or both combined. The persistent high levels of the drugs in peripheral circulation and within local tumor areas were observed. Importantly, the concurrent dual local delivery of celecoxib and PD-1 synergistically elevated the levels of CD4+ IFN-γ+/CD8+ IFN-γ+ T cells in the tumor and the immune system, implying that the combinatorial therapy synergistically enhances antitumor immunity. In addition, this combination treatment induces the production of two antiangiogenic chemokines such as C–X–C motif ligand (CXCL)9 and CXCL10 as well as inhibits the intra-tumoral formation of IL-1, IL-6, and COX2, indicating a diminished pro-cancer angiogenic and inflammatory microenvironment. This celecoxib/PD-1 mAb combination treatment provides a promising regimen for treating human BC.
By combining celecoxib with the HIV protease inhibitor, nelfinavir (Viracept), Cho et al130 explored the aggravation of endoplasmic reticulum stress caused by this combination, which led to apoptosis in chemoresistant BC cells. Furthermore, unmethylated celecoxib (UMC), with superior COX-2 inhibitory efficacy, showed substantially weaker antitumor effect. Therefore, they speculated that the antitumor effect of celecoxib was COX-2 independent in chemoresistant BC. Chloroquine is another material that can play a role in antitumor effect with celecoxib through endoplasmic reticulum stress response.131 In addition, treatment with the combination of anti-IL-17 antibody and celecoxib can significantly decrease bone and lung metastasis in SKG mice with mammary gland tumors and autoimmune arthritis.132 Preclinical studies also suggested that celecoxib could be combined with peroxisome proliferator-activated receptor gamma agonist for the treatment of spontaneous BC133 and minocycline hydrochloride for osseous metastasis in BC.134 These combination treatments were strikingly more effective than celecoxib alone. Moreover, many plant-derived materials were combined with celecoxib and emerged synergistic effect on antitumor effect via VEGF/Akt/NF-κB signaling, including matrine,135 berbamine,136 and luteolin.137,138 Resveratrol139 can also enhance the tumor prevention effect of celecoxib, but the exact mechanism is still under investigated.
Several epidemiological studies have shown that vitamin D has beneficial effects against the carcinogenesis and development of BC.140,141 Recent studies revealed an association between vitamin D and PGE2 metabolism. Thill et al142 demonstrated that a synergistic growth-inhibiting effect in BC cell lines can be elicited by the combination of celecoxib and calcitriol (1,25-dihydroxycholecalciferol or 1,25-[OH]2D3), which is a biologically active form of vitamin D.143 Calcitriol could also inhibit COX-2 expression at both protein and mRNA levels. New perspectives emerge from the growing knowledge of innovative combination of celecoxib, and other anticancer agents, which act in a complementary way, increase the efficacy and minimize toxicity.
Side effects and clinical complications
Celecoxib is the only FDA-approved COX-2 inhibitor for use in the USA. Although celecoxib is usually a well-tolerated drug, it is not harmless. Its typical doses range from 200 to 400 mg/day; nonetheless, the dose for acute gout can reach 800 mg once, followed by 400 mg on the first day, then 400 mg twice daily for 7 days. A higher dose of celecoxib (800 mg per day) might be related to augmented cardiovascular risk according to the Adenoma Prevention with Celecoxib (APC) study.144 The cardiotoxicity side effects may be attributed to its off-target effect, namely modulating calcium levels within the cell, according to the immediate time-dependent cell response profiles (TCRPs) for celecoxib.145 In fact, previous studies have shown that celecoxib therapy induces an immediate increase in intracellular calcium levels.146 In addition, clinical data indicate that chronic use of celecoxib may damage normal skeletal function resulting in reduced BMD in older male patients.147 Serious allergic reactions to celecoxib have also been reported.148
When celecoxib is recommended in advanced cancer patients, the pros and cons need to be considered prudently. A meta-analysis by Chen et al149 suggested that celecoxib has certain benefits in the treatment of cancer, but increases the risk of cardiovascular events. Specifically, they demonstrated an increase in grade 3 and 4 toxicities of cardiovascular events with the incorporation of celecoxib to the treatment of advanced cancers. Other toxicities include rash, hepatotoxicity, and gastrointestinal events, and there is no statistically significant difference among them. Of note, the risk of anemia in the celecoxib group is also significant. Although the risk of grade 3 and 4 cardiovascular events increases by 1.78 times after celecoxib use for a long time, the risk is acceptable, considering that this is a prescription for life-threatening diseases. Nevertheless, clinical monitoring of side effects, for example, cardiovascular events should be strengthened. According to these findings, it is necessary to carefully consider the benefit vs harm when recommending celecoxib in the treatment of patients with advanced cancer, especially those with a history of heart disease. Further studies are needed to confirm these results with large samples.
Conclusion
In addition to being widely used for treating inflammatory diseases such as rheumatoid arthritis and osteoarthritis,150 celecoxib may also play an important role in the cancer prevention and treatment. Preclinical evidence demonstrates that celecoxib seems to suppress the proliferation and growth of different types of cancer through various mechanisms (Figure 1). Results of abundant clinical studies, although unconvincing, suggest that celecoxib administration is related not only to diminished incidence of cancer but also to the better prognosis in cancer patients. In view of the potential variations in response to celecoxib in cancer patients, it seems critical to ascertain target populations for its use. Nevertheless, factors that contribute to better outcome in celecoxib consumers are still to be explicated. The data on the effectiveness of celecoxib as neoadjuvant treatment in cancer patients are deficient. There are substantial clinical studies evaluating the role of celecoxib in the cancer treatment. The results will permit evaluation of the position of celecoxib in cancer prevention and therapy and identify the target populations in the near future. Of note, comparative studies should be designed to ascertain the optimal dosage, duration, side effects (especially the gastrointestinal and cardiovascular systems), and its cost-effectiveness. As was originally pointed out more than 10 years ago, “there exists an urgent need for clinical trials of this compound so as to accelerate its effective application in the chemoprevention and treatment of cancer.” NSAIDs, and especially celecoxib, represent an inspiring proposition for repurposing as anticancer drugs with low toxicity, hence demonstrating how understanding cancer-relevant molecular signaling pathways in combination with clinical data will contribute to further development of oncology.
 | Figure 1 The identified mechanisms of anticancer effect of celecoxib. Abbreviations: EP1, prostaglandin E2 receptor 1; MMP, matrix metalloproteinase; NK, natural killer; PEG2, prostaglandin E2; PGH2, prostaglandin E2; VEGF, vascular endothelial growth factor. |
Acknowledgment
This study was supported in part by NIH-NIMHD U54MD007598, NIH/NCI1 U54CA14393, U56CA101599-01; Department-of-Defense Breast Cancer Research Program (grant BC043180), NIH/NCATS CTSI UL1TR000124 to JV Vadgama, and Accelerating Excellence in Translational Science Pilot (grants G0812D05), NIH/NCI (SC1CA200517) to Y Wu; the National Natural Science Foundation of China 81630049; National Key R&D Program of China 2017YFC0113302; China Scholarship Council 201706165022.
Disclosure
The authors report no conflicts of interest in this work.
References
Evans DG, Howell A. Breast cancer risk-assessment models. Br Cancer Res. 2007;9(5):213. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 20182018;68(1):7–30. | ||
Rayburn E, Ezell SJ, Zhang R. Anti-Inflammatory Agents for Cancer Therapy. Mol Cell Pharmacol. 2009;1(1):29–43. | ||
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420(6917):860–867. | ||
Allen MD, Jones LJ. The role of inflammation in progression of breast cancer: Friend or foe? (Review). Int J Oncol. 2015;47(3):797–805. | ||
Chan DS, Bandera EV, Greenwood DC, Norat T. Circulating C-Reactive Protein and Breast Cancer Risk-Systematic Literature Review and Meta-analysis of Prospective Cohort Studies. Cancer Epidemiol Biomarkers Prev. 2015;24(10):1439–1449. | ||
Landskron G, de La Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014(2):149185–19. | ||
Galliera E, Corsi M, Bonecchi R, Locati M, Mantovani A. Chemokines as pharmacological targets. Mini Rev Med Chem. 2008;8(7):638–646. | ||
Knowles MA. Novel therapeutic targets in bladder cancer: mutation and expression of FGF receptors. Future Oncol. 2008;4(1):71–83. | ||
Bosco JL, Palmer JR, Boggs DA, Hatch EE, Rosenberg L. Regular aspirin use and breast cancer risk in US Black women. Cancer Causes Control. 2011;22(11):1553–1561. | ||
Díaz-Cruz ES, Shapiro CL, Brueggemeier RW. Cyclooxygenase Inhibitors Suppress Aromatase Expression and Activity in Breast Cancer Cells. J Clin Endocrinol Metab. 2005;90(5):2563–2570. | ||
Masferrer JL, Koki A, Seibert K. COX-2 inhibitors. A new class of antiangiogenic agents. Ann N Y Acad Sci. 1999;889(1 CANCER PREVEN):84–86. | ||
Bennett A, Berstock DA, Carroll MA, Stamford IF, Wilson AJ. Breast cancer, its recurrence, and patient survival in relation to tumor prostaglandins. Adv Prostaglandin Thromboxane Leukot Res. 1983;12:299–302. | ||
Lala PK, Al-Mutter N, Orucevic A. Effects of chronic indomethacin therapy on the development and progression of spontaneous mammary tumors in C3H/HEJ mice. Int J Cancer. 1997;73(3):371–380. | ||
Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp Cell Res. 2003;289(2):265–274. | ||
Majumder M, Xin X, Liu L, Girish GV, Lala PK. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014;105(9):1142–1151. | ||
Chang S-H, Liu CH, Conway R, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci U S A. 2004;101(2):591–596. | ||
Bhattacharjee RN, Timoshenko AV, Cai J, Lala PK. Relationship between cyclooxygenase-2 and human epidermal growth factor receptor 2 in vascular endothelial growth factor C up-regulation and lymphangiogenesis in human breast cancer. Cancer Sci. 2010;101(9):2026–2032. | ||
Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988;48(15):4399–4404. | ||
Kang HF, Wang XJ, Liu XX, Dai ZJ, Xue FJ, Xue XH. [Chemopreventive effect of celecoxib against DMBA-induced breast cancer and its mechanism]. Nan Fang Yi Ke Da Xue Xue Bao. 2006;26(11):1599–1602. | ||
Woditschka S, Haag JD, Mau B, Lubet RA, Gould MN. Chemopreventive effects of celecoxib are limited to hormonally responsive mammary carcinomas in the neu-induced retroviral rat model. Breast Cancer Res. 2008;10(1):R18. | ||
Lanza-Jacoby S, Miller S, Flynn J, et al. The cyclooxygenase-2 inhibitor, celecoxib, prevents the development of mammary tumors in Her-2/neu mice. Cancer Epidemiol Biomarkers Prev. 2003;12(12):1486–1491. | ||
Liu Y, Kosaka A, Ikeura M, et al. Premetastatic soil and prevention of breast cancer brain metastasis. Neuro Oncol. 2013;15(7):891–903. | ||
Harris RE, Beebe-Donk J, Alshafie GA. Reduction in the risk of human breast cancer by selective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer. 2006;6(1):27. | ||
Ashok V, Dash C, Rohan TE, Sprafka JM, Terry PD. Selective cyclooxygenase-2 (COX-2) inhibitors and breast cancer risk. Breast. 2011;20(1):66–70. | ||
Dai Z-J, Ma X-B, Kang H-F, et al. Antitumor activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on breast cancer in Vitro and in Vivo. Cancer Cell Int. 2012;12(1):53. | ||
Bocca C, Bozzo F, Bassignana A, Miglietta A. Antiproliferative effects of COX-2 inhibitor celecoxib on human breast cancer cell lines. Mol Cell Biochem. 2011;350(1–2):59–70. | ||
Brandão RD, Veeck J, van de Vijver KK, et al. A randomised controlled phase II trial of pre-operative celecoxib treatment reveals anti-tumour transcriptional response in primary breast cancer. Breast Cancer Res. 2013;15(2):R29. | ||
Basu GD, Pathangey LB, Tinder TL, Gendler SJ, Mukherjee P. Mechanisms underlying the growth inhibitory effects of the cyclo-oxygenase-2 inhibitor celecoxib in human breast cancer cells. Breast Cancer Res. 2005;7(4):R422–435. | ||
Basu GD, Pathangey LB, Tinder TL, Lagioia M, Gendler SJ, Mukherjee P. Cyclooxygenase-2 inhibitor induces apoptosis in breast cancer cells in an in vivo model of spontaneous metastatic breast cancer. Mol Cancer Res. 2004;2(11):632–642. | ||
Barnes NLP, Warnberg F, Farnie G, et al. Cyclooxygenase-2 inhibition: effects on tumour growth, cell cycling and lymphangiogenesis in a xenograft model of breast cancer. Br J Cancer. 2007;96(4):575–582. | ||
Singh B, Irving LR, Tai K, Lucci A. Overexpression of COX-2 in celecoxib-resistant breast cancer cell lines. J Surg Res. 2010;163(2):235–243. | ||
Baumann KH, Klusmeier E, Eggemann I, et al. Effects of celecoxib and ly117018 combination on human breast cancer cells in vitro. Breast Cancer. 2009;3:23–34. | ||
Wang L, Liu LH, Shan BE, Zhang C, Sang MX, Li J. [Celecoxib promotes apoptosis of breast cancer cell line MDA-MB-231 through down-regulation of the NF-kappaB pathway]. Ai Zheng. 2009;28(6):569–574. Chinese. | ||
Jendrossek V. Targeting apoptosis pathways by Celecoxib in cancer. Cancer Lett. 2013;332(2):313–324. | ||
Li Z, Hao Q, Luo J, et al. USP4 inhibits p53 and NF-κB through deubiquitinating and stabilizing HDAC2. Oncogene. 2016;35(22):2902–2912. | ||
Tuynman JB, Vermeulen L, Boon EM, et al. Cyclooxygenase-2 inhibition inhibits c-Met kinase activity and Wnt activity in colon cancer. Cancer Res. 2008;68(4):1213–1220. | ||
Xia J-J, Pei L-B, Zhuang J-P, et al. Celecoxib Inhibits β-Catenin-Dependent Survival of the Human Osteosarcoma MG-63 Cell Line. J Int Med Res. 2010;38(4):1294–1304. | ||
Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16(12):3153–3162. | ||
Fodde R, Brabletz T. Wnt/β-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19(2):150–158. | ||
Maier TJ, Janssen A, Schmidt R, Geisslinger G, Grösch S. Targeting the beta-catenin/APC pathway: a novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005;19(10):1353–1355. | ||
Altieri DC. New wirings in the survivin networks. Oncogene. 2008;27(48):6276–6284. | ||
Kim Y-Y, Lee E-J, Kim Y-K, et al. Anti-cancer effects of celecoxib in head and neck carcinoma. Mol Cells. 2010;29(2):185–194. | ||
Kardosh A, Soriano N, Pyrko P, et al. Reduced survivin expression and tumor cell survival during chronic hypoxia and further cytotoxic enhancement by the cyclooxygenase-2 inhibitor celecoxib. J Biomed Sci. 2007;14(5):647–662. | ||
Hsiao P-W, Chang C-C, Liu H-F, Tsai C-M, Chiu TH, Chao J-I. Activation of p38 mitogen-activated protein kinase by celecoxib oppositely regulates survivin and gamma-H2AX in human colorectal cancer cells. Toxicol Appl Pharmacol. 2007;222(1):97–104. | ||
Fukada K, Takahashi-Yanaga F, Sakoguchi-Okada N, et al. Celecoxib induces apoptosis by inhibiting the expression of survivin in HeLa cells. Biochem Biophys Res Commun. 2007;357(4):1166–1171. | ||
Sakoguchi-Okada N, Takahashi-Yanaga F, Fukada K, et al. Celecoxib inhibits the expression of survivin via the suppression of promoter activity in human colon cancer cells. Biochem Pharmacol. 2007;73(9):1318–1329. | ||
Bai X-M, Jiang H, Ding J-X, et al. Prostaglandin E2 upregulates survivin expression via the EP1 receptor in hepatocellular carcinoma cells. Life Sci. 2010;86(5-6):214–223. | ||
Zhao S, Cai J, Bian H, Gui L, Zhao F. Synergistic inhibition effect of tumor growth by using celecoxib in combination with oxaliplatin. Cancer Invest. 2009;27(6):636–640. | ||
Gaiser T, Becker MR, Habel A, et al. TRAIL-mediated apoptosis in malignant glioma cells is augmented by celecoxib through proteasomal degradation of survivin. Neurosci Lett. 2008;442(2):109–113. | ||
Tran-Thanh D, Buttars S, Wen Y, Wilson C, Done SJ. Cyclooxygenase-2 inhibition for the prophylaxis and treatment of preinvasive breast cancer in a her-2/neu mouse model. Cancer Prev Res. 2010;3(2):202–211. | ||
Langers I, Renoux VM, Thiry M, Delvenne P, Jacobs N. Natural killer cells: role in local tumor growth and metastasis. Biologics. 2012;6:73–82. | ||
Howe L, Subbaramaiah K, Brown AM, Dannenberg AJ. Cyclooxygenase-2: a target for the prevention and treatment of breast cancer. Endocr Relat Cancer. 2001;8(2):97–114. | ||
Markosyan N, Chen EP, Evans RA, Ndong V, Vonderheide RH, Smyth EM. Mammary carcinoma cell derived cyclooxygenase 2 suppresses tumor immune surveillance by enhancing intratumoral immune checkpoint activity. Breast Cancer Res. 2013;15(5):R75. | ||
Kundu N, Walser TC, Ma X, Fulton AM. Cyclooxygenase inhibitors modulate NK activities that control metastatic disease. Cancer Immunol Immunother. 2005;54(10):981–987. | ||
Gallouet A-S, Travert M, Bresson-Bepoldin L, et al. COX-2-independent effects of celecoxib sensitize lymphoma B cells to TRAIL-mediated apoptosis. Clin Cancer Res. 2014;20(10):2663–2673. | ||
Lang S, Picu A, Hofmann T, et al. COX-inhibitors relieve the immunosuppressive effect of tumor cells and improve functions of immune effectors. Int J Immunopathol Pharmacol. 2006;19(2):409–419. | ||
Ferrandina G, et al. Celecoxib Up-Regulates the Expression of the Chain of T Cell Receptor Complex in Tumor-Infiltrating Lymphocytes in Human Cervical Cancer. Clin Cancer Res. 2006;12(7):2055–2060. | ||
Sarkar D, Fisher PB. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 2006;236(1):13–23. | ||
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–252. | ||
Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–329. | ||
Kim S, Kim SH, Hur SM, et al. Silibinin prevents TPA-induced MMP-9 expression by down-regulation of COX-2 in human breast cancer cells. J Ethnopharmacol. 2009;126(2):252–257. | ||
Afsharimani B, Cabot PJ, Parat M-O. Effect of lysine antifibrinolytics and cyclooxygenase inhibitors on the proteolytic profile of breast cancer cells interacting with macrophages or endothelial cells. Br J Anaesth. 2014;113(Suppl 1):i22–i31. | ||
Jaimes EA, Tian R-X, Pearse D, Raij L. Up-regulation of glomerular COX-2 by angiotensin II: Role of reactive oxygen species. Kidney Int. 2005;68(5):2143–2153. | ||
Chang Y-WE, Putzer K, Ren L, et al. Differential regulation of cyclooxygenase 2 expression by small GTPases Ras, Rac1, and RhoA. J Cell Biochem. 2005;96(2):314–329. | ||
Kaul R, Verma SC, Murakami M, Lan K, Choudhuri T, Robertson ES. Epstein-Barr virus protein can upregulate cyclo-oxygenase-2 expression through association with the suppressor of metastasis Nm23-H1. J Virol. 2006;80(3):1321–1331. | ||
Diaz-Cruz E, Brueggemeier R. Interrelationships between cyclooxygenases and aromatase: unraveling the relevance of cyclooxygenase inhibitors in breast cancer. Anticancer Agents Med Chem. 2006;6(3):221–232. | ||
Harris R, Beebe J, Alshafie GA. Reduction in cancer risk by selective and nonselective cyclooxygenase-2 (COX-2) inhibitors. J Exp Pharmacol. 2012;4:91–96. | ||
Brueggemeier RW, Quinn AL, Parrett ML, Joarder FS, Harris RE, Robertson FM. Correlation of aromatase and cyclooxygenase gene expression in human breast cancer specimens. Cancer Lett. 1999;140(1-2):27–35. | ||
Terry MB, Gammon MD, Zhang F, et al. Association of frequency and duration of aspirin use and hormone receptor status with breast cancer risk. JAMA. 2004;291(20):2433–2440. | ||
Holmes MD, Chen WY, Li L, Hertzmark E, Spiegelman D, Hankinson SE. Aspirin intake and survival after breast cancer. J Clin Oncol. 2010;28(9):1467–1472. | ||
Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009;302(6):649–658. | ||
Ursin G, Hovanessian-Larsen L, Parisky YR, Pike MC, Wu AH, Ah W. Greatly increased occurrence of breast cancers in areas of mammographically dense tissue. Breast Cancer Res. 2005;7(5):R605–608. | ||
Boyd NF. Mammographic density and risk of breast cancer. Am Soc Clin Oncol Educ Book. 2013. | ||
Pettersson A, Graff RE, Ursin G, et al. Mammographic density phenotypes and risk of breast cancer: a meta-analysis. J Natl Cancer Inst. 2014;106(5). | ||
Esbona K, Inman D, Saha S, et al. COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 2016;18(1):35. | ||
Li H, Yang B, Huang J, et al. Cyclooxygenase-2 in tumor-associated macrophages promotes breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. Oncotarget. 2015;6(30):29637–29650. | ||
Gupta GP, Nguyen DX, Chiang AC, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446(7137):765–770. | ||
Leahy K, Koki A, Masferrer J. Role of cyclooxygenases in angiogenesis. Curr Med Chem. 2000;7(11):1163–1170. | ||
Zha S, Yegnasubramanian V, Nelson WG, Isaacs WB, de Marzo AM. Cyclooxygenases in cancer: progress and perspective. Cancer Lett. 2004;215(1):1–20. | ||
Fosslien E. Review: molecular pathology of cyclooxygenase-2 in cancer-induced angiogenesis. Ann Clin Lab Sci. 2001;31(4):325–348. | ||
Zhang Y, Daaka Y. PGE2 promotes angiogenesis through EP4 and PKA C pathway. Blood. 2011;118(19):5355–5364. | ||
Basu GD, Liang WS, Stephan DA, et al. A novel role for cyclooxygenase-2 in regulating vascular channel formation by human breast cancer cells. Breast Cancer Res. 2006;8(6):R69. | ||
Fife RS, Stott B, Carr RE. Effects of a selective cyclooxygenase-2 inhibitor on cancer cells in vitro. Cancer Biol Ther. 2004;3(2):228–232. | ||
Hahn T, Alvarez I, Kobie JJ, et al. Short-term dietary administration of celecoxib enhances the efficacy of tumor lysate-pulsed dendritic cell vaccines in treating murine breast cancer. Int J Cancer. 2006;118(9):2220–2231. | ||
Sun TW, Zh W, Weng XS. Celecoxib can suppress expression of genes associated with PGE2 pathway in chondrocytes under inflammatory conditions. Int J Clin Exp Med. 2015;8(7):10902–10910. | ||
Fournier LS, Novikov V, Lucidi V, et al. MR monitoring of cyclooxygenase-2 inhibition of angiogenesis in a human breast cancer model in rats. Radiology. 2007;243(1):105–111. | ||
Han F, Zhang S, Zhang L, Hao Q. The overexpression and predictive significance of MMP-12 in esophageal squamous cell carcinoma. Pathol Res Pract. 2017;213(12):1519–1522. | ||
Kumar BNP, Rajput S, Dey KK, et al. Celecoxib alleviates tamoxifen-instigated angiogenic effects by ROS-dependent VEGF/VEGFR2 autocrine signaling. BMC Cancer. 2013;13(1):273. | ||
Vaish V, Sanyal SN. Role of Sulindac and Celecoxib in the regulation of angiogenesis during the early neoplasm of colon: Exploring PI3-K/PTEN/Akt pathway to the canonical Wnt/β-catenin signaling. Biomed Pharmacother. 2012;66(5):354–367. | ||
Kalalinia F, Elahian F, Behravan J. Potential role of cyclooxygenase-2 on the regulation of the drug efflux transporter ABCG2 in breast cancer cell lines. J Cancer Res Clin Oncol. 2011;137(2):321–330. | ||
Kalalinia F, Elahian F, Mosaffa F, Behravan J. Celecoxib Up Regulates the Expression of Drug Efflux Transporter ABCG2 in Breast Cancer Cell Lines. Iran J Pharm Res. 2014;13(4):1393–1401. | ||
Ratnasinghe D, Daschner PJ, Anver MR, et al. Cyclooxygenase-2, P-glycoprotein-170 and drug resistance; is chemoprevention against multidrug resistance possible? Anticancer Res. 2001;21(3C):2141–2147. | ||
Miller B, Patel VA, Sorokin A. Cyclooxygenase-2 rescues rat mesangial cells from apoptosis induced by adriamycin via upregulation of multidrug resistance protein 1 (P-glycoprotein). J Am Soc Nephrol. 2006;17(4):977–985. | ||
Sorokin A. Cyclooxygenase-2: potential role in regulation of drug efflux and multidrug resistance phenotype. Curr Pharm Des. 2004;10(6):647–657. | ||
Xia W, Zhao T, Lv J, et al. Celecoxib enhanced the sensitivity of cancer cells to anticancer drugs by inhibition of the expression of P-glycoprotein through a COX-2-Independent Manner. J Cell Biochem. 2009;108(1):181–194. | ||
Chen C, Shen HL, Yang J, Chen QY, Xu WL, Wl X. Preventing chemoresistance of human breast cancer cell line, MCF-7 with celecoxib. J Cancer Res Clin Oncol. 2011;137(1):9–17. | ||
Hida T, Kozaki K, Ito H, et al. Significant growth inhibition of human lung cancer cells both in vitro and in vivo by the combined use of a selective cyclooxygenase 2 inhibitor, JTE-522, and conventional anticancer agents. Clin Cancer Res. 2002;8(7):2443–2447. | ||
van Wijngaarden J, van Beek E, van Rossum G, et al. Celecoxib enhances doxorubicin-induced cytotoxicity in MDA-MB231 cells by NF-κB-mediated increase of intracellular doxorubicin accumulation. Eur J Cancer. 2007;43(2):433–442. | ||
Irie T, Tsujii M, Tsuji S, et al. Synergistic antitumor effects of celecoxib with 5-fluorouracil depend on IFN-γ. Int J Cancer. 2007;121(4):878–883. | ||
Young SD, Lafrenie RM, Clemons MJ. Phase ii trial of a metronomic schedule of docetaxel and capecitabine with concurrent celecoxib in patients with prior anthracycline exposure for metastatic breast cancer. Curr Oncol. 2012;19(2):e75–83. | ||
Fabi A, Metro G, Papaldo P, et al. Impact of celecoxib on capecitabine tolerability and activity in pretreated metastatic breast cancer: results of a phase II study with biomarker evaluation. Cancer Chemother Pharmacol. 2008;62(4):717–725. | ||
Perroud HA, Alasino CM, Rico MJ, et al. Metastatic breast cancer patients treated with low-dose metronomic chemotherapy with cyclophosphamide and celecoxib: clinical outcomes and biomarkers of response. Cancer Chemother Pharmacol. 2016;77(2):365–374. | ||
Chow LW, Loo WT, Wai CC, Lui EL, Zhu L, Toi M. Study of COX-2, Ki67, and p53 expression to predict effectiveness of 5-flurouracil, epirubicin and cyclophosphamide with celecoxib treatment in breast cancer patients. Biomed Pharmacother. 2005;59 Suppl 2(Suppl 2):S298–S301. | ||
Chow LW, Tung SY, Ng TY, Ty N, et al. Concurrent celecoxib with 5-fluorouracil/epirubicin/cyclophosphamide followed by docetaxel for stages II - III invasive breast cancer: the OOTR-N001 study. Expert Opin Investig Drugs. 2013;22(3):299–307. | ||
Chow LW, Loo WT, Yip AY, Ng EL. Acceptable cardiac safety profile of neoadjuvant 5-fluorouracil, epirubicin, cyclophosphamide and celecoxib (FEC-C) for breast cancer: a subanalysis of biomarkers for cardiac injury. Int J Biol Markers. 2013;28(1):32–37. | ||
Ueno T, Chow LW, Toi M. Increases in circulating VEGF levels during COX-2 inhibitor treatment in breast cancer patients. Biomed Pharmacother. 2006;60(6):277–279. | ||
Lim JS, Park Y, Lee BM, Kim HS, Yoon S. Co-treatment with Celecoxib or NS398 Strongly Sensitizes Resistant Cancer Cells to Antimitotic Drugs Independent of P-gp Inhibition. Anticancer Res. 2016;36(10):5063–5070. | ||
Webb T, Carter J, Roberts JL, et al. Celecoxib enhances [sorafenib + sildenafil] lethality in cancer cells and reverts platinum chemotherapy resistance. Cancer Biol Ther. 2015;16(11):1660–1670. | ||
Pierga JY, Delaloge S, Espié M, et al. A multicenter randomized phase II study of sequential epirubicin/cyclophosphamide followed by docetaxel with or without celecoxib or trastuzumab according to HER2 status, as primary chemotherapy for localized invasive breast cancer patients. Breast Cancer Res Treat. 2010;122(2):429–437. | ||
Giacchetti S, Hamy AS, Delaloge S, et al. Long-term outcome of the REMAGUS 02 trial, a multicenter randomised phase II trial in locally advanced breast cancer patients treated with neoadjuvant chemotherapy with or without celecoxib or trastuzumab according to HER2 status. Eur J Cancer. 2017;75:323–332. | ||
Subbaramaiah K, Howe LR, Port ER, et al. HER-2/neu status is a determinant of mammary aromatase activity in vivo: evidence for a cyclooxygenase-2-dependent mechanism. Cancer Res. 2006;66(10):5504–5511. | ||
Benoit V, Relic B, Leval Xd X, Chariot A, Merville MP, Bours V. Regulation of HER-2 oncogene expression by cyclooxygenase-2 and prostaglandin E2. Oncogene. 2004;23(8):1631–1635. | ||
Dang CT, Dannenberg AJ, Subbaramaiah K, et al. Phase II study of celecoxib and trastuzumab in metastatic breast cancer patients who have progressed after prior trastuzumab-based treatments. Clin Cancer Res. 2004;10(12 Pt 1):4062–4067. | ||
El-Awady RA, Saleh EM, Ezz M, Elsayed AM. Interaction of celecoxib with different anti-cancer drugs is antagonistic in breast but not in other cancer cells. Toxicol Appl Pharmacol. 2011;255(3):271–286. | ||
Gates MA, Tworoger SS, Eliassen AH, Missmer SA, Hankinson SE. Analgesic use and sex steroid hormone concentrations in postmenopausal women. Cancer Epidemiol Biomarkers Prev. 2010;19(4):1033–1041. | ||
Marshall SF, Bernstein L, Anton-Culver H, et al. Nonsteroidal anti-inflammatory drug use and breast cancer risk by stage and hormone receptor status. J Natl Cancer Inst. 2005;97(11):805–812. | ||
Brueggemeier RW, Díaz-Cruz ES, Li PK, Sugimoto Y, Lin YC, Shapiro CL. Translational studies on aromatase, cyclooxygenases, and enzyme inhibitors in breast cancer. J Steroid Biochem Mol Biol. 2005;95(1-5):129–136. | ||
Sun X, Lv M, Wang B, et al. Comparative pharmacokinetics study of anastrozole after single administration and combination with celecoxib. Xenobiotica. 2018;48(3):1–6. | ||
Chow LW, Cheng CW, Wong JL, Toi M. Serum lipid profiles in patients receiving endocrine treatment for breast cancer--the results from the Celecoxib Anti-Aromatase Neoadjuvant (CAAN) Trial. Biomed Pharmacother. 2005;59(Suppl 2):S302–S305. | ||
Chow LW, Yip AY, Loo WT, Lam CK, Toi M. Celecoxib anti-aromatase neoadjuvant (CAAN) trial for locally advanced breast cancer. J Steroid Biochem Mol Biol. 2008;111(1–2):13–17. | ||
Canney PA, Machin MA, Curto J. A feasibility study of the efficacy and tolerability of the combination of Exemestane with the COX-2 inhibitor Celecoxib in post-menopausal patients with advanced breast cancer. Eur J Cancer. 2006;42(16):2751–2756. | ||
Falandry C, Debled M, Bachelot T, et al. Celecoxib and exemestane versus placebo and exemestane in postmenopausal metastatic breast cancer patients: a double-blind phase III GINECO study. Breast Cancer Res Treat. 2009;116(3):501–508. | ||
Lustberg MB, Povoski SP, Zhao W, et al. Phase II trial of neoadjuvant exemestane in combination with celecoxib in postmenopausal women who have breast cancer. Clin Breast Cancer. 2011;11(4):221–227. | ||
Dirix LY, Ignacio J, Nag S, et al. Treatment of advanced hormone-sensitive breast cancer in postmenopausal women with exemestane alone or in combination with celecoxib. J Clin Oncol. 2008;26(8):1253–1259. | ||
Boneberg EM, Legler DF, Senn HJ, Fürstenberger G. Reduced expression of cyclooxygenase-2 in primary breast cancer. J Natl Cancer Inst. 2008;100(14):1042–1043. | ||
Aristarco V, Serrano D, Gandini S, et al. A Randomized, Placebo-Controlled, Phase II, Presurgical Biomarker Trial of Celecoxib Versus Exemestane in Postmenopausal Breast Cancer Patients. Cancer Prev Res. 2016;9(5):349–356. | ||
Basu GD, Tinder TL, Bradley JM, et al. Cyclooxygenase-2 inhibitor enhances the efficacy of a breast cancer vaccine: role of IDO. J Immunol. 2006;177(4):2391–2402. | ||
Li Y, Fang M, Zhang J, et al. Hydrogel dual delivered celecoxib and anti-PD-1 synergistically improve antitumor immunity. Oncoimmunology. 2016;5(2):e1074374. | ||
Cho HY, Thomas S, Golden EB, et al. Enhanced killing of chemo-resistant breast cancer cells via controlled aggravation of ER stress. Cancer Lett. 2009;282(1):87–97. | ||
Thomas S, Sharma N, Golden EB, et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Lett. 2012;325(1):63–71. | ||
das Roy L, Pathangey LB, Tinder TL, Schettini JL, Gruber HE, Mukherjee P. Breast-cancer-associated metastasis is significantly increased in a model of autoimmune arthritis. Breast Cancer Res. 2009;11(4):R56. | ||
Mustafa A, Kruger WD. Suppression of tumor formation by a cyclooxygenase-2 inhibitor and a peroxisome proliferator-activated receptor gamma agonist in an in vivo mouse model of spontaneous breast cancer. Clin Cancer Res. 2008;14(15):4935–4942. | ||
Niu G, Liao Z, Cai L, Wei R, Sun L. The combined effects of celecoxib and minocycline hydrochloride on inhibiting the osseous metastasis of breast cancer in nude mice. Cancer Biother Radiopharm. 2008;23(4):469–476. | ||
Yu P, Liu Q, Liu K, Yagasaki K, Wu E, Zhang G. Matrine suppresses breast cancer cell proliferation and invasion via VEGF-Akt-NF-kappaB signaling. Cytotechnology. 2009;59(3):219–229. | ||
Wang S, Liu Q, Zhang Y, et al. Suppression of growth, migration and invasion of highly-metastatic human breast cancer cells by berbamine and its molecular mechanisms of action. Mol Cancer. 2009;8:81. | ||
Jeon YW, Ahn YE, Chung WS, Choi HJ, Suh YJ. Synergistic effect between celecoxib and luteolin is dependent on estrogen receptor in human breast cancer cells. Tumour Biol. 2015;36(8):6349– 6359. | ||
Jeon YW, Suh YJ. Synergistic apoptotic effect of celecoxib and luteolin on breast cancer cells. Oncol Rep. 2013;29(2):819–825. | ||
Kisková T, Jendželovský R, Rentsen E, et al. Resveratrol enhances the chemopreventive effect of celecoxib in chemically induced breast cancer in rats. Eur J Cancer Prev. 2014;23(6):506–513. | ||
Kim Y, Je Y. Vitamin D intake, blood 25(OH)D levels, and breast cancer risk or mortality: a meta-analysis. Br J Cancer. 2014;110(11):2772–2784. | ||
Rose AA, Elser C, Ennis M, Goodwin PJ. Blood levels of vitamin D and early stage breast cancer prognosis: a systematic review and meta-analysis. Breast Cancer Res Treat. 2013;141(3):331–339. | ||
Thill M, Reichert K, Woeste A, et al. Combined treatment of breast cancer cell lines with vitamin D and COX-2 inhibitors. Anticancer Res. 2015;35(2):1189–1195. | ||
Thill M, Terjung A, Friedrich M. Breast cancer--new aspects of tumor biology: are calcitriol and cyclooxygenase-2 possible targets for breast cancer? Eur J Gynaecol Oncol. 2014;35(4):341–358. | ||
Howes LG. Selective COX-2 inhibitors, NSAIDs and cardiovascular events - is celecoxib the safest choice? Ther Clin Risk Manag. 2007;3(5):831–845. | ||
Abassi YA, Xi B, Zhang W, et al. Kinetic cell-based morphological screening: prediction of mechanism of compound action and off-target effects. Chem Biol. 2009;16(7):712–723. | ||
Pyrko P, Kardosh A, Liu YT, et al. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib, a non-coxib analogue of celecoxib. Mol Cancer Ther. 2007;6(4):1262–1275. | ||
O’Connor JP, Lysz T. NSAIDs and the skeleton. Drugs Today. 2008;44(9):693–709. | ||
Lee JH, Park HK, Heo J, et al. Drug Rash with Eosinophilia and Systemic Symptoms (DRESS) syndrome induced by celecoxib and anti-tuberculosis drugs. J Korean Med Sci. 2008;23(3):521–525. | ||
Chen J, Shen P, Zhang XC, Zhao MD, Zhang XG, Yang L. Efficacy and safety profile of celecoxib for treating advanced cancers: a meta-analysis of 11 randomized clinical trials. Clin Ther. 2014;36(8):1253–1263. | ||
Megeff CE, Strayer SM. Celecoxib for rheumatoid arthritis. J Fam Pract. 2000;49(2):108–109. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.