Back to Journals » Cancer Management and Research » Volume 11
CDKN3 promotes tumor progression and confers cisplatin resistance via RAD51 in esophageal cancer
Authors Wang J ![]() , Che W, Wang W, Su G, Zhen T, Jiang Z
, Che W, Wang W, Su G, Zhen T, Jiang Z
Received 7 November 2018
Accepted for publication 5 March 2019
Published 15 April 2019 Volume 2019:11 Pages 3253—3264
DOI https://doi.org/10.2147/CMAR.S193793
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Jiansong Wang,1 Wencheng Che,2 Weimin Wang,1 Gongzhang Su,3 Tianchang Zhen,3 Zhongmin Jiang3
1Graduate Department, Weifang Medical University, Weifang, Shandong 261031, People’s Republic of China; 2Department of Thoracic Surgery, Zibo Central Hospital, Zibo, Shandong 255022, People’s Republic of China; 3Department of Thoracic Surgery, Qianfoshan Hospital Affiliated to Shandong University, Jinan, Shandong 250014, People’s Republic of China
Purpose: Esophageal cancer (ESCA) progression and chemoresistance are critical factors that impact the survival of patients with esophageal cancer. Cyclin dependent kinase inhibitor 3 (CDKN3) is an important regulator of the cell cycle that has received little attention, therefore the purpose of this study was to investigate CDKN3 involvement in ESCA.
Methods: We first explored the public database in addition to our cohort to evaluate the expression of CDKN3 in ESCA patients. We performed bioinformative analysis on specific processes regulated by CDKN3, then we investigated the role of CDKN3 in ESCA progression and chemoresistance in vitro and in vivo. Finally, we sought to elucidate the mechanism of CDKN3 regulation of chemoresistance in ESCA.
Results: We discovered that CDKN3 was highly expressed in ESCA and serves as an independent prognostic factor of this disease. Bioinformatic analysis showed CDKN3 involvement in DNA replication, the cell cycle G2/M phase transition, DNA damage repair (DDR) signaling pathways, et al Functional experiments in vitro and in vivo demonstrated that CDKN3 promoted ESCA progression and enhanced cisplatin resistance. Furthermore, CDKN3 inhibition resulted in reduced expression of RAD51, which plays a pivotal role in DDR. Overexpression of RAD51 reversed cisplatin-induced DNA damage and chemosensitivity in CDKN3 inhibited ESCA cell lines.
Conclusion: The present research indicated that CDKN3 promoted ESCA progression and enhanced cisplatin resistance via RAD51, thereby influencing overall patient survival.
Keywords: esophageal cancer, chemoresistance, CDKN3, RAD51, DNA damage repair
Introduction
Esophageal cancer (ESCA) is a clinically challenging disease that requires extensive medical attention. ESCA is ranked as the sixth leading cause of mortality (508,585 deaths per year) and accounts for 6.6% of all cancer-related deaths.1 ESCA progression and chemoresistance are important factors that influence patient survival. In patients with locally advanced ESCA, whose 5-year survival is about 20%, the standard treatment consists of chemotherapy or chemoradiotherapy with surgery.2 Cisplatin belongs to the platinum-based drug category, which is the most prevalent first-line treatment for ESCA. Unfortunately, cisplatin resistance has become increasingly more common and is a major cause of treatment failure in ESCA.2 Thus, it is imperative to elucidate the mechanisms involved in tumor progression and cisplatin resistance in ESCA.
Cyclin dependent kinase inhibitor 3 (CDKN3), a member of the dual-specificity protein phosphatase family, plays a significant role in cell division and cancer progression. CDKN3 has inhibitory effects on the CDK-driven cell cycle, which is a major event for the progression of cancer cells. Binding of CDKN3 to CDK1/CDK2 results in dephosphorylation of the activated residues and an inhibition of CDK activities.3 Despite the negative regulatory effects of CDKN3 on CDK1 and CDK2, aberrant overexpression of CDKN3 has been implicated in numerous human cancers including prostate cancer,4 gastric cancer,5 and nasopharyngeal carcinoma,6 et al CDKN3 promotes tumor growth, migration, invasion and other aggressive biological processes,5 but its precise mechanism in the regulation of ESCA progression remains unknown. Therefore, this study aimed to explore the role of CDKN3 in ESCA.
In the pilot study, we demonstrated that the CDKN3 expression was significantly increased in both esophageal adenocarcinoma (EA) and esophageal squamous cell carcinoma (ESCC) in comparison with normal esophageal epithelial tissues. Elevated levels of CDKN3 is associated with a poor prognosis in ESCA. Interestingly, bioinformatic analysis of The Cancer Genome Atlas (TCGA) database revealed CDKN3 involvement in numerous biological processes including cell cycle transformation, DNA replication and DNA damage repair (DDR), et al Therefore, we hypothesized that CDKN3 promotes tumor progression through cell cycle regulation and contributes to chemoresistance through DDR. We also found that CDKN3 expression was positively correlated with RAD51 expression, which mediates homologous pairing and strand exchange reactions between single and double stranded DNA during repair.7 In all, we determined that CDKN3 can promote cell proliferation by facilitating the cell cycle G2/M transition and influencing chemosensitivity by interacting with RAD51 in ESCA.
Material and methods
ESCA clinical specimens
Patients with pathologically confirmed ESCA who underwent surgery followed by platinum-based adjuvant chemotherapy between September 2016 and September 2017 were enrolled in this study. Patients who received neoadjuvant chemoradiotherapy were excluded. Chemoresistance refers to the occurrence of tumor recurrence or metastasis within one year after surgery, and patients without tumor progression are included in the sensitive group.8 Twenty-three pairs of ESCA and normal tissues were gathered in the Qianfoshan hospital. There were twenty-one ESCC patients and two EA patients with an age range of 61–73. All patients received the same radical esophectomy procedure. Both the clinical stage and the pathological stage were locally advanced, but there was no evidence of metastasis. Among these patients, sixteen patients were in the sensitive group and seven patients were in the resistant group. The study protocol was approved by the Qianfoshan Hospital ethics committee. Signed informed consent was received prior to tissue collection.
Bioinformatic analysis of the TCGA and GEO database
Information regarding 184 ESCA tissues and 11 normal esophageal tissues was extracted from the TCGA database (
Cell lines and cell culture
ESCC cell lines EC109 and kyse150 were purchased from the Chinese Academy of Sciences cell bank (Shanghai, China). EA cell line OE19 and esophageal epithelial cell line Het-1A were purchased from the European Collection of Authenticated Cell Cultures (ECACC) and American Type Culture Collection (ATCC), respectively. All cells were cultured in RPMI-1640 medium with 10% fetal bovine serum and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere containing 5% CO2. The cisplatin-resistant cell lines EC109R and OE19R were generated by exposing the parental cells to cisplatin at increasing drug concentrations for one year. The cell lines were cultured in medium containing 0.5 μM cisplatin to maintain the cisplatin-resistant phenotype.
Plasmid transfection
Cells were cultured in a 6-well plate overnight. 1μg plasmid were transfected with 3μl Lipofectamine 2000 reagent (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. 1 μg/mL G418 was added to eliminate uninfected cells. Transfected cells were used in the subsequent study. Human CDKN3 specific shRNA (shCDKN3) and a scrambled control shRNA (NC) were cloned into pGPU6-neo vector. The sequences were: shCDKN3, 5ʹ- CGGGACAAATTAGCTGCACATC −3ʹ; NC, 5ʹ-TTCTCCGAACGTGTCACGT-3ʹ. Human RAD51 cDNA was constructed into PCDNA3.1(+) plasmid.
Quantitative real-time PCR (qPCR)
According to the manufacturers’ instructions, total RNA was extracted with the R6934 Kit (Omega Bio-tek, Norcross, GA, USA), cDNA was synthesized with the RT kit (Takara, Kusatsu, Shiga, Japan) and mRNA levels were quantified with the qPCR kit (Takara, Kusatsu, Shiga, Japan). Primers were as follows.
CDKN3 (Forward) 5ʹ- GCCCAGTTCAATACAAAC-AAGT −3ʹ
CDKN3 (Reverse) 5ʹ- CAACCTGGAAGAGCACAT-AAAC −3ʹ
GAPDH (Forward) 5ʹ- CAGAACATCATCCCTGCCT-CTAC −3ʹ
GAPDH (Reverse) 5ʹ- ATGAAGTCAGAGGAGACC-ACCTG −3ʹ
Western blots
The cell cultures were lysed in RIPA, then proteins were separated by 12.5% SDS-PAGE and transferred onto PVDF membrane. After being blocked with 5% nonfat milk at room temperature for 2 h, the membrane was incubated with the primary antibody at 4 °C overnight followed by the secondary antibody at room temperature for one hour. Millipore ECL reagent and Bio-Rad ChemiDoc Imaging Systems were used for protein quantification. CDKN3 antibody was purchased from Abcam Technology (Cambridge, Cambridgeshire, UK) and all other antibodies were acquired from Cell Signaling Technology (Danvers, MA, USA).
Co-immunoprecipitation (CoIP)
To determine the interaction between CDKN3 and RAD51, total EC109 and OE19 cell extracts were precleared with 50μL protein A/G agarose beads (Santa Cruz, Dallas, Texas, USA) at 4 °C for 2 hrs. The supernatant was incubated with anti-CDKN3 or anti-RAD51 with gentle shaking overnight at 4 °C, followed by the addition of 50μL of protein A/G agarose beads for 4 hrs. Immobilized protein complexes were denatured in 2×SDS sample buffer at 95 °C for 10min for subsequent western blots.
Cell counting kit 8 assay (CCK8)
For the cell proliferation assay, five thousand stably transfected cells per well were plated into 96-well plates overnight. 10 μl CCK-8 reagent (Dojindo laboratories, Kumamoto, Japan) was added to each well and they were incubated for 2h at 37 °C. Then, the absorbance of each well was examined at 450 nm by the microplate reader (Bio-rad, Hercules, CA, USA). For the drug sensitivity assay, after treating with gradient dilution of cisplatin (0, 1, 2, 4, 8, 16, 32, 64 and 128 μM) for 48 h, the cell viability was tested as above. Cisplatin was purchased form Solarbio (Beijing, China) and was dissolved in normal saline at 1 mg/ml.
Colony formation assay
1×103 stably transfected cells were seeded onto 60-mm culture plates. Cells were incubated for 2 weeks until visible colonies appeared, then fixed with 4% paraformaldehyde followed by staining with 0.25% crystal violet solution. The colonies were photographed and counted.
Cell cycle analysis
1×106 stably transfected cells per well were plated into 6-well plates, then harvested and fixed with 70% cold ethanol overnight at 4 °C. PI-staining solution containing RNase A (BD Biosciences, San Jose, CA, USA) was added and the plates were incubated for 30 min at 37 °C in the dark. FACSAria II cytometry (BD Biosciences, San Jose, CA, USA) was utilized to record the samples, and data was analyzed using FlowJo software (version10.3, BD Biosciences, San Jose, CA, USA).
Apoptosis analysis
Apoptosis was measured by Annexin V-PE/7-AAD kit (BioLegend, San Diego, CA, USA) according to the supplier’s instructions. Next, cells were plated into 6-well plates and then treated with 5 μM cisplatin for 48 h. Cells were stained with 5 μl Annexin V-PE and 5 μl 7-AAD for 15 min at room temperature, then samples were recorded with FACSAria II cytometry (BD Biosciences, San Jose, CA, USA). Data was analyzed using FlowJo software (version10.3, BD Biosciences, San Jose, CA, USA).
Immunofluorescence
Cells were placed on glass slides at the density of 1 × 105 cells/ml overnight at 37 °C. Following incubation with 5 μM cisplatin for 48 h, cells were fixed in methanol for 10min, permeabilized with 0.5% Triton X-100 for 5 min and blocked in 5% goat serum for 60min at room temperature. Cells were then incubated with γH2AX (1:100, CST, Danvers, MA, USA) overnight at 4 °C, followed by incubation with the Alexa Fluor 594 – conjugated Goat Anti-Rabbit IgG (1:100, Proteintech, Rosemont, IL, USA) for 1h at room temperature in the dark. Last, the cells were stained with 5 μg/ml DAPI (Solarbio, Beijing, China) and immunofluorescence was visualized using the Carl Zeiss Confocal Microscope LSM780 (Oberkochen, Germany). Frozen tissues were cut into 6μm sections and incubated with Ki67 (1:100, CST, Danvers, MA, USA). The other procedures were identical to cellular immunofluorescence.
Xenograft tumor models
Male BALB/C nude mice were purchased from Vital River Laboratory (Beijing, China) and housed in specific pathogen free conditions. All mice were randomly grouped, then cancer cells (5×106) were resuspended in 100 μl PBS and were injected into the double dorsal flank of the mice. For the cisplatin drug injection group, after the tumor volume reached 200 mm3, cisplatin (5 mg/kg) was administered intraperitoneally every three days. After one month, mice were euthanized and the tumors were dissected. All animal experiments were carried out in accordance with the National Institute of Health guidelines for the care and use of laboratory animals. The xenograft tumor model experiments were approved by the Qianfoshan hospital Animal Care and Use Committee.
Statistical analysis
SPSS (version 20.0) was used to perform the statistical analysis. Quantitative data were showed as mean ± standard deviation, paired Student's t test was used for comparison between the paired groups, and unpaired Student's t test was used for comparison between the unpaired groups. Multiple sets of quantitative data were analyzed by one-way ANOVA, and the Tukey method was used for pairwise comparison. Survival curves were plotted using the Kaplan-Meier method and compared using the log rank test. Univariate Cox proportional hazards regression models were used for univariate analysis of the impact of CDKN3 expression on overall survival (OS). The Pearson correlation coefficient was used to evaluate the degree of linear correlation between two variables. All in vitro experiments were performed at least three times. All statistical tests were two-sided and a P-value <0.05 was considered statistically significant.
Results
CDKN3 was upregulated in ESCA and was associated with a poor prognosis
To explore the role of CDKN3 in ESCA, we first compared CDKN3 expression between esophageal cancer tissues and normal tissues. TCGA analysis showed a higher expression of CDKN3 mRNA in cancer tissues compared with normal tissues (Figure 1A) in both the EA dataset GSE92396 (Figure 1B) and the ESCC dataset GSE17351 (Figure 1C). The next step was to assess CDKN3 expression in the 23 paired esophageal cancer tissues and normal tissues in our cohort. The ESCC group consisted of 21 patients and the EA group consisted of 2 patients. The expression of CDKN3 mRNA in ESCA was more than double the expression of matched adjacent tissues (Figure 1D). Western blot analysis also revealed a significantly higher CDKN3 protein expression in ESCA tissues compared with paired normal tissues (Figure 1E and F).
 | Figure 1 The association between CDKN3 overexpression and ESCA. (A–D) CDKN3 mRNA expression analysis in the TCGA dataset (A), EA dataset GSE92396 (B), ESCC dataset GSE17351 (C) and our cohort (D). (E–F) CDKN3 protein expression analysis in our cohort by western blot analysis (E) and the intensity results (F). (G) The prognostic significance of CDKN3 expression was analyzed by Kaplan–Meier survival analysis in the TCGA database. *P<0.05, **P<0.01, ***P<0.001. |
Moreover, we explored the prognostic value of CDKN3 using the TCGA ESCA dataset. ESCA patients were divided into groups of high and low expression using X-tile software.10 Patients in the high CDKN3 expression group (n=79) had a significantly worse prognosis than those in the low CDKN3 expression group (n=105) (median survival time 553 vs 987 days, P=0.0153) (Figure 1G). Furthermore, CDKN3 expression was validated as an independent prognostic indicator of OS (HR=1.82, 95%CI: 1.141–2.904, P=0.012) via univariate cox regression analysis.
These findings demonstrated high expression of CDKN3 in both EA and ESCC and that elevated CDKN3 may predict a poor outcome for ESCA patients.
Bioinformatic analysis of CDKN3 and co-regulated genes
To further explore potential biological functions regulated by CDKN3 in ESCA, correlation analysis of CDKN3 expression in whole-genome profiling was performed by LinkedOmics.11 A total of 989 positively related genes and 456 negatively related genes was excavated. The GO biological process analysis revealed that these genes were involved in a variety of processes, including DNA replication, cell cycle G2/M phase transition, double strand break repair, recombinational repair (Figure 2A). KEGG enrichment analysis also demonstrated involvement in similar pathways (Figure 2B). GSEA analysis revealed that high CDNK3 expression is strongly associated with the cell cycle phase transition, DNA replication, interstrand crosslink repair, et al (Figure 2C). Altogether, these data further implicated the malignant role of CDKN3 in ESCA.
 | Figure 2 The correlation between CDKN3 expression with the cell cycle and DNA damage repair. (A–B) GO (A) and KEGG (B) enrichment analysis with CDKN3 strongly correlated genes. The X axis represents counts. (C) Representative GSEA enriched gene sets within CDKN3 co-regulated genes.Abbreviations: FDR, false discovery rate; NES, normalized enrichment score. |
CDKN3 promoted tumor progression in vitro and in vivo
Enrichment analysis and GSEA suggested that CDKN3 may be involved in DNA replication and cell cycle regulation in ESCA, but further research was necessary to determine if CDKN3 overexpression would facilitate ESCA progression. ESCC cell lines (EC109, kyse150) and EA cell line OE19 displayed a significantly higher expression of CDKN3 mRNA and proteins than the esophageal epithelial cell line Het-1A (Figure 3A–C). Due to the significant elevation of CDKN3 in ESCC and EA, ESCC cell line EC109 and EA cell line OE19 were chosen to further investigate the role of CDKN3 in these two different histological types of ESCA in the following study.
 | Figure 3 CDKN3 facilitated tumor progression in vitro and in vivo. (A–C) CDKN3 mRNA (A) and protein (B–C) expression in ESCA and esophageal epithelial cells. (D) CDKN3 protein expression decreased in shCDKN3 transfected cells, as determined by western blots. (E) The cell viability reduced in CDKN3 inhibited cells by CCK8. (F–G) Cell colonies reduced in CDKN3 inhibited cells as determined by colony formation assay (F) and the quantitative results (G). (H–I) CDKN3 knockdown affected cell cycle distribution (H) and the quantitative results (I). The number in the picture is the percentage of time spent in G2/M of the cell cycle. (J–L) CDKN3 knockdown decreased tumor volumes (J–K) and Ki-67 intensity (L) in xenograft tumors. Scale bar=5 μm. **P<0.01, ***P<0.001.Abbreviations: NC, negative control; shCDKN3, cells transfected with shCDKN3 plasmid. |
Western blot analysis confirmed that transfection with the shCDKN3 plasmid reduced CDKN3 expression to appr-oximately 30% after 72 h compared with the NC (Figure 3D). Thereafter, we compared the proliferation rate among ESCA cells transfected with shCDKN3, NC and untransfected cells (BLANK). Downregulation of CDKN3 suppressed cell viability in both EA and ESCC cell lines, compared with NC and BLANK cells (Figure 3E). Similar results were shown in the colony formation assay (Figure 3F–G). To explore the mechanism by which CDKN3 impacts cell proliferation, we conducted a study of the cell cycle. Cell cycle analysis showed that CDKN3 knockdown led to a decrease in the time spent in the G2/M phase transition, compared to NC and BLANK (Figure 3H–I). Furthermore, CDKN3 knockdown significantly decreased tumor volume using a xenograft tumor model in vivo (Figure 3J–K), which was accompanied by decreasing intensity of Ki-67 fluorescence (Figure 3L). Collectively, these data verified our speculation that CDKN3 promoted ESCA progression by regulating the cell cycle in vitro and in vivo.
CDKN3 contributed to cisplatin resistance of ESCA in vitro and in vivo
As shown in Figure 2, CDKN3 is involved in the regulation of the DDR signaling pathway. Our pilot study discovered that CDKN3 mRNA is significantly increased in resistant patients (Figure 4A). Based on the above findings and the relationship between chemoresistance and DDR,12 we hypothesized that CDKN3 can mediate the chemosensitivity of ESCA.
 | Figure 4 CDKN3 increased ESCA resistance to cisplatin in vitro and in vivo. (A) Comparison of CDKN3 mRNA between sensitive and resistant tumors. (B–C) EC109R (B) and OE19R (C) cells displayed greater resistance to cisplatin than their parental cells. (D) Comparison of CDKN3 mRNA between resistant cells and their parental cells. (E) CDKN3 protein expression was inhibited in cisplatin-resistant ESCA by shCDKN3 as determined by western blots. (F–G) Decreased CDKN3 expression weakened ESCA chemoresistance in EC109R (F) and OE19R (G) as determined by CCK8 assay. (H–I) CDKN3 Knockdown enhanced cisplatin-induced ESCA apoptosis (H) and the quantitative results (I). (J–K) CDKN3 inhibition regulated cisplatin-induced xenograft tumor volumes (J) and the quantitative results (K). **P<0.01, ***P<0.001. Abbreviations: EC109R, cisplatin-resistant EC109 cell line; OE19R, cisplatin-resistant OE19 cell line; NC, negative control; shCDKN3, cells transfected with shCDKN3 plasmid; IC50, half maximal inhibitory concentration. |
EC109R and OE19R cells were identified via CCK8 assay as cisplatin-resistant ESCA cell lines, compared with their parental cells (Figure 4B and C). We discovered that CDKN3 mRNA is higher in resistant cells than their parental cells (Figure 4D). Next, drug-sensitivity assay was utilized to verify the contribution of CDKN3 to cisplatin resistance in ESCA by measuring the effect of CDKN3 on chemosensitivity. Western blot assays were used to assure the knockdown performance of shCDKN3 in EC109R and OE19R (Figure 4E). CDKN3 knockdown significantly decreased the IC50 over twofold in EC109R and OE19R cells (Figure 4F and G, Table 1). Similarly, the cisplatin-induced apoptotic rate of EC109R or OE19R cells with shCDKN3 was higher than that of the NC and BLANK cells (Figure 4H and I). Xenograft models were established to verify the correlation of CDKN3 and cisplatin resistance in vivo. Knockdown of CDKN3 improved cisplatin chemosensitivity in both ESCC and EA compared with the controls (Figure 4J and K).
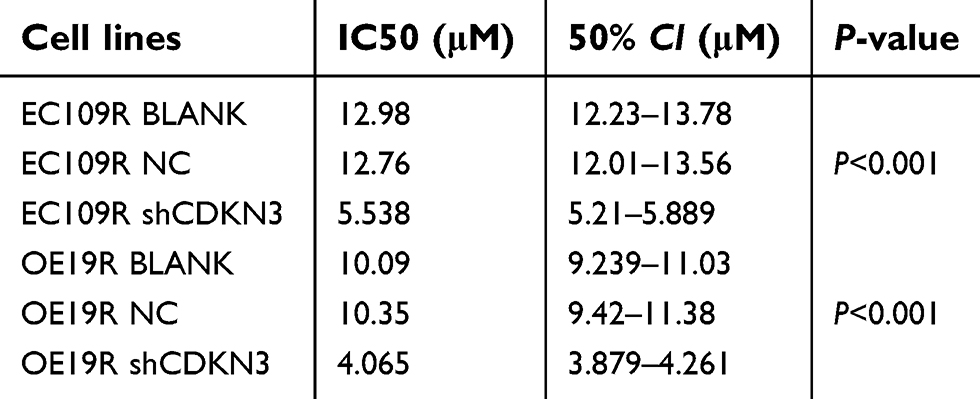 | Table 1 CDKN3 knockdown decreased the IC50 in EC109R and OE19R cells |
These results provided evidence of the correlation between CDKN3 overexpression and resistance to cisplatin in ESCA and that CDKN3 inhibition can restore cisplatin sensitivity in cisplatin-resistant ESCA cells.
CDKN3 interacted with DDR related protein RAD51 in ESCA
We next sought to elucidate the mechanism of CDKN3 involvement in CDKN3-mediated cisplatin resistance of ESCA. While there are numerous pathways involved in cisplatin resistance, the best understood mode of action includes increased efflux, reduced uptake, and enhanced DDR. To examine the effect of CDKN3 on DNA damage, we examined the expression of DNA damage marker γH2AX. CDKN3 inhibition was able to increase γH2AX foci counts greatly induced by cisplatin (Figure 5A and B). Next, we examined the expression of CTR1, MRP2, ERCC1, and RAD51 proteins, which play important roles in cisplatin efflux, uptake of cisplatin, and DDR, respectively. As shown in Figure 5C, CDKN3 alteration had no effect on the expression of CTR1 and MRP2, while CDKN3 inhibition reduced the expression of ERCC1 and RAD51 significantly. To further investigate the mechanism by which CDKN3 affects chemoresistance in ESCA, we screened for genes co-expressed with CDKN3 in the TCGA database. Unexpectedly, we found a positive correlation between the expression of CDKN3 and RAD51 mRNA (Pearson r=0.5419, P<0.001; Figure 5D). We hypothesized that RAD51 may be a downstream target of CDKN3. To determine the specific binding between CDKN3 and RAD51, we next performed CoIP assays in EC109R and OE19R cells. We extracted the total protein and performed immunoprecipitation using anti-CDKN3 or anti-RAD51 antibodies, followed by western blot analysis. The results indicated that endogenous CDKN3 could interact with RAD51 in both EC109R cells (Figure 5E) and OE19R cells (Figure 5F). The next step was to assess whether CDKN3 facilitated chemoresistance via RAD51.
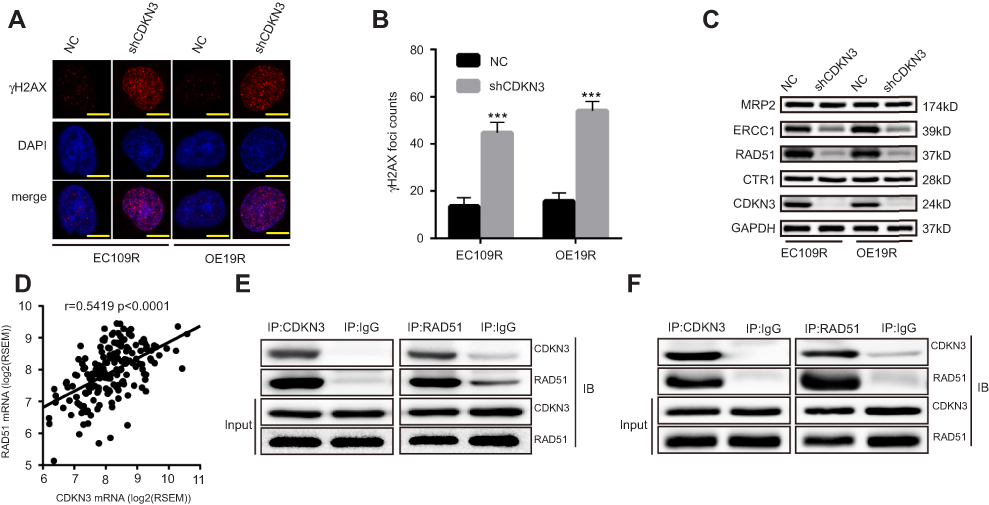 | Figure 5 The interaction between CDKN3 and RAD51 in ESCA. (A–B) CDKN3-regulated cisplatin-induced γH2AX foci (A) and quantitative results (B) in ESCA. Scale bar=5 μm. (C) CDKN3 knockdown regulated cisplatin chemoresistance-related proteins. (D) Correlation between CDKN3 and RAD51 mRNA expression in TCGA ESCA dataset. (E–F) Interaction between endogenous CDKN3 and RAD51 in EC109R (E) and OE19R (F) cell lines by Co‑IP. ***P<0.001. Abbreviations: EC109R, cisplatin-resistant EC109 cell line; OE19R, cisplatin-resistant OE19 cell line; NC, negative control; shCDKN3, cells transfected with shCDKN3 plasmid; RSEM, RNA-Seq by Expectation-Maximization. |
CDKN3 regulated chemosensitivity of cisplatin resistant ESCA by RAD51
To further elucidate the function of RAD51 in CDKN3-mediated cisplatin resistance, we examined a series of cellular effects in CDKN3 inhibited cells followed by overexpression of RAD51. RAD51 protein overexpression in CDKN3 inhibited cisplatin-resistant EC109 (EC109R/shCDKN3) and OE19 (OE19R/shCDKN3) cells was confirmed by western blot analysis (Figure 6A). Following RAD51 overexpression, cell viability increased to approximately 5 μM cisplatin in both EC109R/shCDKN3 cells and OE19R/shCDKN3 cells, compared with the NC group (Figure 6B and C, Table 2). Cisplatin-induced apoptotic cells were significantly reduced in RAD51 overexpression EC109R/shCDKN3 cells and OE19R/shCDKN3 cells (Figure 6D and E), which were similar to those of the untreated EC109R and OE19R cells. Meanwhile, we determined that cisplatin-induced γH2AX foci was significantly reduced after rescuing RAD51 expression, compared with the NC group (Figure 6F and G). These results indicated that ectopic RAD51 expression can reverse cisplatin resistance, which can be regulated by inhibition of CDKN3. Collectively, CDKN3 regulated chemosensitivity by DDR related protein RAD51 in both ESCC and EA.
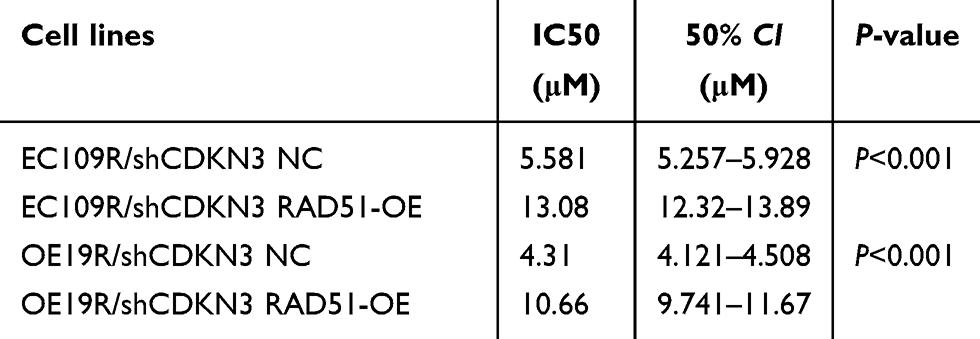 | Table 2 RAD51 overexpression reversed chemosensitivity in CDKN3 inhibited EC109R and OE19R cells |
 | Figure 6 CDKN3-regulated cisplatin chemoresistance by RAD51. (A) The results of western blot analyses demonstrated the effective ectopic expression of Rad51 in EC109R/shCDKN3 cells and OE19R/shCDKN3 cells. (B–C) Cell viability of EC109R/shCDKN3 cells (B) and OE19R/shCDKN3 cells (C) with or without RAD51 ectopic expression. (D–E) Apoptosis rate of EC109R/shCDKN3 cells and OE19R/shCDKN3 cells with or without RAD51 overexpression (D) and quantitative results (E). (F–G) γH2AX foci counts of EC109R/shCDKN3 cells and OE19R/shCDKN3 cells with or without RAD51 overexpression (F) and quantitative results (G). Scale bar=5 μm. ***P<0.001. Abbreviations: NC, negative control; RAD51-OE, RAD51 overexpression; EC109R/shCDKN3, CDKN3 inhibited cisplatin-resistant EC109 cells; OE19R/shCDKN3, CDKN3 inhibited cisplatin-resistant OE19 cells; IC50, half maximal inhibitory concentration. |
Discussion
In the last ten years, the incidence ranking of ESCA has increased from the ninth to the seventh of all malignant tumors.1 The standard treatment for ESCA consists of adjuvant chemoradiotherapy combined with surgery. There are numerous recommended chemotherapy regimens such as platinum, which is highly utilized. Unfortunately, most patients will experience the pain of platinum resistance after a short period of relief.2 Resistance to platinum-based chemoradiotherapy is a major reason for treatment failure in ESCA and the mechanism needs elucidation.
The CDKN3 gene encodes a dual-specificity protein phosphatase that used to be considered a tumor suppressor by controlling mitosis through CDK1/CDK2. CDKN3 is responsible for the dephosphorylation of Thr-161/Thr-160 in CDK1/CDK2 and thus prevents CDK1/CDK2 activation that occurs via phosphorylation.3 However, recent studies have shown that CDKN3 can promote tumorigenesis and cancer progression, but the mechanism remains unclear. Nalepa. et al discovered that CDK1 inactivation mediates mitotic exit and that CDKN3 inhibits residual CDK1 activity to ensure normal mitotic exit.13 Srinivas V’s study showed that CDKN3 is essential for maintaining the appropriate number of centrosomes by regulating the centrosomal stability of MPS1. Therefore, CDKN3 inhibition can result in spindle assembly checkpoint failure and inhibition of cell growth.14 These findings indicate that CDKN3 has a positive role in regulating cell proliferation, which is consistent with our conclusion that CDKN3 promotes the progression of ESCA through cell cycle regulation.
Overexpression of CDKN3 has been found in numerous human cancers,4,5,6 but the mechanism of CDKN3 overexpression has not been clear. Chau-Ting Yeh discovered that multiple forms of CDKN3 mutants and aberrant transcripts were present in hepatocellular cancerous tissues, but not in hepatitis tissues.15 The same research team found that truncated CDKN3 mutant prevents CDKN3 from exerting its inhibitory effect on CDK2,16 which suggested that dominant-negative CDKN3 mutants could interfere with normal CDKN3 function and inversely increase CDKN3 expression. However, subsequent studies have failed to find any evidence of CDKN3 mutation in hepatocellular carcinoma.17 Studies have shown that CDKN3 gene amplification is limited to 2% of lung adenocarcinomas. Thus, it is unlikely for CDKN3 overexpression to be caused by gene mutation or amplification in human cancer.17 FAN’s study suggested that CDKN3 protein expression was increased in M phase and overexpression of CDKN3 may be attributed to the fact that more cancer cells are undergoing mitosis.18 In our study, we found that CDKN3 was upregulated in ESCA and analyzed the whole genomic profile of the TCGA database through cbioportal.19 However, the CDKN3 mutation was not found in ESCA. CDKN3 copy number variations accounted for 0.54%, and gene amplification accounted for 1.08% in ESCA (data not shown). It is gratifying that our study discovered an association between CDKN3 hypomethylation and CDKN3 mRNA expression (Pearson r=−0.39263, P<0.0001). These results provide a new impetus towards studying the cause of CDKN3 overexpression in ESCA.
CDKN3 has been associated with a poor prognosis in various cancers. The specific mechanism regarding the impact of CDKN3 overexpression on patient survival necessitates further exploration. Most studies have shown that CDKN3 overexpression is associated with the cell cycle, DNA replication, et al, thereby promoting tumor progression.4,6 Our study also showed that CDKN3 plays an important role in the proliferation of ESCA cells. CDKN3 overexpression promoted ESCA progression by increasing the proportion of time spent in the G2/M phase of ESCA cells. Previous studies have shown that high expression of CDKN3 can promote tumor invasion and metastasis;5,17 however, we didn’t observe a clear correlation between high CDKN3 expression and metastasis of ESCA. To our surprise, we found that the expression of CDNK3 has a positive correlation with the chemoresistance of ESCA patients. We are the first to make this finding. In the cisplatin-resistant ESCC and EA cell lines, we also observed a significant increase in CDKN3 expression. Furthermore, functional experiments indicated that CDKN3 inhibition can recover cisplatin sensitivity of cisplatin-resistant ESCA cells, accompanied by the increase of γH2AX foci counts. In addition, CDKN3 interacted with RAD51 in ESCA. RAD51 was noted to be recruited to sites of DNA damage to mediate repair and appears to be related to chemotherapy resistance.7 RAD51 overexpression can increase cell viability, decrease the rate of cell apoptosis, lower DNA damage in CDKN3 inhibited cisplatin-resistant ESCA cells. These data suggested that CDKN3 impacted chemosensitivity of ESCA through RAD51, which is a DDR related protein.
Our study demonstrated, for the first time, that CDKN3 was highly expressed in both ESCC and EA. CDKN3 was an independent indicator for ESCA prognosis. CDKN3 promoted tumor progression and facilitated chemoresistance by RAD51 in ESCA (Figure 7). Taken together, we demonstrated the role of CDKN3 in regulating ESCA progression and chemosensitivity, indicating that CDKN3 is a potential target in ESCA and should be further studied. The shortcomings of this research include the unclear mechanism of CDKN3 regulation of RAD51.
 | Figure 7 CDKN3 promoted esophageal cancer progression and enhanced cisplatin resistance through RAD51. |
Conclusion
In summary, we have demonstrated that CDKN3 played a critical role in the progression and chemoresistance of ESCA. CDKN3 has the potential to become a new target for cancer therapy and to overcome chemotherapy resistance in ESCA.
Acknowledgments
We appreciated technical supports provided by teachers of the Shandong Qianfoshan Hospital medical research center.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Lagergren J, Smyth E, Cunningham D, Lagergren P. Oesophageal cancer. Lancet. 2017;390(10110):2383–2396. doi:10.1016/S0140-6736(17)31462-9
3. Poon RY, Hunter T. Dephosphorylation of Cdk2 Thr160 by the cyclin-dependent kinase-interacting phosphatase KAP in the absence of cyclin. Science. 1995;270(5233):90–93. doi:10.1126/science.270.5233.90
4. Yu C, Cao H, He X, et al. Cyclin-dependent kinase inhibitor 3 (CDKN3) plays a critical role in prostate cancer via regulating cell cycle and DNA replication signaling. Biomed Pharmacother. 2017;96:1109–1118. doi:10.1016/j.biopha.2017.11.112
5. Li Y, Ji S, Fu LY, Jiang T, Wu D, Meng FD. Knockdown of cyclin-dependent kinase inhibitor 3 inhibits proliferation and invasion in human gastric cancer cells. Oncol Res. 2017;25(5):721–731. doi:10.3727/096504016X14772375848616
6. Chang SL, Chen TJ, Lee YE, Lee SW, Lin LC, He HL. CDKN3 expression is an independent prognostic factor and associated with advanced tumor stage in nasopharyngeal carcinoma. Int J Med Sci. 2018;15(10):992–998. doi:10.7150/ijms.25065
7. Gachechiladze M, Skarda J, Soltermann A, Joerger M. RAD51 as a potential surrogate marker for DNA repair capacity in solid malignancies. Int J Cancer. 2017;141(7):1286–1294. doi:10.1002/ijc.30764
8. Liu YB, Mei Y, Tian ZW, Long J, Luo CH, Zhou HH. Downregulation of RIF1 enhances sensitivity to platinum-based chemotherapy in epithelial ovarian cancer (EOC) by regulating nucleotide excision repair (NER) pathway. Cell Physiol Biochem. 2018;46(5):1971–1984. doi:10.1159/000489418
9. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
10. Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10(21):7252–7259. doi:10.1158/1078-0432.CCR-04-0713
11. Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46(D1):D956–d963. doi:10.1093/nar/gkx1090
12. Sakthivel KM, Hariharan S. Regulatory players of DNA damage repair mechanisms: role in cancer chemoresistance. Biomed Pharmacother. 2017;93:1238–1245. doi:10.1016/j.biopha.2017.07.035
13. Nalepa G, Barnholtz-Sloan J, Enzor R, et al. The tumor suppressor CDKN3 controls mitosis. J Cell Biol. 2013;201(7):997–1012. doi:10.1083/jcb.201205125
14. Srinivas V, Kitagawa M, Wong J, Liao PJ, Lee SH. The tumor suppressor Cdkn3 is required for maintaining the proper number of centrosomes by regulating the centrosomal stability of Mps1. Cell Rep. 2015;13(8):1569–1577. doi:10.1016/j.celrep.2015.10.039
15. Yeh CT, Lu SC, Chen TC, Peng CY, Liaw YF. Aberrant transcripts of the cyclin-dependent kinase-associated protein phosphatase in hepatocellular carcinoma. Cancer Res. 2000;60(17):4697–4700.
16. Yeh CT, Lu SC, Chao CH, Chao ML. Abolishment of the interaction between cyclin-dependent kinase 2 and Cdk-associated protein phosphatase by a truncated KAP mutant. Biochem Biophys Res Commun. 2003;305(2):311–314.
17. Fan C, Chen L, Huang Q, et al. Overexpression of major CDKN3 transcripts is associated with poor survival in lung adenocarcinoma. Br J Cancer. 2015;113(12):1735–1743. doi:10.1038/bjc.2015.378
18. Cress WD, Yu P, Wu J. Expression and alternative splicing of the cyclin-dependent kinase inhibitor-3 gene in human cancer. Int J Biochem Cell Biol. 2017;91(Pt B):98–101. doi:10.1016/j.biocel.2017.05.013
19. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.